
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Covalent non-fused tetrathiafulvalene–acceptor systems
Flavia
Pop†
* and
Narcis
Avarvari
*
Université d'Angers, CNRS, Laboratoire MOLTECH-Anjou, UMR 6200, UFR Sciences, Bât. K, 2 Bd. Lavoisier, 49045 Angers, France. E-mail: narcis.avarvari@univ-angers.fr
First published on 25th April 2016
Abstract
Covalent donor–acceptor (D–A) systems have significantly contributed to the development of many organic materials and to molecular electronics. Tetrathiafulvalene (TTF) represents one of the most widely studied donor precursors and has been incorporated into the structure of many D–A derivatives with the objective of obtaining redox control and modulation of the intramolecular charge transfer (ICT), in order to address switchable emissive systems and to take advantage of its propensity to form regular stacks in the solid state. In this review, we focus on the main families of non-fused TTF–acceptors, which are classified according to the nature of the acceptor: nitrogen-containing heterocycles, BODIPY, perylenes and electron poor unsaturated hydrocarbons, as well as radical acceptors. We describe herein the most representative members of each family with a brief mention of their synthesis and a special focus on their D–A characteristics. Special attention is given to ICT and its modulation, fluorescence quenching and switching, photoconductivity, bistability and spin distribution by discussing and comparing spectroscopic and electrochemical features, photophysical properties, solid-state properties and theoretical calculations.
 Flavia Pop | Flavia Pop obtained her PhD in 2009 (Babeş-Bolyai University, Cluj-Napoca, Romania and University of Angers, France) under the joint supervision of Prof. I. Grosu and Dr J. Roncali. She continued her research with a postdoc position in the field of molecular materials (with Dr N. Avarvari), focusing on tetrathiafulvalene-based chiral conducting materials, covalent donor–acceptor systems and chiral supramolecular aggregates. Flavia is currently a postdoctoral researcher working on chiral chromophores and their aggregation for active materials in solar cells (with Prof. D. B. Amabilino, The University of Nottingham, UK). |
 Narcis Avarvari | Narcis Avarvari was born in Romania. He received his PhD in Chemistry in 1998 at the Ecole Polytechnique, France. After one-year postdoctoral stay at the ETH Zürich, he obtained in 1999 a permanent research position with the CNRS in Nantes, and then he moved in 2001 to the University of Angers, laboratory MOLTECH-Anjou. He was awarded the 2007 Prize of the French Chemical Society, Coordination Chemistry Division. In 2010, he was promoted to CNRS director of research. He is currently heading a research team dealing with molecular materials, crystal engineering and coordination chemistry, as well as self-assembly and chirality. |
1. Introduction
Covalent donor–acceptor systems have long attracted significant interest due to their involvement in electron transfer processes in biology1–3 and as they constitute valuable precursors for many organic materials, with applications in fields such as photovoltaics, nonlinear optics and molecular electronics.4 One of the most important electron donors, especially in the field of molecular materials, is the tetrathiafulvalene (TTF) unit,5 which has provided many organic metals and superconductors6 and, more recently, multifunctional materials combining conductivity and magnetism,7 conductivity and chirality,8 or based on luminescent single molecule magnets9 or electroactive ligands and metal complexes.10 TTF has also been associated with electron poor units and thus has been explored in donor–acceptor (D–A) systems. The manifold interest in TTF–acceptor systems is primarily at the stage of fundamental research, as in such compounds there is, for example, the possibility to modulate the intramolecular charge transfer with the oxidation state of TTF, which can afford electrochromic and/or tuneable emission properties, or can allow obtaining peculiar architectures in the solid state with either segregation or alternation of the donors and acceptors.11 The interest in the latter feature is related to the possibility of observing charge separation and photoconductivity,12 and hence has potential in the fields of photovoltaics13,14 or molecular electronics.11,15 However, the first highlighted potential application of a covalent donor–acceptor derivative was the molecular rectifier,16 theoretically postulated by Aviram and Ratner in a hypothetical system consisting of a TTF donor and a tetracyanoquinodimethane (TCNQ) acceptor separated by a σ-bridge (Scheme 1).17 Because of some synthesis issues, it took more than twenty years after this theoretical prediction for covalently linked TTF–TCNQ or TTF–TCNAQ (TCNAQ = tetracyanoanthraquinodimethane) compounds to be described and properly characterized. Some of the first such examples consist of TTF–TCNAQ systems 2 and derivatives, as reported by Bryce et al.,18 and 3, as reported by Liu et al.,19 while in compound 4, as described by Martin et al.,20 TTF was replaced by extended-TTF (ext-TTF) and a conjugated linkage between the two units was preserved. Both flexible 5 and rigid 6 TTF–σ–TCNQ systems possessing small HOMO–LUMO gaps were described by Bryce and Perepichka et al.21 and Khodorkovsky et al.,22 respectively. Finally, Rovira et al. reported the first fully conjugated TCNQ–TTF–TCNQ 7,23 in which the radical anion obtained by a one-electron reduction was a class II mixed valence species thanks to communication through the TTF bridge.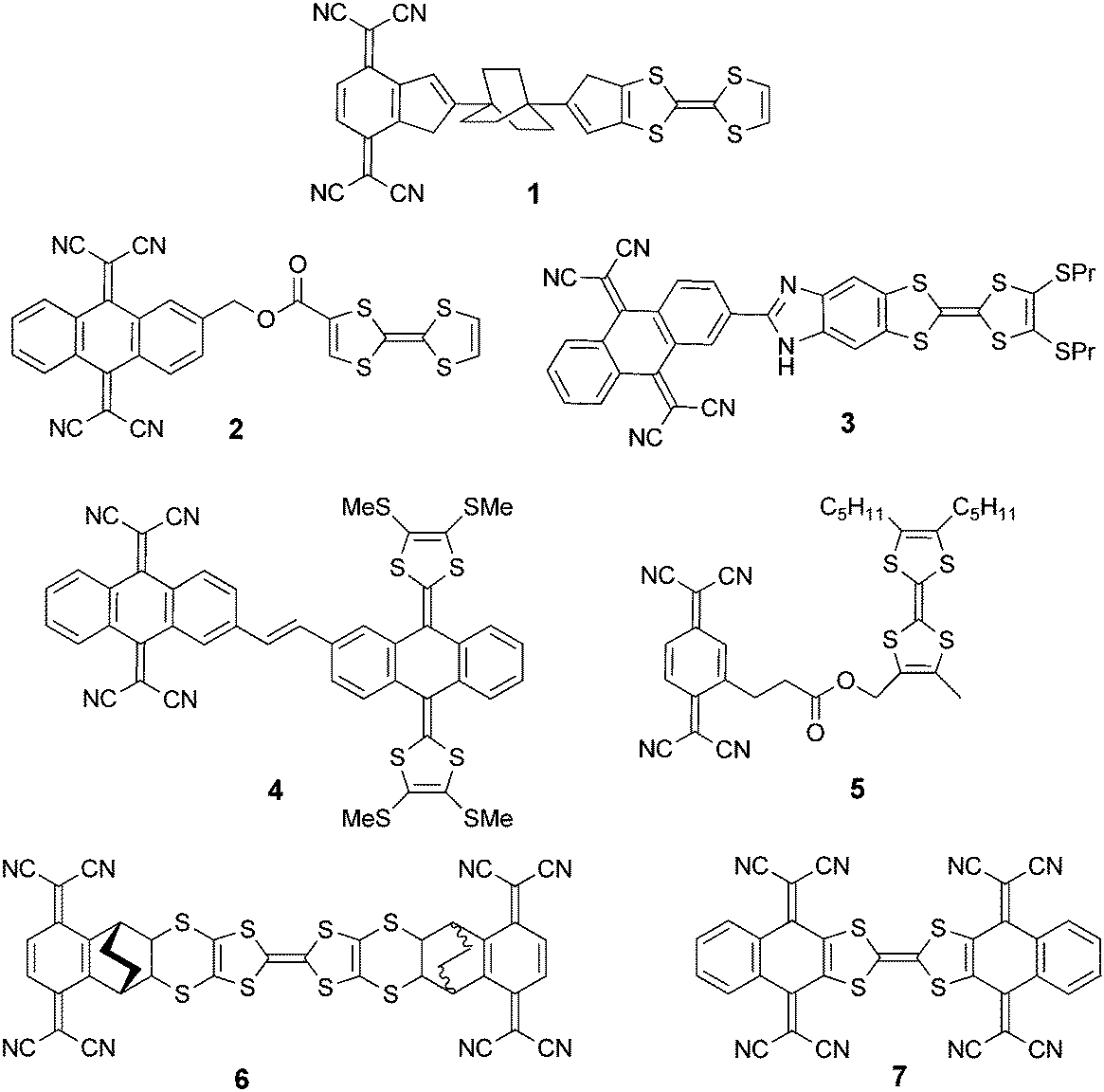 | ||
| Scheme 1 Aviram and Ratner's theoretical molecular rectifier 1 and experimental systems based on TTF and TCNQ units. | ||
Across this short historical saga of TTF–TCNQ, we wanted to point out a very illustrative scientific process, originating from a hypothesis that evolved towards other numerous other directions, constituting the molecular electronics field,24 and motivated an enormous amount of experimental work25 that eventually provided several covalent TTF–TCNQ systems. Although molecular rectification was not reported in any of the latter reports, their synthesis and investigations stimulated along the years many attempts at the covalent association of TTF and ext-TTF units to a large diversity of acceptors within fused and non-fused derivatives.11,26–28 Remarkably, inspired by Aviram and Ratner's prediction, Bryce, Perepichka and colleagues prepared a TTF–σ–trinitrofluorene system29 showing molecular rectification behaviour.30 TTF–acceptor systems published before 2004 were reviewed by Bendikov, Wudl and Perepichka,11 while the more recent reviews by Martín et al.27 and Liu et al.28 deal with ext-TTF–acceptors and fused TTF–acceptors. In this review, we focus on several recent representative families of non-fused covalently linked TTF–acceptors, classified according to the chemical nature of the acceptor, with an aim to highlight some of their most peculiar features.
2. Nitrogen-containing six-membered ring acceptors
The most representative unit of the nitrogen based six-membered rings is pyridine (Py), which has been attached to TTF in various ways (Scheme 2). Simple pyridines are poor electron acceptors and therefore they are not expected to favour massive intramolecular charge transfer when attached to TTF units. This was confirmed by the relatively high energy charge transfer bands at λ = 400–440 nm (Table 1), indicative of large HOMOTTF–LUMOA gaps, which were observed in the directly attached TTF–Py 8a–b,318c32 and Py–TTF–Py 9,33 and in derivatives with ethenyl or ethynyl bridges, such as mono(Py)–TTF 1034,35 and 1136 or bis(Py)–TTF 12a35 and 12b.37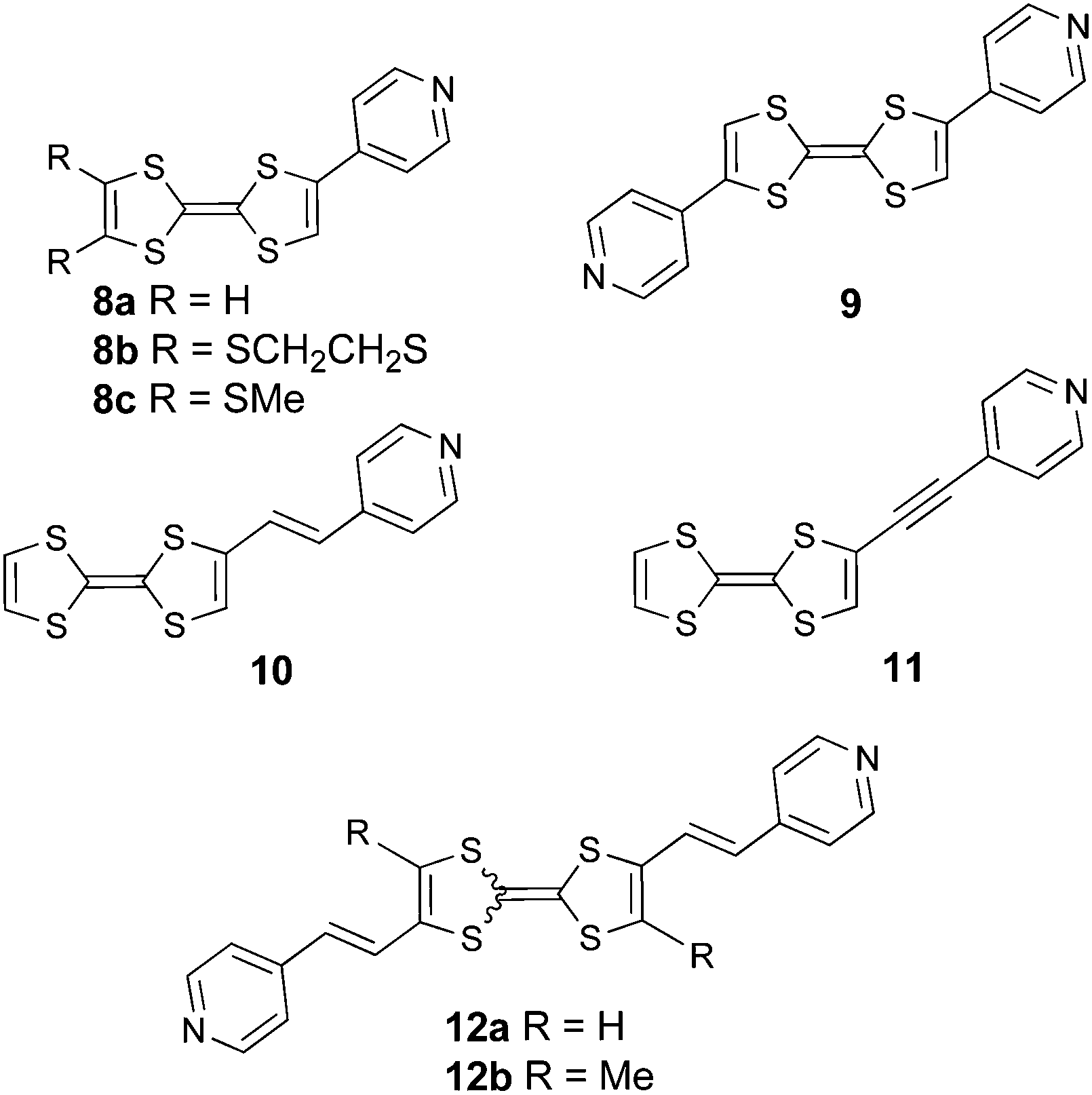 | ||
| Scheme 2 TTF–pyridines showing the modulation of ICT upon protonation and/or coordination. | ||
| Compound | E 11/2,ox (V vs. SCE) | E 21/2,ox (V vs. SCE) | λ max ICT (nm) | ΔEHOMO–LUMO (eV) |
|---|---|---|---|---|
| a In CH3CN. b In CH2Cl2/CH3CN 1/1. c In CH2Cl2. d B3LYP/6-31G(d) with PCM. e B3LYP/6-31G. f B3LYP/6-31G(d). g B3LYP/6-31G(d,p). | ||||
| 8a | 0.439 | 0.777 | 428a | 3.253d |
| 8a-H+ | 0.529 | 0.827 | 574a | 2.150d |
| 8b | 0.529 | 0.804 | 414a | 3.389d |
| 8b-H+ | 0.603 | 0.851 | 551a | 2.332d |
| 8c | 0.494 | 0.797 | 425b | 3.529e |
| 8c-H+ | 0.571 | 0.797 | 561b | 1.445e |
| 9 | 0.508 | 0.835 | 403a | 3.03e |
| 9-H+ | 0.679 | 0.918 | 539a | 1.43e |
| 10 | 0.440 | 0.805 | 444a | 2.858f |
| 10·Pb2+ | 0.510 | 0.885 | 555a | |
| 11 | 0.484 | 0.839 | 432a | 2.898f |
| 11·Pb2+ | 0.520 | 0.879 | 550a | |
| 12a | 0.578 | 0.945 | 440a | |
| 12a·Pb2+ | 0.695 | 0.980 | 540a | |
| 12b | 0.583 | 1.049 | 450c | 2.9g |
The position of these bands slightly shifts with the polarity of the solvent and the connectivity of pyridine in the ortho, meta or para position, with the latter generally showing the strongest effect. The corresponding transitions are assigned to HOMO → LUMO excitations, with the HOMO clearly based on TTF, while the LUMO spans over the Py units, with a non-negligible contribution from the TTF half bearing the acceptor (Fig. 1 for 8a–b). The calculated HOMO–LUMO gaps depend on the basis set and the inclusion or not of a solvation model (Table 1) and correlate more or less with the experimental values.
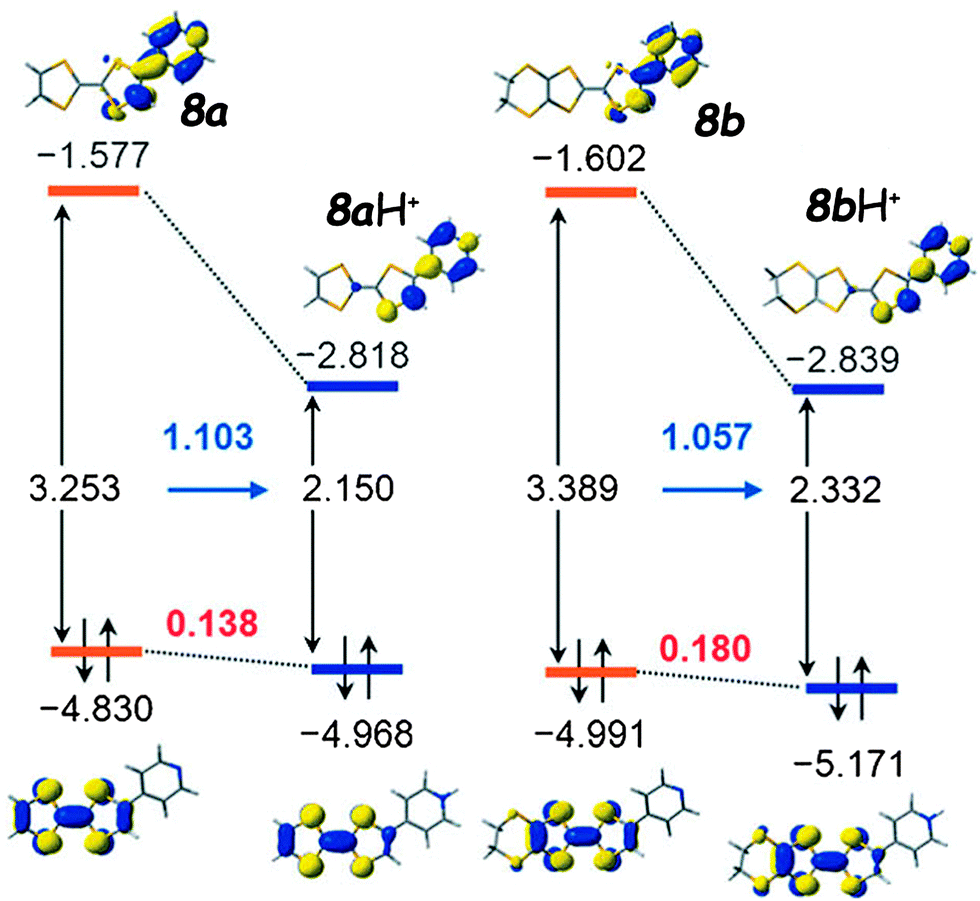 | ||
| Fig. 1 HOMO and LUMO orbitals with the associated gaps for the TTF–pyridines 8a (left) and 8b (right) together with their protonated forms. Adapted with permission from ref. 31b; Copyright 2014, John Wiley and Sons. | ||
The optimized structures show co-planarity between the TTF and Py units, thus allowing conjugation and a more efficient charge transfer. When conjugated spacers are introduced, as in 10 and 11, the theoretical HOMO–LUMO gap and the maximum of the CT band differ very little when comparing the ethenyl and ethynyl bridges (Table 1 and Fig. 2).
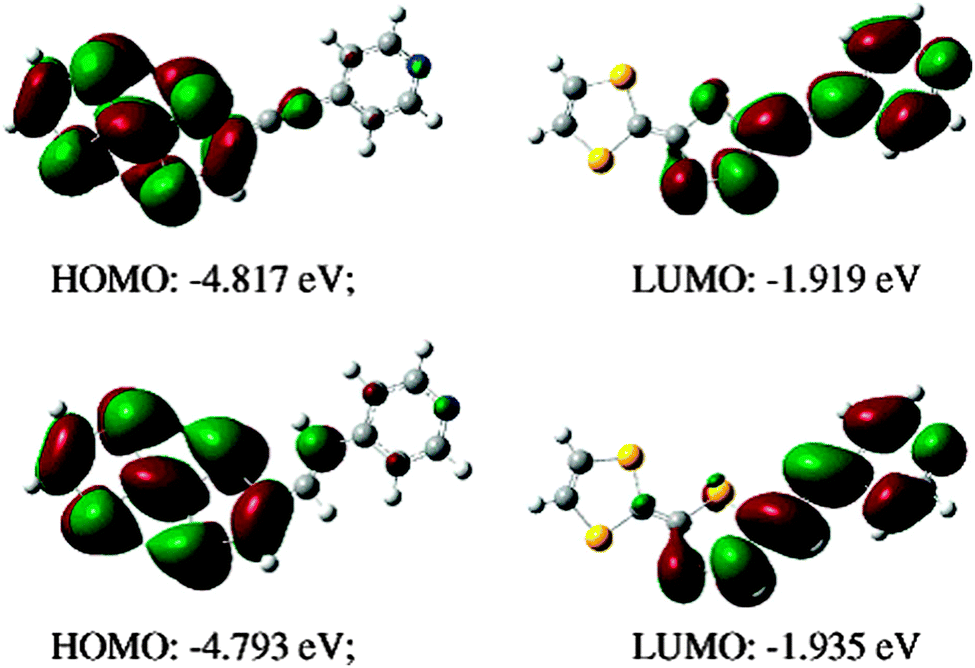 | ||
| Fig. 2 HOMO and LUMO orbitals for the TTF–pyridines 10 (top) and 11 (bottom). Reprinted with permission from ref. 36; Copyright 2007, American Chemical Society. | ||
Cyclic voltammetry measurements show the classical pair of reversible oxidation waves at potentials anodically shifted with respect to the unsubstituted TTFs (Table 1). However, the common feature of these systems is the massive bathochromic shift (∼135–150 nm) of the ICT band upon protonation, mainly as a consequence of strongly decreasing the energy of LUMO, which is now mostly developed over the pyridinium unit (Fig. 1). For example, the titration of 8c with p-toluene–sulfonic acid resulted in a red-shift of 136 nm for the ICT (Fig. 3),32 which is in agreement with the much smaller calculated HOMO–LUMO gap, although the latter seems underestimated (Table 1) when compared to the experimental value. More accurate values were obtained for 8a–b using a solvation model.31b
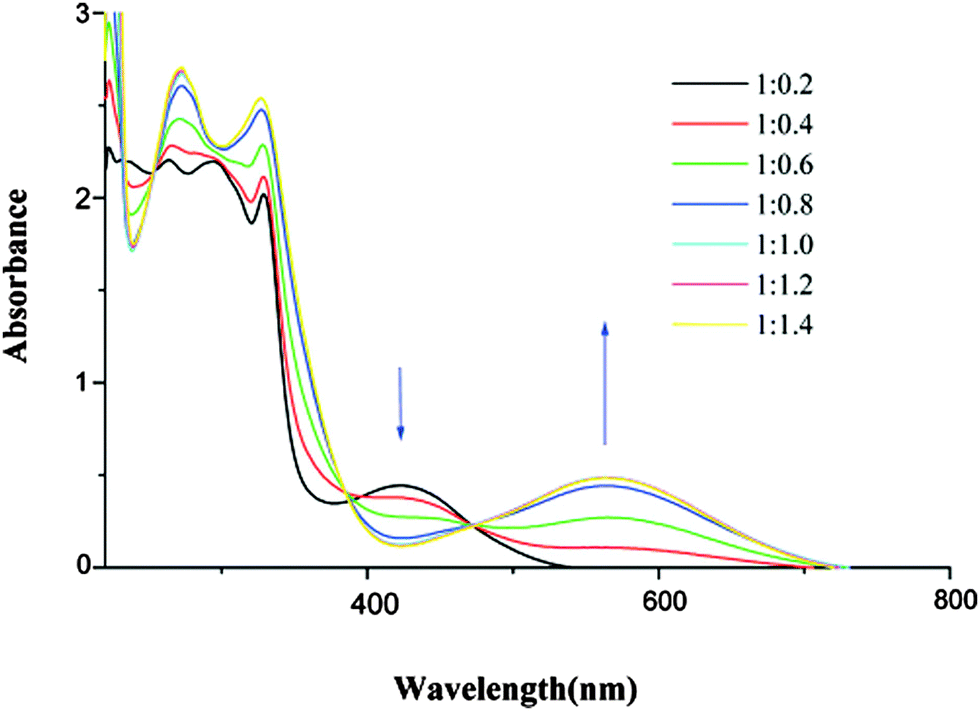 | ||
| Fig. 3 Protonation of 8c (10−4 mol L−1 in CH2Cl2/CH3CN 1/1) with increasing concentration of p-toluene–sulfonic acid (molar ratio). Adapted with permission from ref. 32; Copyright 2007, American Chemical Society. | ||
Interestingly, compounds 10, 11 and 12a proved to be very efficient for the selective colorimetric detection of Pb2+, showing bathochromic shifts of 70, 36 and 117 nm, respectively (Table 1 and Fig. 4 for 10), while much weaker variation was observed for other transition metal cations, such as Zn2+, Cd2+ or Ag+.35,36
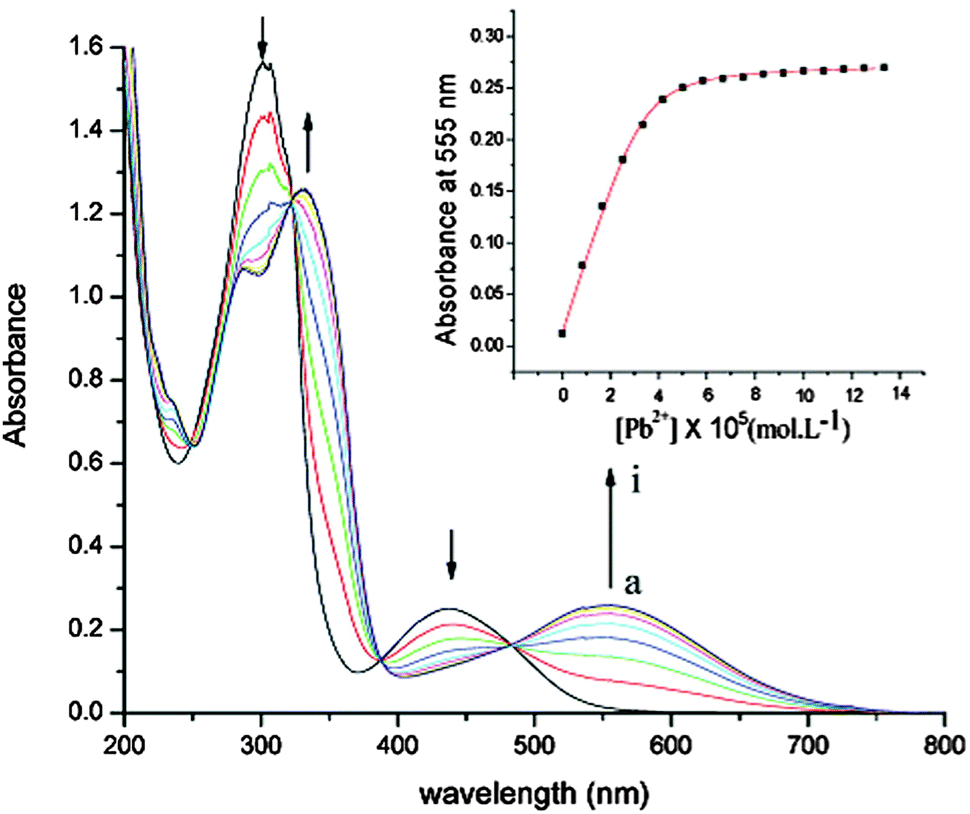 | ||
| Fig. 4 Variation of absorption spectra of 10 (5.2 × 10−5 mol dm−3 in CH3CN, 0.1 mol dm−3 (nBu4N)PF6) with the addition of Pb(ClO4)2; [Pb2+]: a to i, 0 to 6.6 × 10−5 mol dm−3; inset: absorbance at 555 nm against [Pb2+]. Adapted with permission from ref. 35; Copyright 2005, American Chemical Society. | ||
When the pyridine unit is connected to TTF via an electron withdrawing linker, such as an amide in 1338 and 14,39 an imine in 1640 or an azine in 1741 (Scheme 3), the ICT is due to the linker rather than the Py, as the LUMO is mainly based on the former, albeit with some participation of the latter, and the band is generally red-shifted when compared to the directly linked compounds previously discussed.
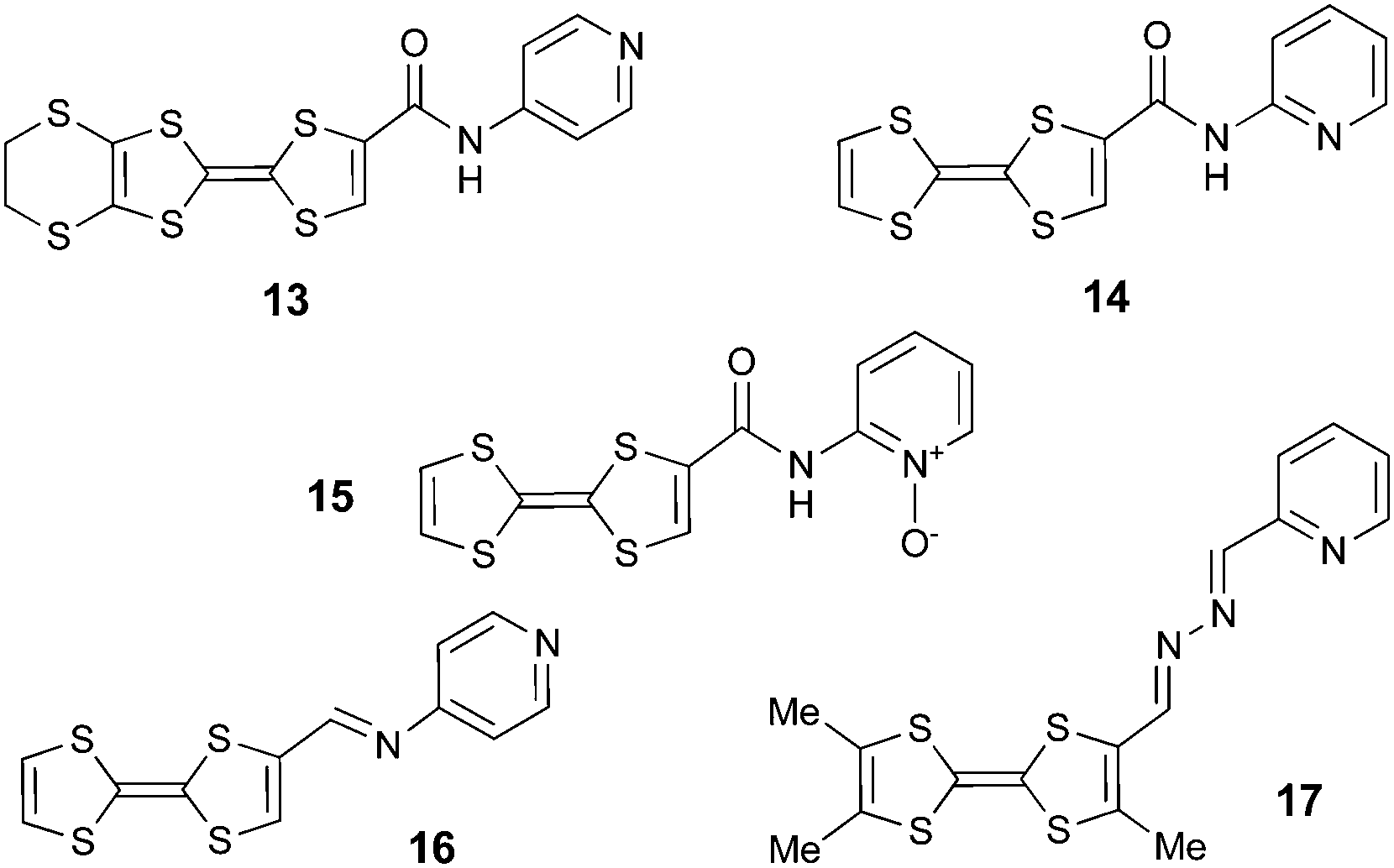 | ||
| Scheme 3 TTF–pyridines with electron withdrawing bridges. | ||
The replacement of Py by Py-oxide in 15 provokes a slight bathochromic shift of the ICT, supported by DFT calculations,39 due to a lowering of the LUMO level (Fig. 5). Note that TTF–Py-oxide 15 was successfully used as an antenna ligand for the sensitization of Yb(III) luminescence through charge transfer.42
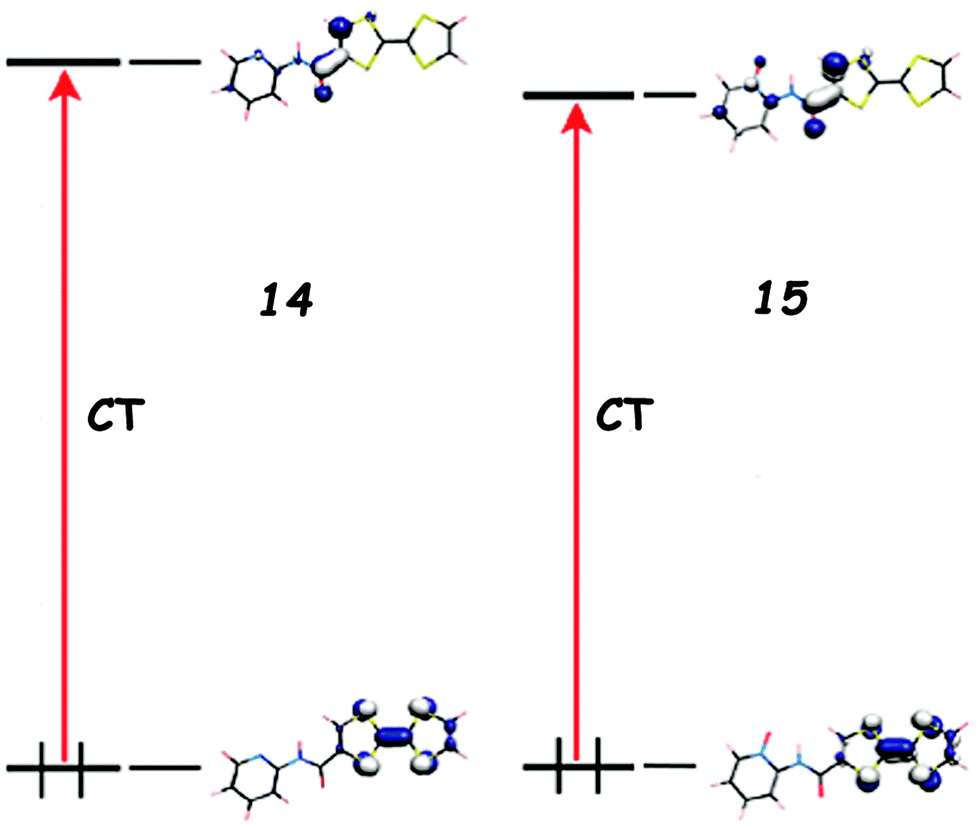 | ||
| Fig. 5 Frontier orbitals for 14 and 15 and the corresponding ICT transitions computed at λ = 468 nm and 489 nm, respectively. Adapted with permission from ref. 39; Copyright 2010, American Chemical Society. | ||
The first oxidation potentials are sensitive to the additional substituents, with an anodic shift for 13 because of the ethylenedithio bridge, a trend also observed in the cases of 8a and 8b. On the contrary, the methyl groups in 17 induce a cathodic shift, and thus the formation of the radical cation species takes place at a lower potential (Table 2).
The solid-state structure of compound 13 shows the formation of stacks, with molecules disposed in a head-to-tail fashion and with alternation of the donors and acceptors, where the mean plane to plane distance is 3.7 Å (Fig. 6).38 These intermolecular D–A interactions seem to be the driving force for the crystallization of 13, together with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯HCvinyl intermolecular hydrogen bonding, while the NPy atom does not engage in any H bonding. A similar type of architecture was observed in the structure of the meta isomer of 13, where the D–A alternation favoured an eclipsed arrangement of the TTF outer CC double bonds bearing the amide-Py substituent. Single crystal irradiation of this isomer led to the formation of a cyclobutane unit by a fully regio- and stereospecific [2+2] cycloaddition.43
O⋯HCvinyl intermolecular hydrogen bonding, while the NPy atom does not engage in any H bonding. A similar type of architecture was observed in the structure of the meta isomer of 13, where the D–A alternation favoured an eclipsed arrangement of the TTF outer CC double bonds bearing the amide-Py substituent. Single crystal irradiation of this isomer led to the formation of a cyclobutane unit by a fully regio- and stereospecific [2+2] cycloaddition.43
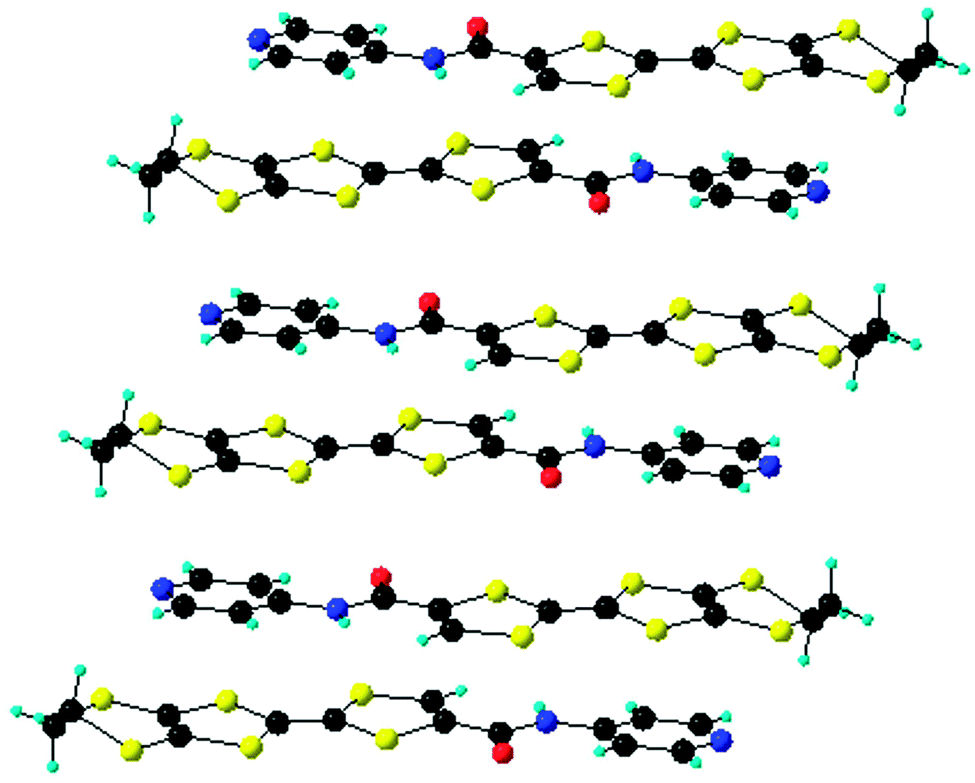 | ||
| Fig. 6 Head-to-tail stack of molecules 13 in the solid state, with D–A alternation. | ||
Triazines are better electron acceptors than pyridines as the electron acceptor ability of the heterocycle increases with the number of N atoms. Indeed, 1,3,5-triazines can be reduced between −1.1 and −2 V vs. SCE, depending on the substituents, yet the process is hardly reversible (Scheme 4).44
 | ||
| Scheme 4 TTF-1,3,5-triazines (TTF-TZ). | ||
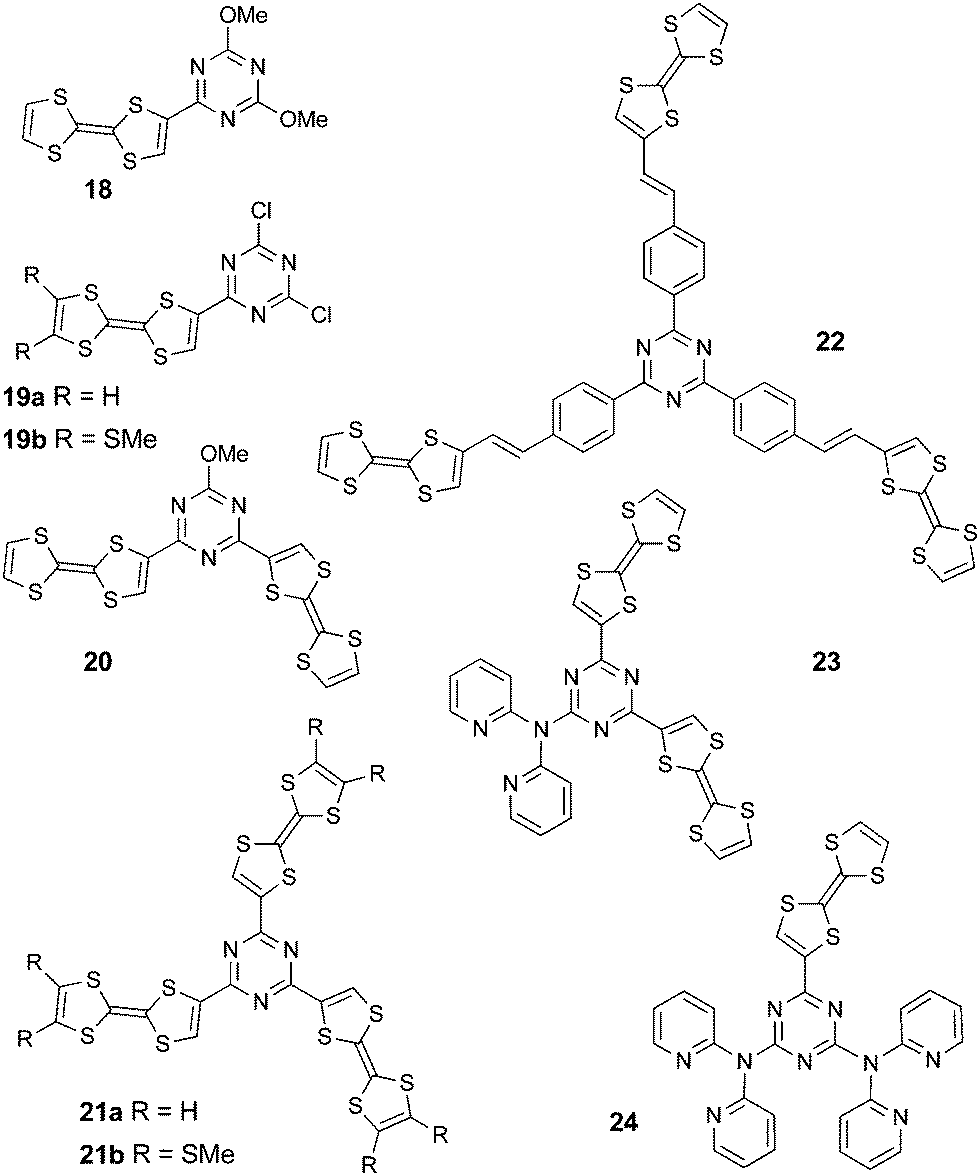
The interest in this C3 symmetric platform, which has been used in recent years in the structure of a number of optoelectronic materials,45 also relies on its facile functionalization by stepwise nucleophilic substitution starting from cyanuric chloride.46 Accordingly, the reaction of methoxy-chloro-triazines (MeO-Cl-TZ) with the TTF anion allowed the preparation of the mixed methoxy-TTF-TZ 18 and 20 in relatively low yields, i.e. 11% and 21%, respectively, while the tris(TTF)-TZ 21a has only been produced in traces.47 Alternatively, the Stille-type coupling reaction between the MeO-Cl-TZ precursors and TTF–SnMe3 provided 18, 20 and 21a in much higher yields (75%, 78% and 30%, respectively).48 Additionally, the TTF-TZ-Cl2 derivatives 19a–b and tris(TTF)-TZ 21b were prepared by the same strategy,48 while the tris(TTF)-TZ 22, with conjugated spacers between TTF and the central platform, was obtained by a Wittig–Horner olefination reaction49 from tris(phosphonate)-TZ and TTF-aldehyde. As 1,3,5-triazines constitute attractive building blocks for the preparation of mono, di or tritopic ligands,50 dipyridylamine (dpa) units have been introduced together with TTF into the electroactive ligands (TTF)2-TZ-dpa 23 and TTF-TZ-(dpa)224.51
When considering their oxidation potentials (Table 3), the donor character is clearly diminished compared to pristine TTF, especially in compounds 18–21 and 23–24 with direct linkages between the TTF and TZ units, because of the electron withdrawing effect of the latter. Moreover, the broadened first oxidation process of bis(TTF) 20 and tris(TTF) 21b suggests an electronic communication between TTFs across the triazine platform. The reduction potentials vary in a larger range, with the most anodic being those for 19a–b, which contain Cl substituents. However, in all cases, the reduction process was irreversible.
| Compound | E 11/2,ox (V) | E 21/2,ox (V) | E 1red (V) | λ max ICT (nm) | ΔEHOMO–LUMO (eV) |
|---|---|---|---|---|---|
a In THF vs. SCE.
b In THF.
c DFT/PBE0/6-311++G(3df,2pd).
d In MeCN/CH2Cl2 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vs. SCE.
e In CH2Cl2.
f DFT/B3LYP/6-31+G(d).
g In CHCl3.
h In THF vs. Ag/AgNO3.
i In MeCN.
j DFT/PBE0/6-311+G(2df,pd). :1 vs. SCE.
e In CH2Cl2.
f DFT/B3LYP/6-31+G(d).
g In CHCl3.
h In THF vs. Ag/AgNO3.
i In MeCN.
j DFT/PBE0/6-311+G(2df,pd).
|
|||||
| 18 | 0.41a | 0.79a | −1.98a | 492b | 3.03c |
| 19a | 0.54d | 0.96d | −1.15d | 592e | 2.51c |
| 19b | 0.60d | 0.92d | −1.18d | 571e | 2.66c |
| 20 | 0.43a | 0.76a | −1.65a | 530b | 2.42f |
| 21a | 2.61c | ||||
| 21b | 0.59a | 0.87a | −1.33a | 552g | |
| 22 | 0.240h | 0.532h | −1.853h | 461g | |
| 23 | 0.45d | 0.89d | −1.43d | 504i | 2.74j |
| 24 | 0.50d | 0.95d | −1.75d | 473i | 3.06j |
ICT bands ranging from 473 nm to 592 nm observed for the directly connected compounds were related to the values of the HOMO–LUMO gaps, as calculated by DFT (Table 3). For example, in the series 18, 19b and 19a, where the same level of theory was employed, the decrease in the theoretical gaps is in agreement with the red-shift of the ICT transition (Table 3). This band is strongly blue-shifted in 22 when compared to 21b as a consequence of the separation between the donor and acceptor units in the former. Upon chemical or electrochemical oxidation, the ICT bands gradually disappear to the profit of the typical bands of TTF+˙. The variation is particularly easily monitored for compound 19b, where the two bands of TTF+˙ centred at ν = 23040 cm−1 (434 nm) and ν = 12625 cm−1 (792 nm) are well separated from the initial ICT band at 571 nm (Fig. 7).48
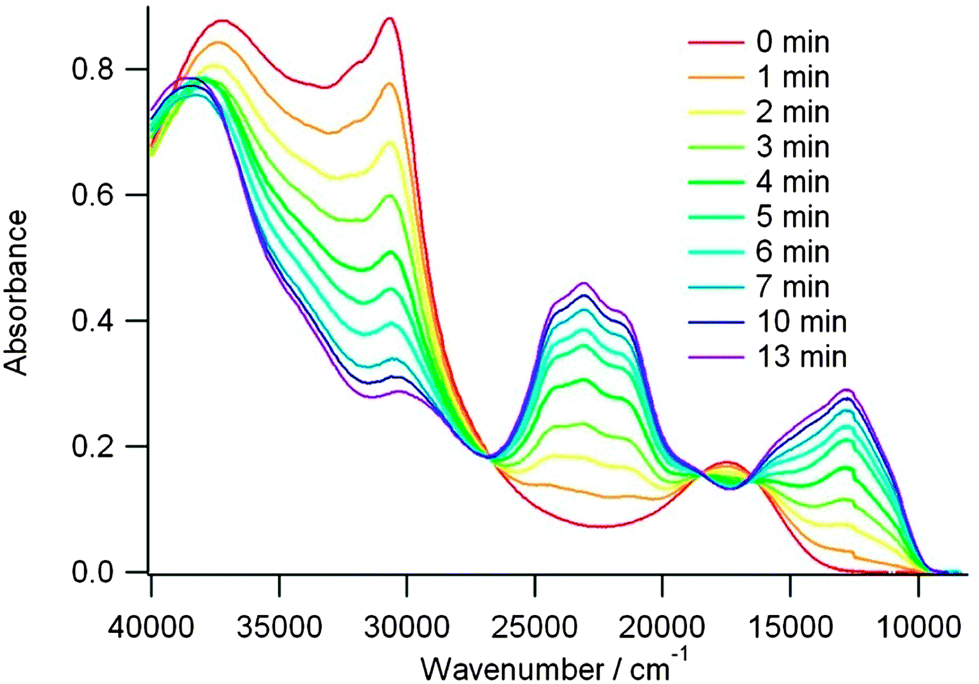 | ||
| Fig. 7 UV-vis absorption spectra of 19b (7.6 × 10−4 M) upon electrochemical oxidation (0.69 V vs. SCE, DCM at room temperature, cell length: 0.7 mm). Reprinted with permission from ref. 48; Copyright 2013, American Chemical Society. | ||
The case of tris(TTF)-TZ 21b is very interesting as upon the gradual addition of FeCl3, a band appears at 2000 nm, reaches a maximum for one equivalent of oxidant, then it decreases, while the intensity of the other TTF+˙ centred bands continue to increase (Fig. 8). This near IR band was assigned to an intervalence transition and indicates the occurrence of mixed valence species.52 The use of NOBF4 as an oxidant produced similar variations, but the oxidized species were less stable.
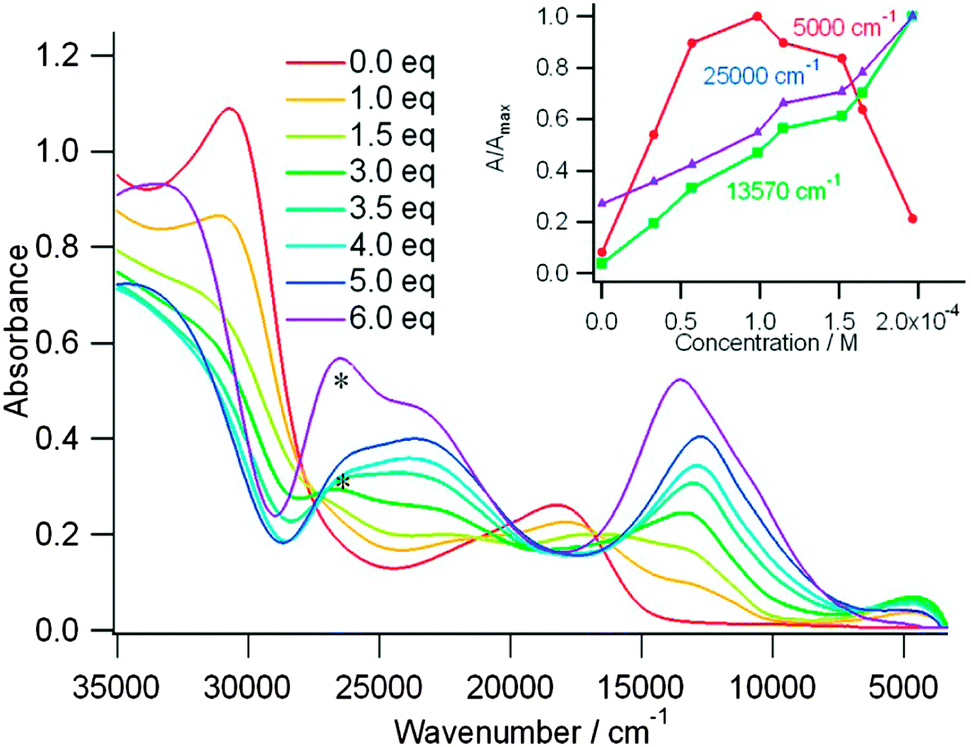 | ||
| Fig. 8 UV-vis absorption spectra of 21b (3.3 × 10−5 M) during chemical oxidation by addition of the oxidant FeCl3 in CHCl3 at room temperature. Inset: Absorbance changes at different wavenumbers. “*” represents unreacted FeCl3. Reprinted with permission from ref. 48; Copyright 2013, American Chemical Society. | ||
Interestingly, compound 21b is poorly emissive when excited in the ICT band in chloroform solution, with the emission band centred at 766 nm (Fig. 9), and its oxidized species are not luminescent, since the ICT transition has been suppressed. Moreover, when considering their multiple broad absorption bands (Fig. 8), a quenching of any luminescence by energy transfer is likely to occur.
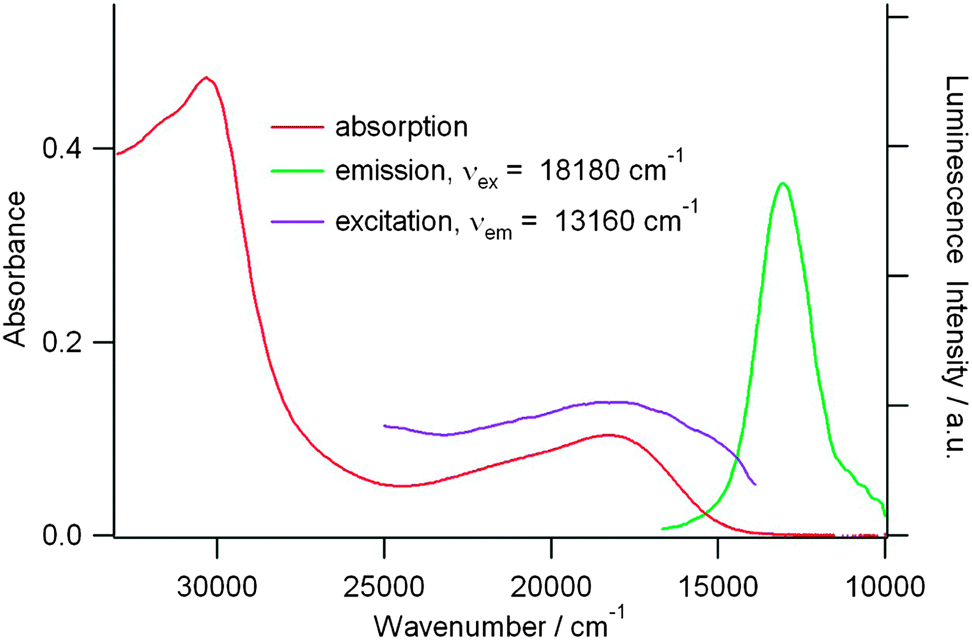 | ||
| Fig. 9 Absorption, emission, and excitation spectra of 21b (9.0 × 10−6 M, CHCl3, room temperature). Reprinted with permission from ref. 48; Copyright 2013, American Chemical Society. | ||
Note that apart from 21b, no other TTF-triazine derivative has been found to be luminescent.
The solid-state structures of 18, 19a–b, 20, 23 and 24 show co-planarity between the TTF units and triazine, with a segregation of the donors and acceptors in the packing of 18,47 while for all the other compounds, alternating D⋯A stacks are observed.48,51
Compounds 23 and 24 contain one or two dipyridylamine (dpa) chelating units and were therefore investigated as ligands towards Zn(II) with either ZnCl2 or Zn(ClO4)2 precursors. With ZnCl2, neutral mono or bimetallic complexes were isolated and structurally characterized, while with the perchlorate salt, a dimeric bimetallic tetracationic complex crystallized. In the latter, the TZ(dpa)2 platforms acted as bridges between the metal centres, which were each coordinated by four pyridines from two dpa units and two water molecules in a octahedral geometry.51 The most peculiar feature was the involvement of two ClO4− anions in π–anion interactions with the triazine rings, as attested by the short distance of 3.00 Å between one oxygen atom (O1) and the triazine centroid (Fig. 10).
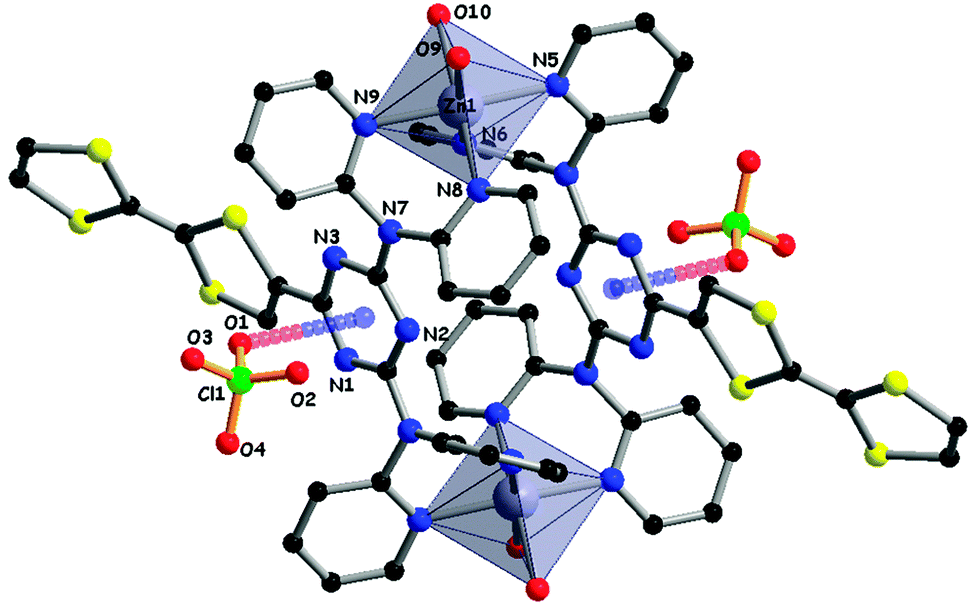 | ||
| Fig. 10 Dimeric structure of (24) Zn(ClO4)2 in the solid state, with an emphasis on the ClO4−⋯TZ anion–π interactions. H atoms and additional ClO4− anions have been omitted for clarity. | ||
Anion–π interactions have been recognized and increasingly investigated over the last decade as important motifs in crystal engineering53 and supramolecular chemistry.54
Finally, the strongest electron acceptor of the nitrogen six-membered series is the tetrazine unit.55 Besides their reversible reduction into radical anions at relatively easy accessible potentials,56 due to the low lying π* LUMO, many 1,2,4,5-tetrazine derivatives show fluorescence, arising from a symmetry forbidden n–π* transition.57 Nevertheless, only two TTF-tetrazine compounds have been described to date, namely TTF-tetrazine-Cl 25 and TTF-tetrazine-dipicolylamine 26, synthesized from 3,6-dichlorotetrazine (TTZ-Cl2) by a Stille-type coupling with TTF–SnMe3 (Scheme 5).58
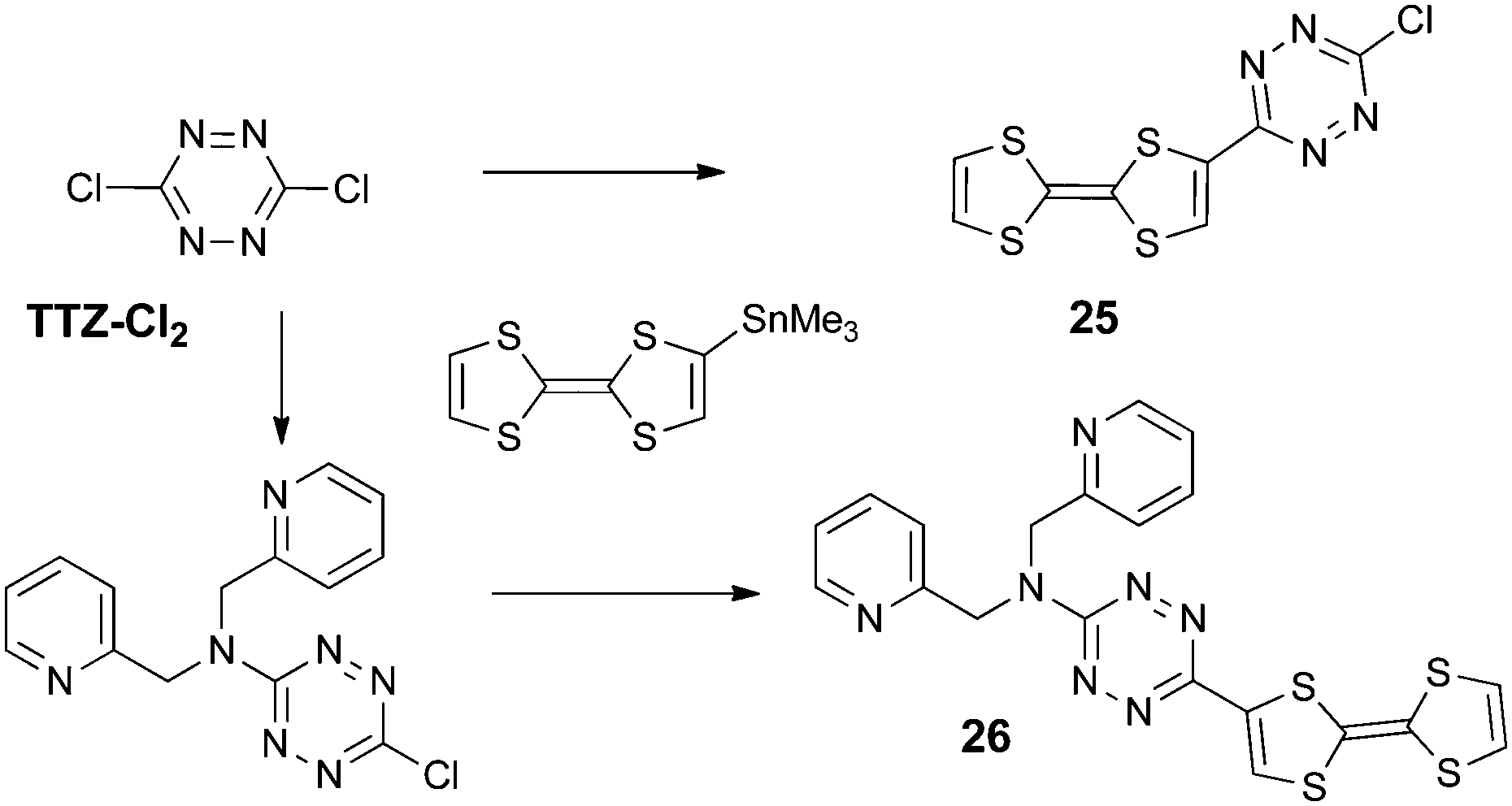 | ||
| Scheme 5 Synthesis of TTF-tetrazines. | ||
Contrary to the TTF-triazine derivatives, TTF-TTZ 25 and 26 show, beside a pair of one-electron oxidation processes at +0.50 and +0.93 V vs. SCE, a reversible reduction of the acceptor into a radical anion at −0.45 and −0.93 V vs. SCE, respectively. The cathodic shift in the latter is imputable to the electron releasing amine substituent. Theoretical calculations on 25 were in agreement with a low lying π* LUMO (−3.363 eV) located on the acceptor and with a TTF-based HOMO of the π-type (Fig. 11). The n type orbital is only the HOMO−2 (Fig. 11), whereas in fluorescent tetrazine derivatives, it corresponds to the HOMO.
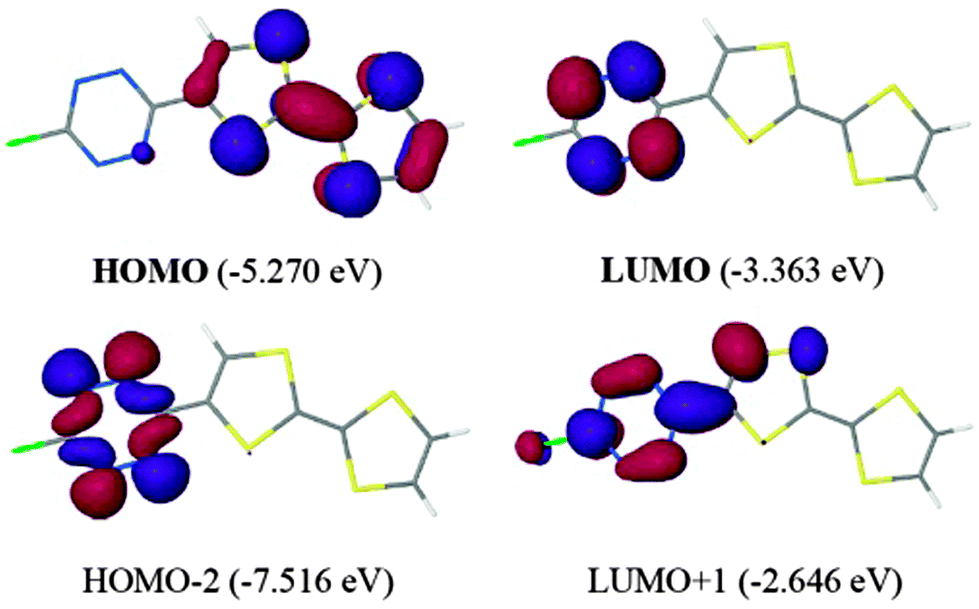 | ||
| Fig. 11 Frontier orbitals of TTF-TTZ 25. Reproduced from ref. 58 by permission of The Royal Society of Chemistry. | ||
TD DFT calculations indicated that the lowest energy singlet transition occurs at 10388 cm−1 (963 nm), being an S0 → S1 excitation, corresponding to an ICT from TTF to TTZ. This calculated transition appears in the experimental UV-vis spectrum as a weakly intense tail centred at 12000 cm−1 (833 nm). Then, the more intense band at 526 nm is also an ICT transition from HOMO to LUMO+1, while the TTZ-based symmetry forbidden transition, calculated at 532 nm, is very likely hidden by the more intense, former band. As observed in other TTZ-donor systems, the fluorescence of the tetrazine unit is reductively quenched.59 Ligand 26 has been coordinated to a ZnCl2 fragment, and in the solid-state structure of the resulting neutral complex (26) ZnCl2, one of the Cl ligands interacts with the TTZ ring through an anion–π interaction, as previously discussed for triazine-based complexes.
3. Nitrogen-containing five-membered ring acceptors
Among the azole series, only imidazole (Im) and 1,2,3-triazole (Trz) rings have been directly connected to TTF units in non-fused dyads. Although these heterocycles are not particularly strong electron acceptors, their basicity permits the modulation of the ICT by protonation and alkylation. Moreover, they can coordinate transition metals, especially within chelating ligands. TTF-imidazoles 27 and 28, prepared by a Pd(II)-catalyzed Stille coupling of TTF–SnBu3 with iodo-imidazoles, were described by Morita et al.60 The same strategy allowed the preparation of TTF-purine nucleobases, such as TTF-adenine 29 and TTF-guanine 30 derivatives (Scheme 6).61 | ||
| Scheme 6 TTF-imidazoles and TTF-nucleobases. | ||
TTF-imidazoles are good electron donors, with the oxidation potentials anodically shifted for the TTF-nucleobases as a consequence of the stronger electron acceptor ability of the purine moiety (Table 4). The latter indeed show ICT bands at around 430 nm.61b TTF-Im 27 has been extensively exploited in combination with a number of diverse electron acceptors for the preparation of conducting charge transfer complexes, in which the intermolecular hydrogen bonding between imidazole and the acceptors directs the solid-state architectures and triggers the charge and proton transfer.62
| Compound | E 11/2,ox (V) | E 21/2,ox (V) | λ max ICT (nm) | ΔEHOMO–LUMO (eV) |
|---|---|---|---|---|
| a In DMF vs. SCE, (TBA)ClO4. b In THF. c In CH2Cl2. d In CH2Cl2/CH3CN, (TBA)PF6. e DFT/PBE0/6-311++G(3df,2pd). f In CH2Cl2, (TBA)PF6. | ||||
| 27 | 0.345a | 0.605a | ||
| 28 | 0.295a | 0.555a | ||
| 29 | 0.435a | 0.655a | 430b | |
| 30 | 0.405a | 0.635a | 430c | |
| 31 | 0.39d | 0.83d | 390;c 412e | 3.89e |
| 32 | 0.31d | 0.74d | 388;c 410e | 3.87e |
| 33 | 0.49d | 0.90d | 394;c 432e | 3.71e |
| 34 | 0.44d | 0.85d | 394;c 434e | 3.68e |
| 35 | 0.45f | 0.85f | 388c | |
| 36 | 0.57f | 0.96f | 415c | |
| 37 | 0.38e | 0.78e | 386c | |
| 38 | 0.34; 0.44e | 0.81e | 397c | |
TTF-1,2,3-triazoles (TTF-Trz) as 1,4- (31–32) and 1,5-isomers (33–34) have been synthesized by a click chemistry strategy, involving azide alkyne cycloaddition reactions from TTF-alkynes and benzyl azide, catalyzed either by Cu(I) or Ru(II) catalysts (Scheme 7).63
 | ||
| Scheme 7 TTF-1,2,3-triazoles. | ||
In compound 35, the benzyl substituent was replaced by n-butyl by using butyl azide, while the bis(Trz) derivative 36 was obtained starting from TTF-bis(alkyne).64 The chelating ligands Py-Trz 37 and Py(Trz)238 were prepared from the corresponding pyridine azides by the same Ru(II)-based cycloaddition strategy.65 The attachment position of TTF to the triazole ring is important, making the 1,4-isomers easier to oxidize than the 1,5-isomers (Table 4). Interestingly, bis(TTF-Trz)-Py 38 shows a split of 100 mV between the first and second oxidation, indicating a through space communication between the two TTF units. This peculiar feature can be nicely correlated with its solid-state structure, in which the molecule adopts a pincer-like shape with the two TTF units facing each other (Fig. 12). Intramolecular hydrogen bonding between the N pyridine atom and vinylic TTF protons may be at the origin of this conformation.
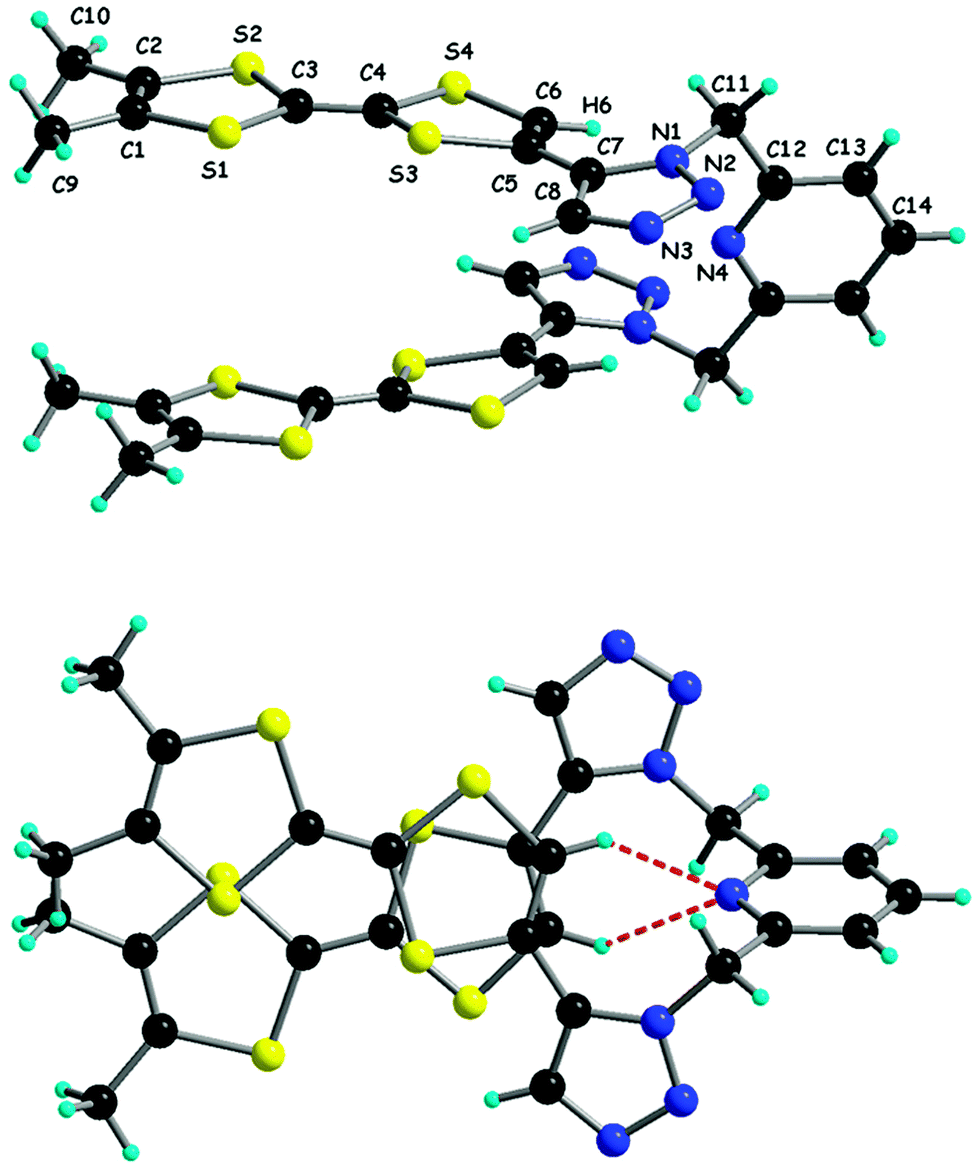 | ||
| Fig. 12 Solid-state conformation of 38, with an emphasis on the NPy⋯HTTF hydrogen bonds. Reproduced from ref. 65 by permission of The Royal Society of Chemistry. | ||
In the UV-vis spectra of all the TTF-Trz, rather high energy ICT bands are observed at 386–415 nm, and were nicely reproduced by TD DFT calculations on 31–34, indicating a moderate electron acceptor ability of the triazol ring (Table 4). On the other hand, DFT calculations on the same series clearly showed that the LUMOs, relatively high in energy, were only partially delocalized over the triazol rings.63 However, the basic properties of the sp2 N atoms allow for protonation and alkylation, thereby inducing a red-shift of the ICT band. For example, TTF-bis(Trz) 36 showed a first bathochromic shift of 14 nm when 1 equivalent of acid was added, followed by a second shift of 22 nm for the second equivalent of HBF4 (Fig. 13).
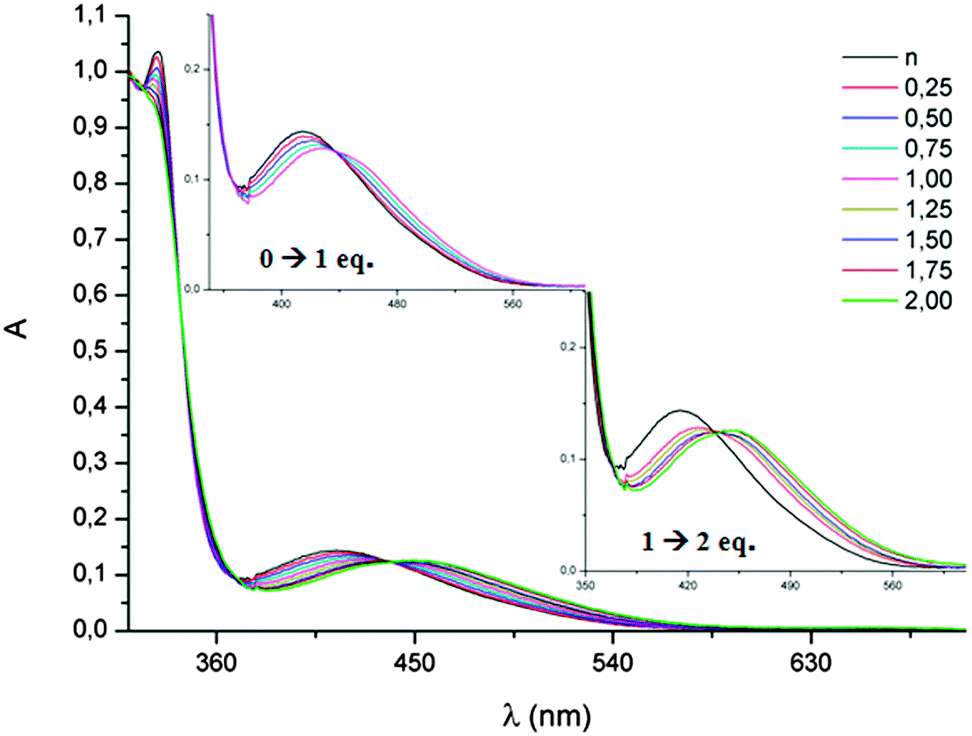 | ||
| Fig. 13 Protonation of 36 (10−4 mol L−1 in CH2Cl2) with the increasing concentration of HBF4; in inset, zoom in on the ICT band. Reproduced from ref. 64 by permission of The Royal Society of Chemistry. | ||
TTF-Trz and TTF-Trz–Py have proven to be suitable ligands towards Cu(II) (33–34),63 Co(II) (37) and Cd(II) (38) centres.65
Heteroazoles, such as benzothiazoles, benzothiadiazoles and oxadiazoles, are stronger electron acceptors compared to imidazoles and triazoles and, besides, many of their derivatives are fluorescent, thus motivating their association to TTF. Benzothiazoles 39 and 40 (Scheme 8) have both been prepared by Fujiwara et al. from TTF-aldehyde either by condensation with amino-thiophenol or by a Wittig coupling with a phosphonium salt.66
 | ||
| Scheme 8 TTF-benzothiazoles (TTF-BT) 39–40, TTF-benzothiadiazoles (TTF-BTD) 41–43 and TTF-rhodanine 44. | ||
Directly linked TTF-benzothiadiazoles (TTF-BTD) 41a–c and the bis-derivative 43 were synthesized by a Stille-type coupling between TTF–SnMe3 and BTD-Br2.67,68 Compounds 42a–b, with an alkyne bridge between the donor and acceptor, were prepared by Sonogashira coupling.68 While all the derivatives showed the two reversible TTF-based oxidation processes at potential values of 0.40–0.48 V vs. SCE for the generation of TTF+˙ species, reduction has only been reported for the BTD derivatives. Thus, formation of the radical anions of BTD-TTF occurs between −1.17 and −1.36 V vs. SCE (Table 5).
| Compound | E 11/2,ox (V) | E 21/2,ox (V) | E 1red (V) | λ max ICT (nm) | ΔEHOMO–LUMO (eV) |
|---|---|---|---|---|---|
| a In PhCN vs. SCE, (TBA)ClO4. b In CHCl3. c In CH2Cl2vs. SCE, (TBA)PF6. d In CH2Cl2. e DFT/PBE/aug-cc-pVTZ. f In THF vs. SCE, (TBA)PF6. g In THF. | |||||
| 39 | 0.48a | 0.88a | ∼460b | ||
| 40 | 0.46a | 0.83a | ∼490b | ||
| 41a | 0.42c | 0.83c | −1.19c | 565d | 2.07e |
| 41b | 0.45c | 0.82c | −1.21c | 551d | |
| 42a | 0.40c | 0.87c | −1.36c | 481d | |
| 42b | 0.42c | 0.88c | −1.19c | 501d | |
| 43 | 0.47f | 0.89f | −1.17f | 609g | |
Clearly, the BTD unit has a stronger electron acceptor character than BT, as evidenced by the red-shift of the ICT band (Table 5), but this feature has to be also correlated with the different attachment position of TTF to the acceptor and with the presence of a Br substituent on BTD in compounds 41a–b and 42b. Indeed, for compound 42a, with R = H, the ICT band is blue-shifted and the reduction potential is cathodically shifted when compared to 42b. Interestingly, compounds 41a–c show emission bands centred at λem = 734 nm (41a) and 715 nm (41b–c) (Fig. 14) upon irradiation at λex = 575 nm, in the 1ICT region.68
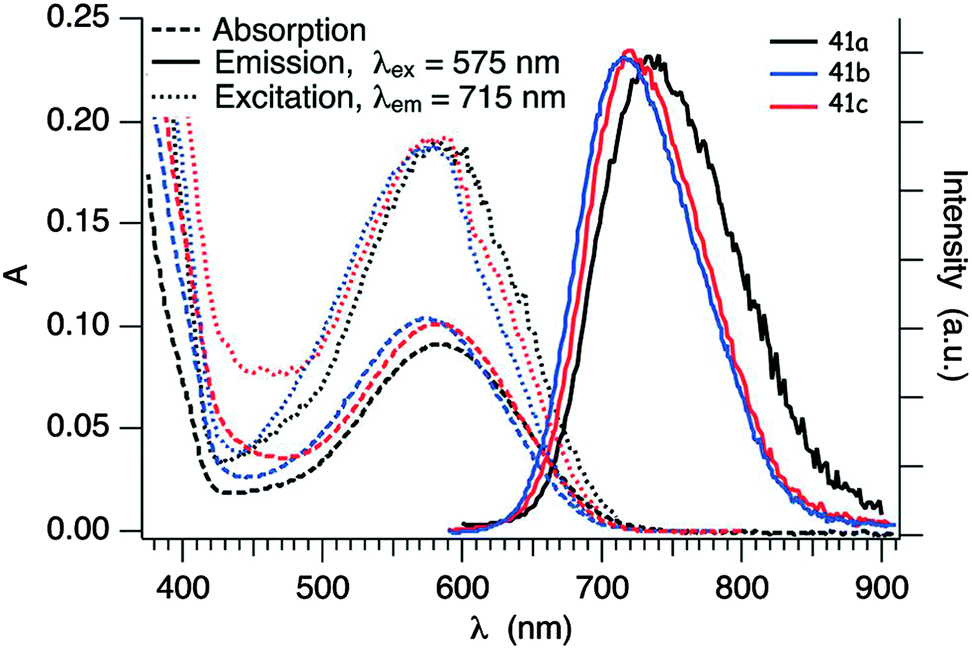 | ||
| Fig. 14 Absorption, emission and excitation spectra of 41a, 41b and 41c in degassed cyclohexane solution (c ≈ 2.5 × 10−5 M) in the region of the ICT transition at room temperature; λex = 575 nm for the emission spectra, λem = 715 nm for the excitation spectra. Adapted from ref. 68 by permission of The Royal Society of Chemistry. | ||
Insertion of an alkyne bridge in 42a, together with the different substitution of the BTD unit, provokes a massive blue-shift of the emission band to λem = 620 nm, besides that of the ICT band. The presence of the second TTF unit induces a self-quenching of the fluorescence of 43, which is not emissive in THF solution.68
The solid-state structures of 40, 41a and 42a show a similar pattern, with segregation of the donor and acceptor parts, a feature of much interest for photoconductivity. Indeed, single crystals of 40 show a slight increase in the current upon irradiation with white light.66 The packing observed in 41a is driven, beside by π–π stacking between the segregated D and A parts, by the formation of dyads through anti-parallel N⋯S contacts (Fig. 15),67 as commonly observed in the solid-state structures of the BTD derivatives.69
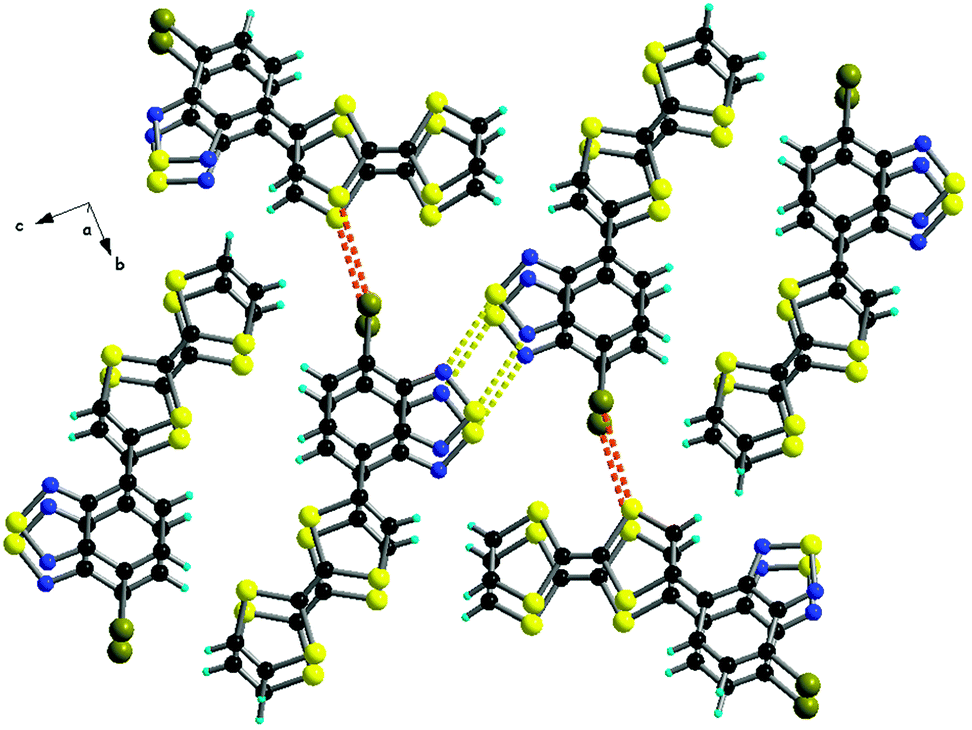 | ||
| Fig. 15 Packing along a in the solid-state structure of 41a, showing the D–A segregation, formation of N2S2 supramolecular dyads (yellow dotted lines) and short Br⋯S contacts (red dotted lines). Reprinted with permission from ref. 67; Copyright 2013, John Wiley and Sons. | ||
The coordinating ability of sp2 N atoms was put to work for TTF-BT 40 in a paramagnetic Cu(II)-based complex, which provided by electrocrystallization crystalline radical cation salts with either AsF6− or ReO4− anions.70 Rhodanine, which contains a thiazolidine cycle, has also been well used as an electron acceptor moiety in NLO active chromophores.71 Martín et al.72 described a TTF-rhodanine derivative (44) bearing a cyanomethylene group, which showed an ICT band at λmax = 498 nm, oxidation potentials comparable to those of pristine TTF and an irreversible reduction of the acceptor.
Fujiwara et al. attached TTF to 2,5-diphenyl-1,3,4-oxadiazole (PPD), a well-known fluorescent unit incorporated into electroluminescent devices,73 within the D–A dyads 45–46 (Scheme 9).
 | ||
| Scheme 9 TTF-oxadiazoles (TTF-PPD) 45–46. | ||
Compounds 45a–b have been prepared by Stille coupling between TTF–SnBu3 and PPD-Br precursors.74 Oxidation potential values of 0.43–0.44 and 0.83–0.84 V have been reported for the generation of TTF+˙ and TTF2+, respectively, together with ICT bands at λmax = 453 nm. These derivatives become emissive upon oxidation of TTF to the dicationic state TTF2+, with the corresponding emission band centred at 390 nm. The solid-state structure of 45a shows the formation of head-to-head dimers that further interact via the acceptor to afford segregation of the donor and acceptor parts (Fig. 16).
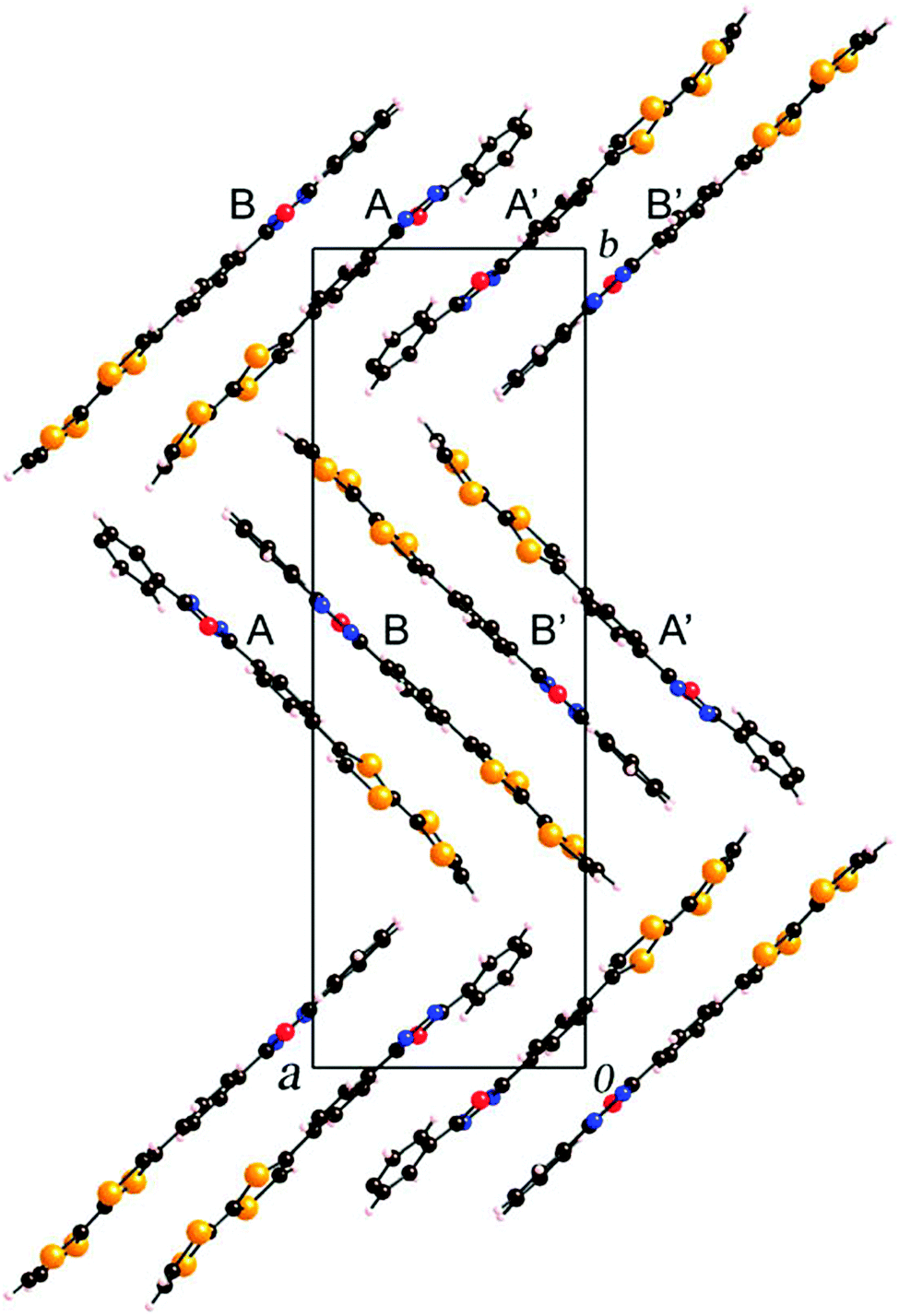 | ||
| Fig. 16 Packing in the solid-state structure of 45a. Reprinted with permission from ref. 74a; Copyright 2008, Elsevier. | ||
Consequently, photoconductivity measurements have been attempted on thin films of 45a. A cathodic photocurrent was observed, with a photo-electric conversion of 0.26%. Then, the spin dynamics of the photoexcited states of 45a and 46 were investigated by time-resolved electron spin resonance (TR-ESR) spectroscopy in frozen solutions.75 This showed that a triplet state T1 is populated via inter-system crossing (ISC) from the S1 state, without any indication of a charge separated (CS) state under those conditions.
4. BODIPY derivatives
Boron-dipyrromethene difluoride (BODIPY) is a very interesting unit that has been included in the structure of numerous compounds possessing fluorescent properties. BODIPY is a chemically robust unit with an intense absorption band at around 500 nm, originating from a π–π* transition, and a corresponding strong fluorescence emission with high quantum yields.76 Nevertheless BODIPY has only relatively recently been associated to TTF in compounds such as 47–50 (Scheme 10). Interestingly, depending on the linkage between the two units, variable ICT and fluorescence quenching are observed. | ||
| Scheme 10 TTF–BODIPY derivatives 47–50. | ||
Compound 47 was prepared by condensation between (MeS)2TTF-COCl and H2N-Ph-BODIPY.77 Its D–A character has not been specifically discussed, yet, the lack of conjugation between the two units because of the amide linker makes any ICT from TTF to BODIPY rather unlikely. However, upon the addition of fluoride ions, the emergence of a new broad band centred at 580 nm was observed. The association constant, for a 1:1 stoichiometry, determined through UV-vis titration reached a value of 1.2 × 103, thus demonstrating the very good affinity of 47 for fluoride binding. The new band at 580 nm might have its origin in a charge transfer from the electron enriched phenyl-amide moiety interacting with F− to the BODIPY unit. Interestingly, this compound shows modulation of the fluorescence quenching with the addition of fluoride. It has also been observed that most of the typical BODIPY emission was quenched by intramolecular photoinduced electron transfer (PET) from TTF to BODIPY. In the F− adduct, the efficiency of the PET was improved, possibly thanks to the stabilization of the hole on TTF, thus inducing a significant decrease in the emission.
As mentioned above, an excellent affinity and selectivity are observed for the fluoride anion, which engages, according to 1H NMR titration experiments and theoretical calculations, in N–H⋯F and C–H⋯F hydrogen bonding. In cyclic voltammetry, a cathodic shift of the oxidation waves accompanies the addition of fluoride (Table 6), as the positive charge on TTF tends to be stabilized.
| Compound | E 11/2,ox (V) | E 21/2,ox (V) | E 1red (V) | λ max (nm) BODIPY | ΔEHOMO–LUMO (eV) | λ em (nm) |
|---|---|---|---|---|---|---|
|
a In CH2Cl2vs. Ag/Ag+, (TBA)ClO4.
b DFT/B3LYP/6-31+G(d,p).
c In DMSO:H2O 95:5.
d In CH2Cl2vs. SCE, (TBA)PF6.
e In CH2Cl2.
f In PhCN vs. Ag/Ag+, (TBA)ClO4.
g In CHCl3.
h DFT/B3LYP/6-31+G(d) with PCM (CHCl3).
|
||||||
| 47 | 0.402a | 0.787a | 502c | 2.059b | 527c | |
| 47·F− | 0.385a | 0.70a | 502c | 1.94b | 524c | |
| 48 | 0.38d | 0.92d | −1.1 irrd | 545e | 581e | |
| 49a | 0.43f | 0.82f | −0.69f | 505g | 1.77h | 513g |
| 49b | 0.51f | 0.85f | −0.68f | 505g | 510g | |
| 49c | 0.45f | 0.80f | −0.66f | 505g | 512g | |
| 50a | 0.43f | 0.82f | −0.70f | 505g | 1.77h | 511g |
| 50b | 0.50f | 0.84f | −0.70f | 505g | 510g | |
| 50c | 0.45f | 0.80f | −0.66f | 505g | 510g | |
Compound 48 was synthesized by condensation between trimethyl-TTF aldehyde and 2,4-dimethyl-3-ethyl-pyrrole, followed by oxidation with DDQ and reaction with BF3·Et2O. Although the two units are directly linked, they are very likely perpendicular to each other because of the steric hindrance, as observed in the X-ray structure of the dithiolethione connected to the same BODIPY moiety.78 This feature has a determinant role in the absorption properties of 48, since no sizeable ICT occurs because of the orthogonality of HOMOTTF and LUMOBODIPY. Nevertheless, despite the perpendicular arrangement of the TTF and BODIPY units, partial CT should take place, as a red-shift of about 0.15 eV for absorption and 0.22 eV for the emission of BODIPY is observed in 48 when compared to the 8-Ph-BODIPY derivatives.76 Indeed, a strong fluorescence for 48 with a quantum efficiency of 0.43 has been observed at λem = 581 nm, originating from the BODIPY-based S0 → S1 excitation appearing at λmax = 545 nm (Fig. 17), thus indicating that fluorescence quenching by PET is thermodynamically unfavourable. The partial ICT very likely induces the stabilization of the excited state of 48.
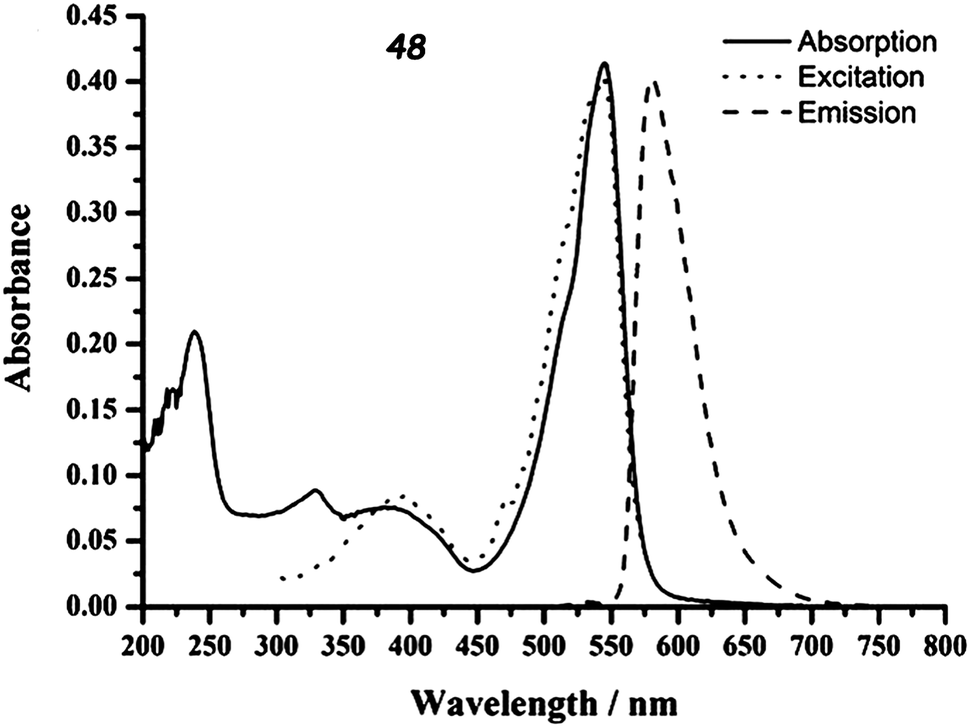 | ||
| Fig. 17 Absorption, excitation and emission spectra of TTF–BODIPY 48 (in CH2Cl2, 1.06 × 10−5 M). Adapted with permission from ref. 78; Copyright 2011, Elsevier. | ||
TTF–BODIPY 48 presents the classical TTF two reversible oxidation waves at +0.38 and +0.92 V vs. SCE, but also an irreversible reduction centred on BODIPY. Interestingly, the oxidation of TTF does not affect the emission properties of the compound, thus further strengthening the conclusion that the orthogonal units have reduced communication. This could constitute a key element in the preparation of fluorescent molecular conductors by the oxidation of TTF into crystalline radical cation salts.
Compounds 49–50 were reported by Fujiwara et al. and were synthesized either by Stille coupling between p-Br-phenyl-BODIPY and the corresponding TTF–SnBu3 precursors (49a–c) or by a Wittig-type reaction between TTF aldehydes and the p-tolyl-BODIPY phosphonium salt (50a–c).79 They all showed the pair of reversible oxidation waves of TTF and reversible reduction of the BODIPY unit at around −0.66 to −0.70 V vs. Ag/Ag+ (Table 6). ICT was observed as a broad very weakly intense shoulder at 550–700 nm, while the typical BODIPY very strong absorption, originating from HOMO−1/HOMO−2 → LUMO excitations, appeared at λmax = 505 nm (Fig. 18 and Table 6). The compounds showed fluorescence, with an emission band centred at λem = 510–513 nm, yet the quantum efficiency was less than half when compared to that of p-Br-phenyl-BODIPY (Fig. 18).
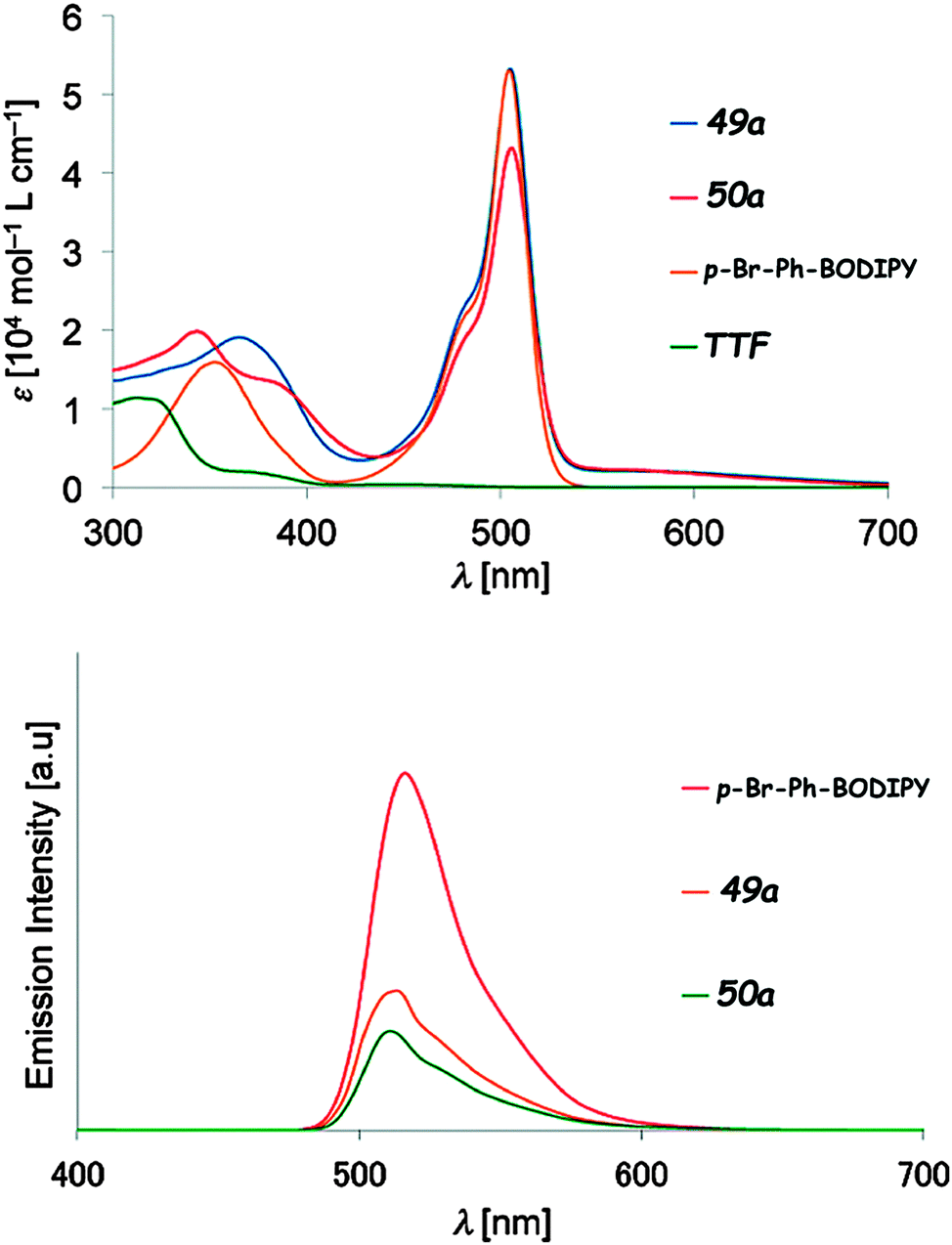 | ||
| Fig. 18 Absorption (up) and emission (bottom) spectra of TTF–BODIPY 49a and 50a, together with those for p-Br-Ph-BODIPY for comparison (in CHCl3, 10−5 M for absorption and 10−6 M for emission). Adapted with permission from ref. 79b; Copyright 2014, John Wiley and Sons. | ||
Similar to the above-mentioned BODIPY derivative 47, fluorescence quenching takes place by intramolecular PET, indicating the possibility of accessing a charge separated state in the D–A dyad. The most interesting aspect reported by Fujiwara et al. within this series of compounds concerned the photo-activated conductivity, which the authors investigated by a photo-electrochemical method with an experimental set-up involving a working electrode, a platinum counter electrode and Ag/AgCl as the reference electrode.
Upon irradiation of thin films of the dyads 49 and 50 prepared on ITO/glass substrates which served as working electrodes, the generation of a photocurrent between the working and platinum counter electrode was observed, with values of Imax = 1.89 μA cm−2 (49a, 510 nm) and 1.10 μA cm−2 (50a, 520 nm) under a potential of −0.30 V vs. Ag/AgCl (Fig. 19). No photocurrent was measured for independent thin films of TTF and Ph-BODIPY, clearly indicating the electron transfer from TTF to BODIPY. The photo-electric conversion at 0 V potential and 510 nm was ηmax = 0.67% for 49a, compared with that for TTF-PPD (vide supra) of 0.26% at 330 nm. The efficiency of the photo-conductor 50a was lower than that of 49a, i.e. ηmax = 0.24%, due to the less favourable electron transfer through the longer bridge. A conductivity increase was also observed upon photo-irradiation of single crystals of 49b, 50a and 50b. TTF–BODIPY 49a was successfully electrocrystallized into crystalline mixed valence salts, formulated as (49a)2PF6 and (49a)2AsF6. The PF6− salt has a half-filled band structure and shows a semiconducting behaviour, with a room temperature conductivity σ = 3.0 × 10−4 S cm−1, probably a consequence of a Mott-type localization; the conductivity was further increased by about 14% upon irradiation with white light.79b
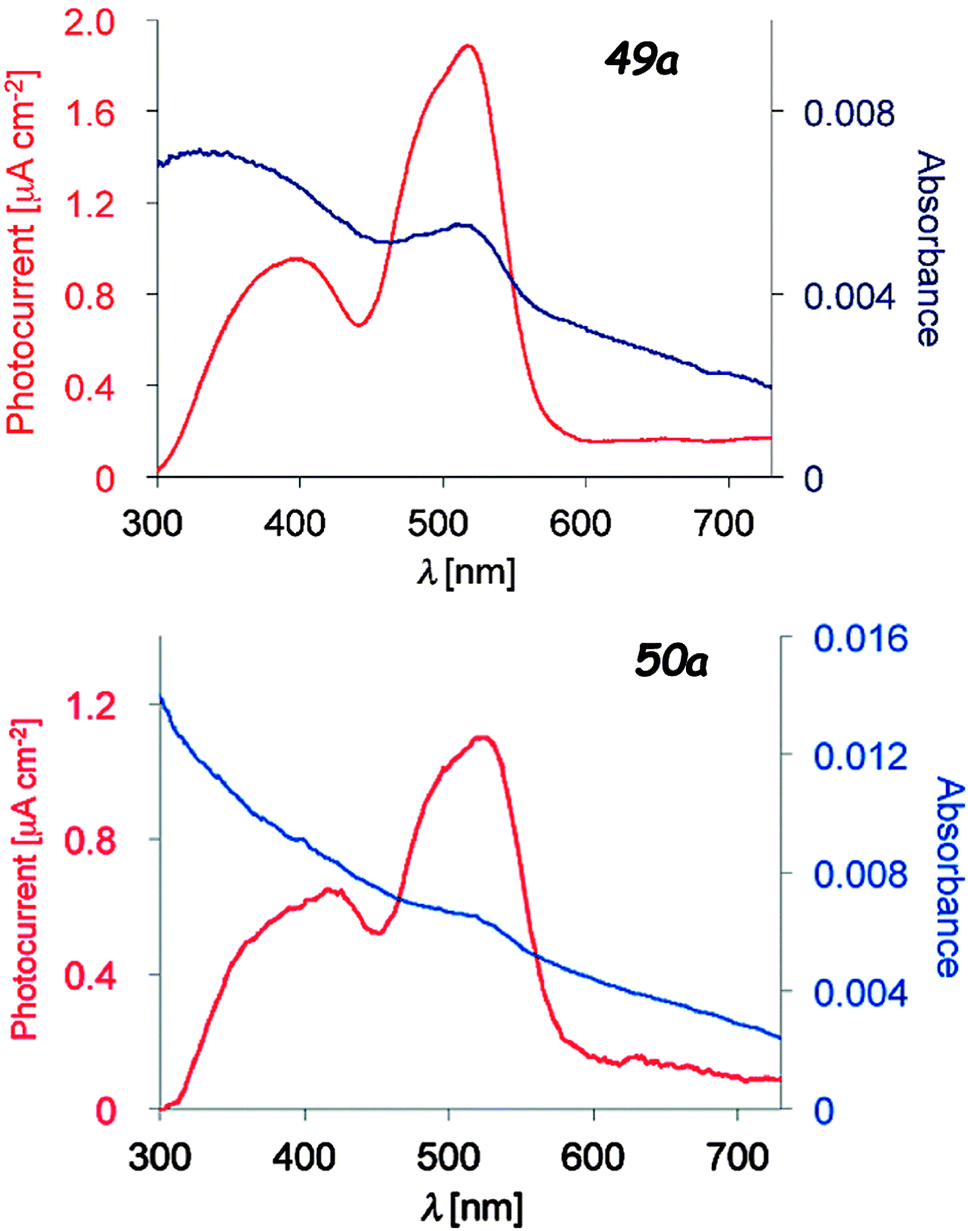 | ||
| Fig. 19 Photocurrent action spectra under a potential of −0.30 V vs. Ag/AgCl (red) and absorption spectra of the thin films spin-coated on to the ITO electrode (blue) of 49a and 50a. Adapted with permission from ref. 79b; Copyright 2014, John Wiley and Sons. | ||
All the above-mentioned observations suggest that the association of TTF and BODIPY appears to be an efficient strategy to access electroactive fluorescent precursors. Some of these dyads also showed the partial quenching of fluorescence depending on the degree of communication between the units and an interesting photo-activated conductivity.
5. Perylene and perylene-diimide (PDI) derivatives
Perylene and especially its bis(imide) (PDI) derivatives have largely been demonstrated to have wide potential as fluorescent dyes80 with excellent chemical and photo-stability in various fields, such as organic electronics,81 photovoltaics,82 electron transfer processes,83 supramolecular assemblies,84etc. Associated to TTF units, perylene and PDI are expected to act as electron acceptor counterparts, as their oxidation potentials are over +1 V vs. SCE, while the reduction potentials for PDI are −0.7 to −0.8 V vs. SCE.84aIt is thus not surprising that perylene and PDI have been attached to TTF via covalent saturated flexible bridges of variable length or fused to the “imide”85 or “bay”86 region. A recent literature survey did not show any examples of non-fused TTF–PDI directly linked or involving a conjugated bridge. Consequently, the general feature of the described TTF–perylene or TTF–PDI systems (Schemes 11 and 12) is the absence of an ICT band in the UV-vis spectra as a result of the lack of conjugation between the donor and acceptor units. Furthermore, massive fluorescence quenching of perylene is observed, very likely due to photoinduced electron transfer (PET).
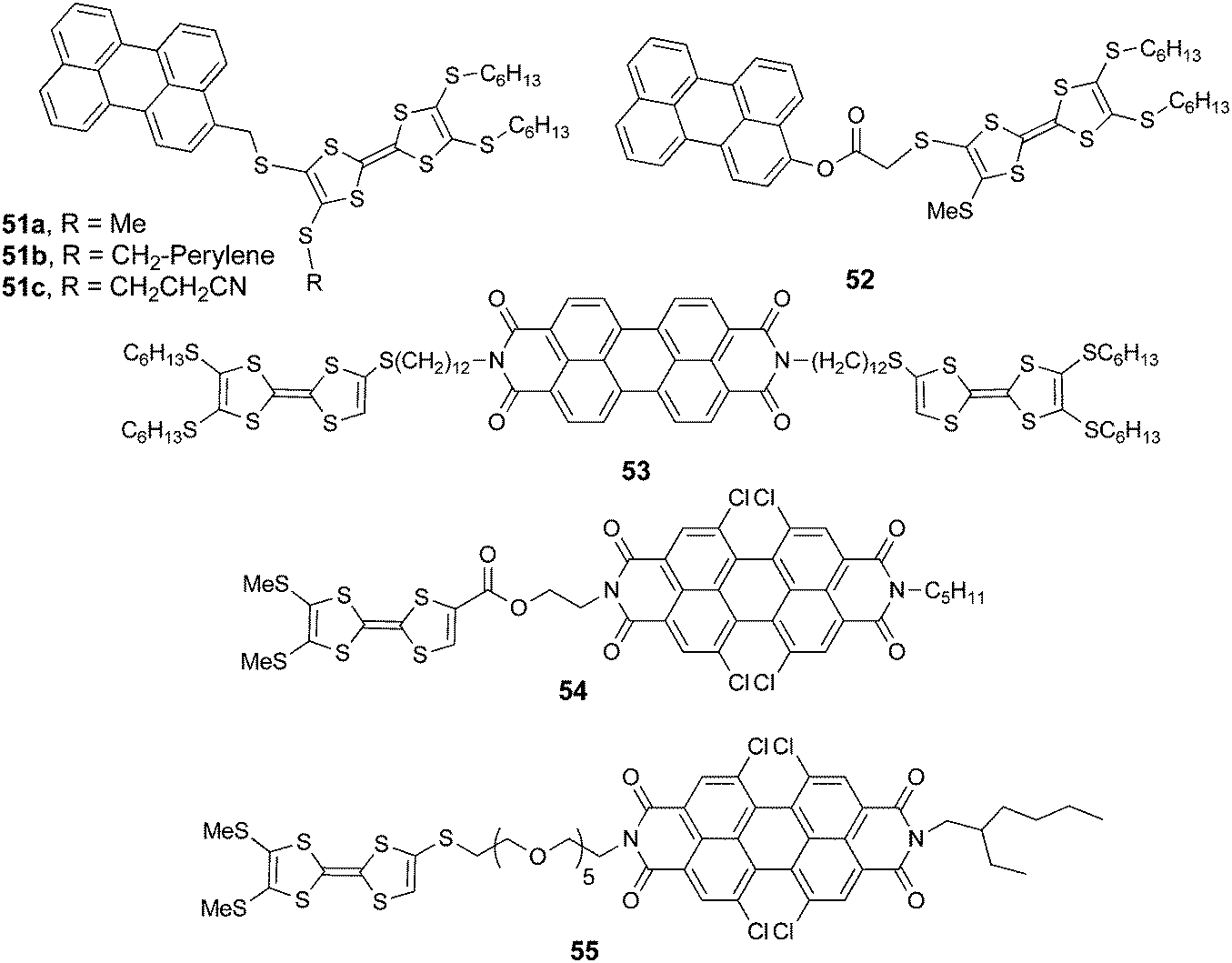 | ||
| Scheme 11 TTF–perylenes 51–52 and TTF–PDI derivatives 53–55. | ||
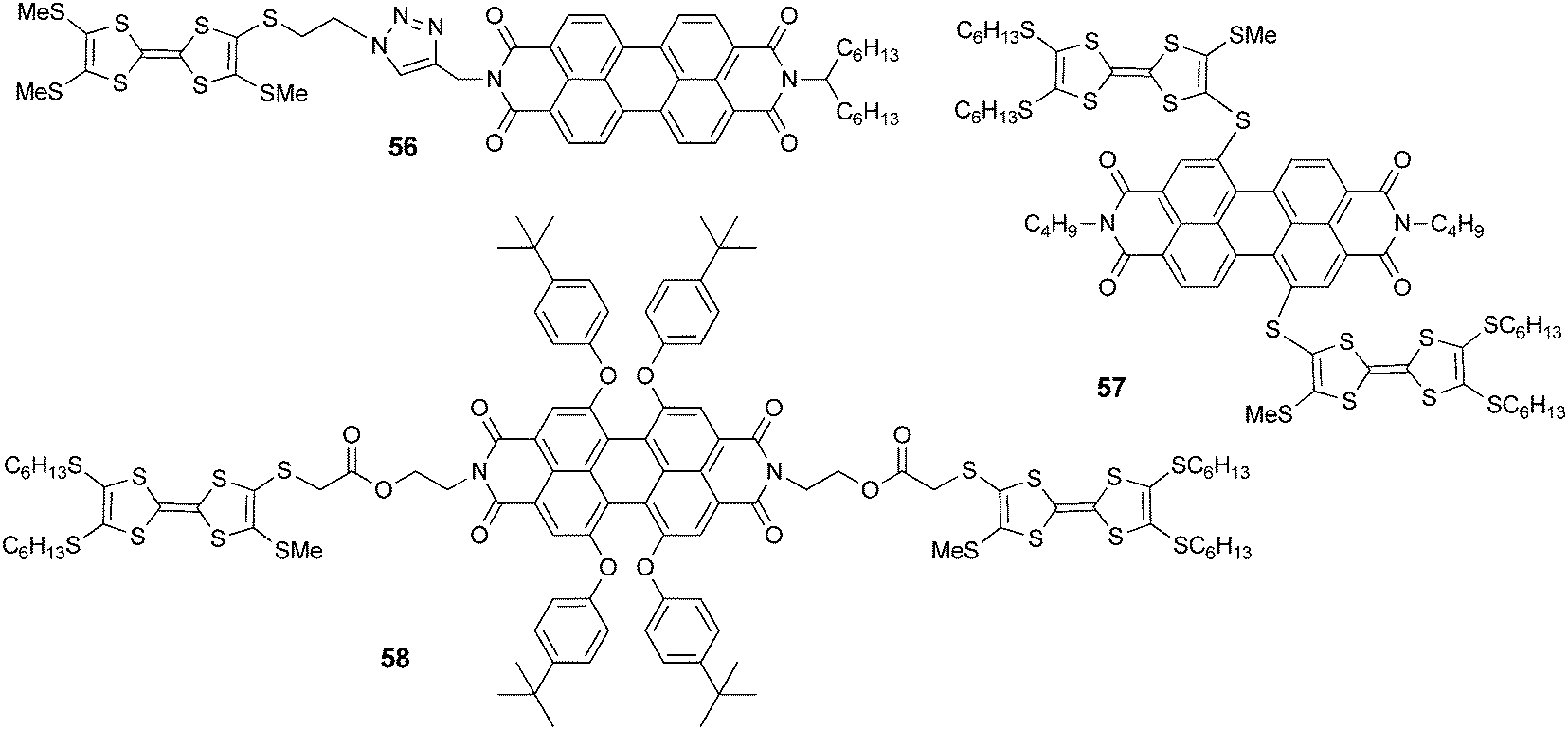 | ||
| Scheme 12 TTF–PDI dyad 56 and the triads 57–58. | ||
TTF–perylene dyads 51a–c87 and 5288 were synthesized, respectively, by the nucleophilic substitution of appropriately substituted TTF-thiolate and bromomethyl-perylene and by esterification between TTF acid and hydroxymethyl-perylene.
Compounds 51 and 52 oxidize reversibly at 0.61–0.65 V vs. Ag/AgCl for the first oxidation process (Table 7), values which are anodically shifted when compared to pristine TTF, because of the four thioalkyl substituents. The irreversible reduction of perylene occurs at a relatively low potential, which is in agreement with the moderate electron acceptor character of the unit. As mentioned before, no ICT was observed in the absorption spectra, and the fluorescence was strongly quenched (90–95%) with respect to perylene. The chemical oxidation of TTF into TTF2+ did not allow the fluorescence switch on, a result which can be explained on the basis of a reverse photoinduced electron transfer from perylene to TTF dication. PDI is a stronger electron acceptor compared to perylene, with the longest wavelength absorption band red-shifted by about 100 nm. The first TTF–PDI derivatives were reported by Zhang and Zhu et al. within triads, such as 53.89 It has been shown that the two electroactive units behave completely independently, as the oxidation potentials to TTF+˙ and TTF2+ and the reduction potential of PDI, which is strongly anodically shifted compared to perylene in 51–52 (Table 7), have exactly the same values as for the separated reference compounds. Moreover, the absorption spectrum shows the characteristic features of PDI and TTF, with no evidence of any ICT band. Thus, here again, no interaction takes place in the ground state between the donors and acceptor. In the excited state, however, photoinduced electron transfer occurs as suggested by the strong decrease in the emission band and in the lifetime of the excited state. In the radical cation state generated by the chemical oxidation of TTF with Fe(ClO4)3, no enhancement of the emission was observed, probably because of the spectral overlap between the TTF+˙ absorption and PDI emission favouring intramolecular energy transfer. The first TTF–PDI dyad 54, reported by Hudhomme et al.,90 was prepared by an esterification reaction between the corresponding TTF carboxylic acid and PDI alcohol. The reversible oxidation of TTF and the reduction of PDI occur at usual potential values (Table 7), and the maximum of the PDI absorption is at λmax = 517 nm. The absorption spectrum practically corresponds to the sum of the separated units, as no ICT band is observed. As for the triad 53, massive fluorescence quenching takes place (∼99.7%) through an allowed PET mechanism. The compound was electrochemically and chemically oxidized in order to study the redox modulation of the emission. A slight decrease was observed for oxidation to TTF+˙, as an energy transfer from 1PDI* to TTF+˙ can occur (Fig. 20). However, the fluorescence could only be partially restored upon oxidation to TTF2+, either electrochemically (Fig. 20) or chemically by the use of PhI(OAc)2/CF3SO3H as an oxidizing reagent, up to 16% of the intensity of the reference PDI compound.
| Compound | E 11/2,ox (V) | E 21/2,ox (V) | E 1red (V) | λ max (nm) perylene | λ em (nm) |
|---|---|---|---|---|---|
| a In CH2Cl2vs. Ag/AgCl, (TBA)PF6. b In CH2Cl2. c In CHCl3vs. Ag/AgCl, (TBA)PF6. d In CHCl3. e In CH2Cl2vs. SCE, (TBA)PF6. | |||||
| 51a | 0.62a | 0.99a | −1.59 irra | 447b | 448b |
| 51b | 0.65a | 1.00a | −1.75 irra | 447b | 458b |
| 51c | 0.61a | 1.00a | −1.58 irra | 447b | 450b |
| 52 | 0.62a | 1.01a | −1.58a | 443b | 451b |
| 53 | 0.48c | 0.86c | −0.67c | 420, 550d | 540, 570d |
| 54 | 0.65a | 1.05a | −0.32a | 517b | 545b |
| 55 | 488, 524b | 533, 572b | |||
| 56 | 0.48e | 0.81e | −0.60e | 489, 525b | |
| 57 | 0.70a | 1.01a | −0.50a | 540, 760 (ICT)b | |
| 58 | 0.62a | 0.96a | −0.59a | 544, 583b | 616b |
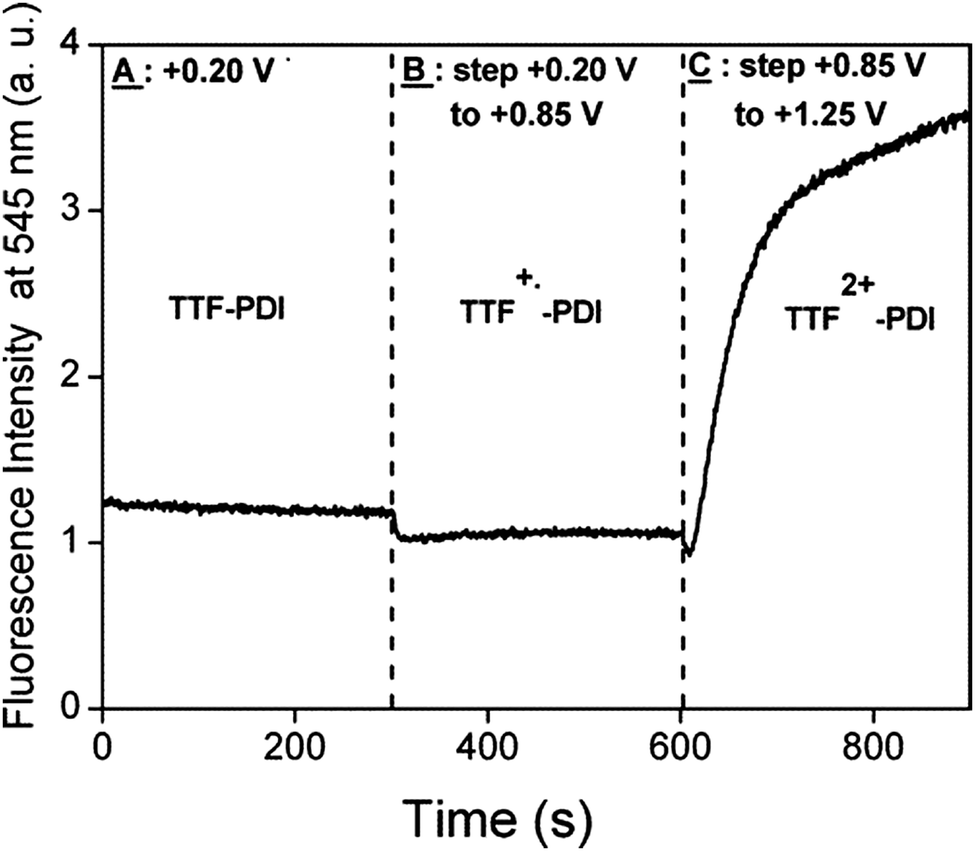 | ||
| Fig. 20 Evolution of the fluorescence intensity at 545 nm for λexc = 490 nm (10−3 M in CH2Cl2) of dyad 54 for different oxidation states versus time. Reprinted with permission from ref. 90a; Copyright 2005, American Chemical Society. | ||
The electrochemical reduction of PDI in 54 led to a total quenching of the fluorescence, this process being then fully reversible upon repetitive oxidation/reduction cycles.
In the dyad 55 containing an oligoethyleneglycol linker, the modulation of fluorescence was achieved by coordination with Ca2+ ions.91 A red-shift of the emission bands and a decrease in the intensity were observed upon the addition of Ca2+, a variation which was ascribed to the intermolecular coordination of the metal ion and aggregation, resulting in further fluorescence quenching.
Dyad 56 (Scheme 12) was prepared by Nielsen et al. using the “click” chemistry strategy of a Cu(II)-catalyzed azide alkyne cycloaddition (CuAAC) between TTF-azide and PDI-alkyne.92 While the emission properties for 56 were not reported, its self-assembly on mica surfaces was investigated by AFM, which provided evidence of the formation of microscopic wires, probably driven by π–π stacking of the PDI units.
TTF–PDI 57 with a connection in the “bay” region of PDI was reported by Shen et al.93 Interestingly, because of the close connection between the units, a broad ICT band centred at ∼760 nm appears in the UV-vis spectrum. The fluorescence of this D–A–D triad is completely quenched, very likely by photoinduced electron transfer. However, upon oxidation to the tetracationic state, the emission was reversibly switched on to reach an intensity of almost 25% of that of a reference PDI compound. Finally, in triad 5894 with a connection in the “imide” region, no ICT was observed in its UV-vis spectrum. In this case the emission was quenched by up to about 93% only because of intramolecular PET. As for 57, chemical oxidation with PhI(OAc)2/CF3SO3H to the tetracationic state induced a reversible switch on/off of the fluorescence, which was restored by up to 35% compared to that of the reference PDI compound.
In conclusion of this section, covalent TTF–PDI derivatives possessing long flexible linkers have shown efficient fluorescence quenching through a photoinduced electron transfer mechanism, as a charge transfer process generally does not occur. In some cases, partial reversible restoration of the emission has been reported upon the oxidation of TTF to the dication state.
6. TTFs with electron deficient hydrocarbon units
Unsaturated hydrocarbons may constitute suitable electron acceptor units, especially when substituted with electron withdrawing groups such as −CO, −CN or –NO2. However, polycondensed conjugated carbocycles already possess a low lying LUMO, ensuring that they have a moderate to strong electron acceptor character. The archetype of the latter is C60 and its derivatives, which have been extensively associated with TTF and ext-TTF and were largely discussed in a recent review.27 Later, a few other covalent TTF–C60 systems, in which the donor and acceptor either communicate95 or behave as isolated units,96 were reported but will not be further detailed herein. Less extended π-systems, such as perylenes, discussed above, as well as fluorenes have a weaker electron acceptor character, yet they are easy to functionalize and present strong fluorescence. Therefore, fluorenes, for example, have been incorporated in electroluminescent and photoactive materials.97The association of TTF and fluorene units was recently reported by Fujiwara et al. in dyads 59a–d, possessing a short saturated linker, and 60a–b, involving a conjugated linker (Scheme 13).98 The former were prepared by the nucleophilic substitution of TTF-thiolate on bromomethyl-fluorene, while the latter were prepared by coupling TTF aldehyde and the phosphonium bromide salt of methyl-fluorene under Wittig conditions. Both series are good electron donors, with oxidation potential values in the usual range (Table 8 for 59a and 60a), while the reduction centred on fluorene is irreversible.
 | ||
| Scheme 13 TTF–acceptors 59–67. | ||
| Compound | E 11/2,ox (V) | E 21/2,ox (V) | E 1red (V) | λ max (nm) ICT | ΔEHOMO–LUMO (eV) | λ em (nm) |
|---|---|---|---|---|---|---|
| a In PhCN vs. Ag/AgCl, (TBA)ClO4. b DFT/B3LYP/6-31G**. c In CHCl3. d In MeCN vs. SCE, (TBA)ClO4. e In MeCN vs. SCE, (TBA)PF6. f In MeCN. g In CH2Cl2vs. SCE, (TBA)PF6. h In CH2Cl2. | ||||||
| 59a | 0.44a | 0.81a | −1.1 irra | 3.57b | 336c | |
| 60a | 0.40a | 0.79a | −1.1 irra | 438c | 2.92b | 397, 416c |
| 61a | 0.48d | 0.80d | 549c | |||
| 61b | 0.55d | 0.82d | 529c | |||
| 62 | 0.45e | 0.80e | 480f | |||
| 63 | 0.64g | 1.07 irrg | −0.23g | 767h | ||
| 64 | 0.43g | 0.94g | −0.88 irrg | 677c | ||
| 65 | 0.56g | 1.02g | −0.64 irrg | 743c | ||
| trans-66 | 0.41g | 0.92g | 490c | |||
| cis-66 | 0.53g | 1.01g | 497c | |||
| 67 | 0.40g | 0.88g | 661c | |||
UV-vis absorption measurements on 59–60 clearly showed the role of the bridge on the electronic communication between the two electroactive units. For example, 59a–d presented only bands resulting from the superposition of TTF and fluorene absorption, while in 60a–b new absorption bands centred at 438 nm (60a) and 425 nm (60b), assigned to ICT, could be observed (Fig. 21).
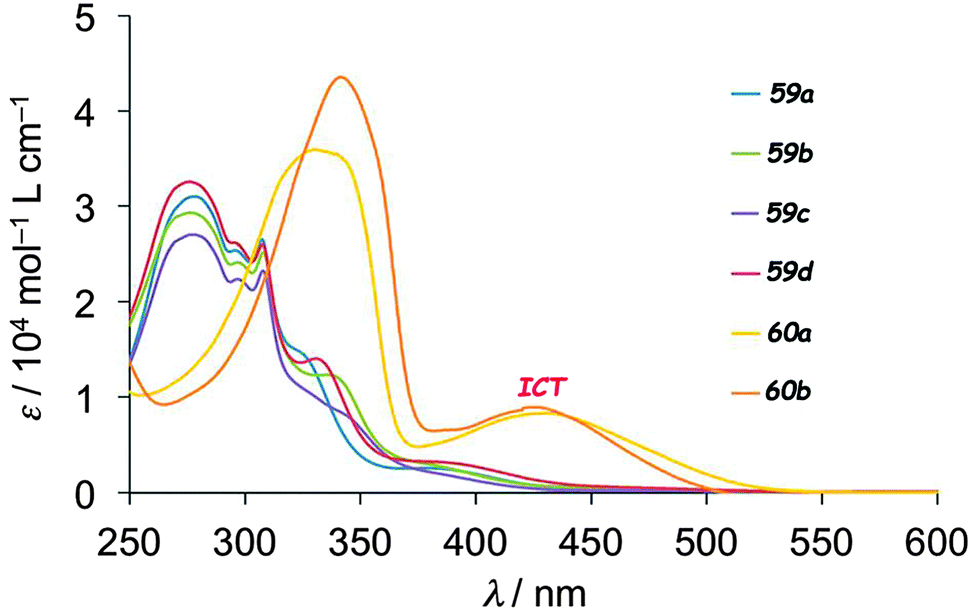 | ||
| Fig. 21 UV-vis absorption spectra of 59a–d and 60a–b in CHCl3 solution (c = 10−5 M). Adapted from ref. 98 with permission from the Centre National de la Recherche Scientifique (CNRS) and The Royal Society of Chemistry. | ||
The occurrence of ICT for 60a–b is also supported by the weaker HOMO–LUMO gap than in 59 (Table 8 for 59a and 60a), resulting from the massive lowering of the LUMO in the former, which was strongly based on the fluorene unit and the conjugated bridge. The emission intensity of fluorene was only slightly decreased in 59a when compared to pristine fluorene due to negligible intramolecular interaction between the units, while quenching up to 88% occurred for 60a as a consequence of the favourable photoinduced electron transfer (PET). The generation of a photocurrent upon irradiation was investigated on thin films of all the dyads deposited on ITO-glass substrates. The highest efficiencies were observed for 59a (0.38%) and 60a (0.49%) under a −0.30 V bias voltage. Conductivity enhancement upon irradiation was also observed for single crystals of 59d, which presented a segregated D–A structure. The electrocrystallization of 60b was particularly successful as it provided the mixed valence crystalline isostructural salts (60b)2Ag(CN)2 and (60b)2Au(CN)2, which showed semiconducting behaviour and a slight increase in conductivity upon irradiation.
In compounds 61a–b, the acceptor unit is a semisquarate fragment, owing the electron poor character to the carbonyl groups. For both derivatives, ICT bands were observed at 549 nm (61a) and 529 nm (61b) (Table 8) and, interestingly, they showed non-negligible conductivity in the neutral state on a single crystal (0.5 × 10−6 S cm−1 for 61a) as well as on compressed pellets (61b).99 These results open ways for the further development of semisquarate–TTF dyads conducting materials.
Dihydroazulene (DHA) was attached to TTF via an ethynyl-phenyl spacer in compound 62 in order to investigate the redox modulation of the photoswitching process to provide a ring opening of DHA towards the metastable vinyl-heptafulvene (VHF) species (Scheme 14).100
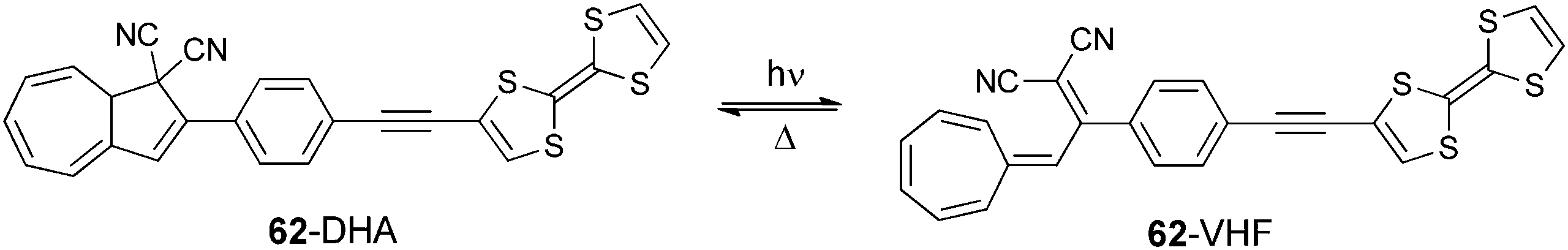 | ||
| Scheme 14 Dihydroazulene (DHA)–vinyl-heptafulvene (VHF) equilibrium in 62. | ||
TTF-DHA 62 was prepared by a Sonogashira coupling between TTF acetylene and the iodo-phenyl derivative of dicyano-DHA. The efficiency of the light induced ring opening of DHA to VHF was reduced more than twice in the radical cation 62+˙ compared to the neutral species, possibly because of a photoinduced electron transfer from DHA to TTF+˙. This hypothesis was supported by the presence of an ICT at 480 nm in 62 (Table 8), which is indicative of the coupling between the two units.
Cyano-rich conjugated fragments constitute excellent electron acceptors, as exemplified with compounds 63–65 (Scheme 13). The TTF-appended 1,1,4,4-tetracyanobuta-1,3-diene (TCBD) derivative 63 was obtained as a deep blue solid by an interesting [2+2] cycloaddition–electrocyclic ring opening sequence between TTF alkyne and tetracyanoethylene (TCNE).101 The single crystal X-ray structure showed an orthogonal arrangement of the two dicyanovinyl (DCV) moieties, with, however, co-planarity between TTF and the directly connected dicyanovinyl, thus ensuring efficient charge transfer. The broad ICT band centred at 767 nm in the absorption spectrum (Table 8) was attributed to a HOMOTTF to LUMOTCBD (Fig. 22) transition based on the TD DFT calculations.
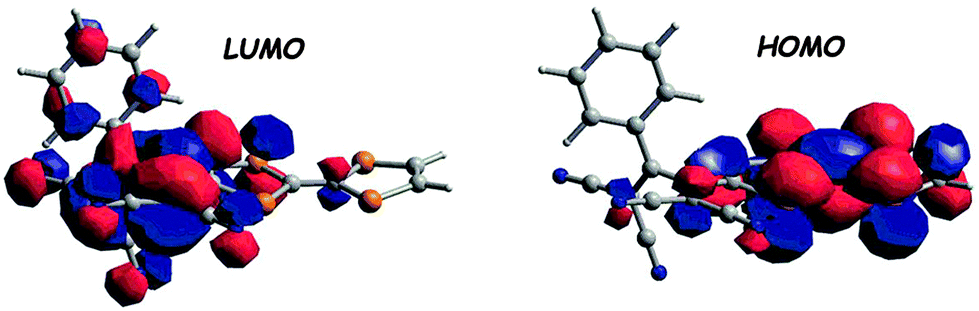 | ||
| Fig. 22 Frontier orbitals of 63 (DFT/B3LYP/6-311+G*). Adapted with permission from ref. 101; Copyright 2009, John Wiley and Sons. | ||
The close proximity of the acceptor to TTF induces a strong anodic shift of the TTF oxidation potentials to +0.64 and +1.07 V, while the first reduction potential of the donor appears at −0.23 V (Table 8). DCV units were attached to TTF at the end of acetylenic spacers in derivatives 64–65 prepared by Nielsen et al. via a Sonogashira coupling of TTF iodo or diiodo derivatives and the protected alkyne-aldehydes, followed by Knoevenagel condensation with malononitrile.102 The longer separation between DCV and TTF led to a cathodic shift of the TTF first oxidation potentials when compared to 63 (Table 8) and a blue-shift of the ICT bands, which still appeared at a rather low energy, i.e. 677 nm (64) and 743 nm (65).
The push–pull chromophores 66–67 containing p-nitro-phenyl units were prepared by Nielsen et al. using a Sonogashira cross-coupling between TTF-ethynyl and appropriate bromo-vinyl precursors.103 The bis(TTF) compound 67 exhibited reversible two electron oxidation processes and a strong red-shift of the ICT band, appearing at 661 nm, when compared to both the isomers of 66 (Table 8). Most interestingly, the former showed a high third-order optical nonlinearity, comparable to other aryl tetraethynylethene (TEE) derivatives.
7. TTF–radical dyads
Connecting TTF to a radical electron acceptor species either by a direct link or across a conjugated bridge affords dyads in which thermo- or solvent-controlled equilibrium between the neutral radical and zwitterion radical forms may occur as a consequence of an internal electron transfer (IET) process. Additionally, depending on the experimental conditions, the formation of intermolecular TTF⋯TTF dimers may take place, thus leading to exceptionally rich systems, in which the spin distribution and supramolecular aggregation are finely tuned (Fig. 23).104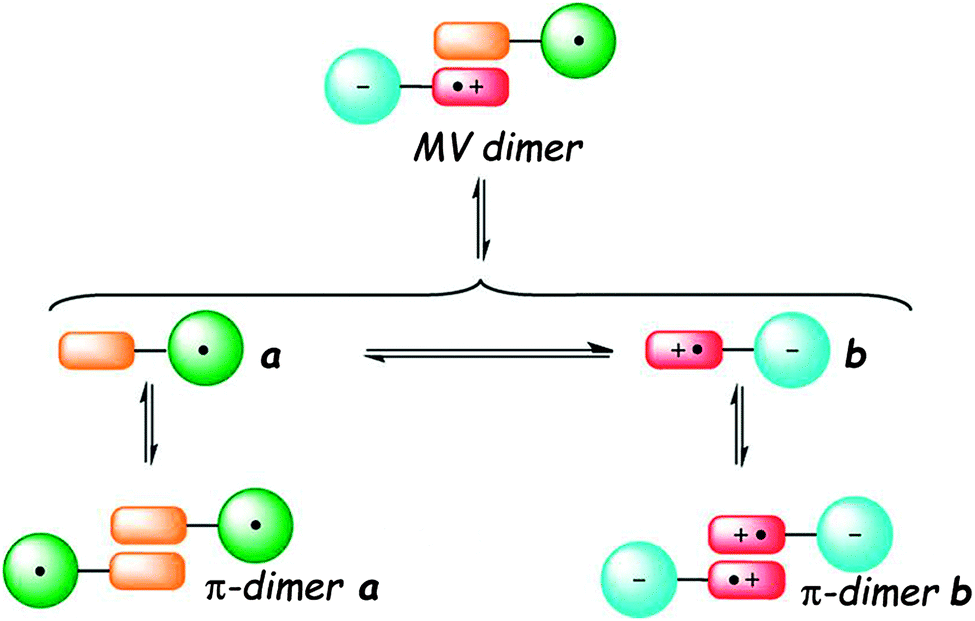 | ||
| Fig. 23 Equilibria occurring in solution between monomeric and dimeric species of TTF–radical acceptors dyads (MV = mixed valence). Adapted with permission from ref. 104; Copyright 2012, John Wiley and Sons. | ||
Stable radical units particularly adapted to covalent association with TTF within D–A dyads, such as 2,5-di-tert-butyl-6-oxophenalenoxyl (6OP),105 are strongly stabilized by delocalization and steric hindrance thanks to the t-butyl groups, and perchlorotriphenylmethyl (PTM),106 one of the stable derivatives of Gomberg's radical triphenylmethyl. Note that TTF–verdazyl dyads have been reported as well;107 however, donor–acceptor issues have not been addressed in this case as the verdazyl radical does not possess an electron acceptor character.
TTF–6OP radical 68 (Scheme 15) was prepared by Morita et al. through a Stille coupling reaction between TTF–SnBu3 and iodo-dimethoxy-phenalenone, followed by deprotection and oxidation steps.108
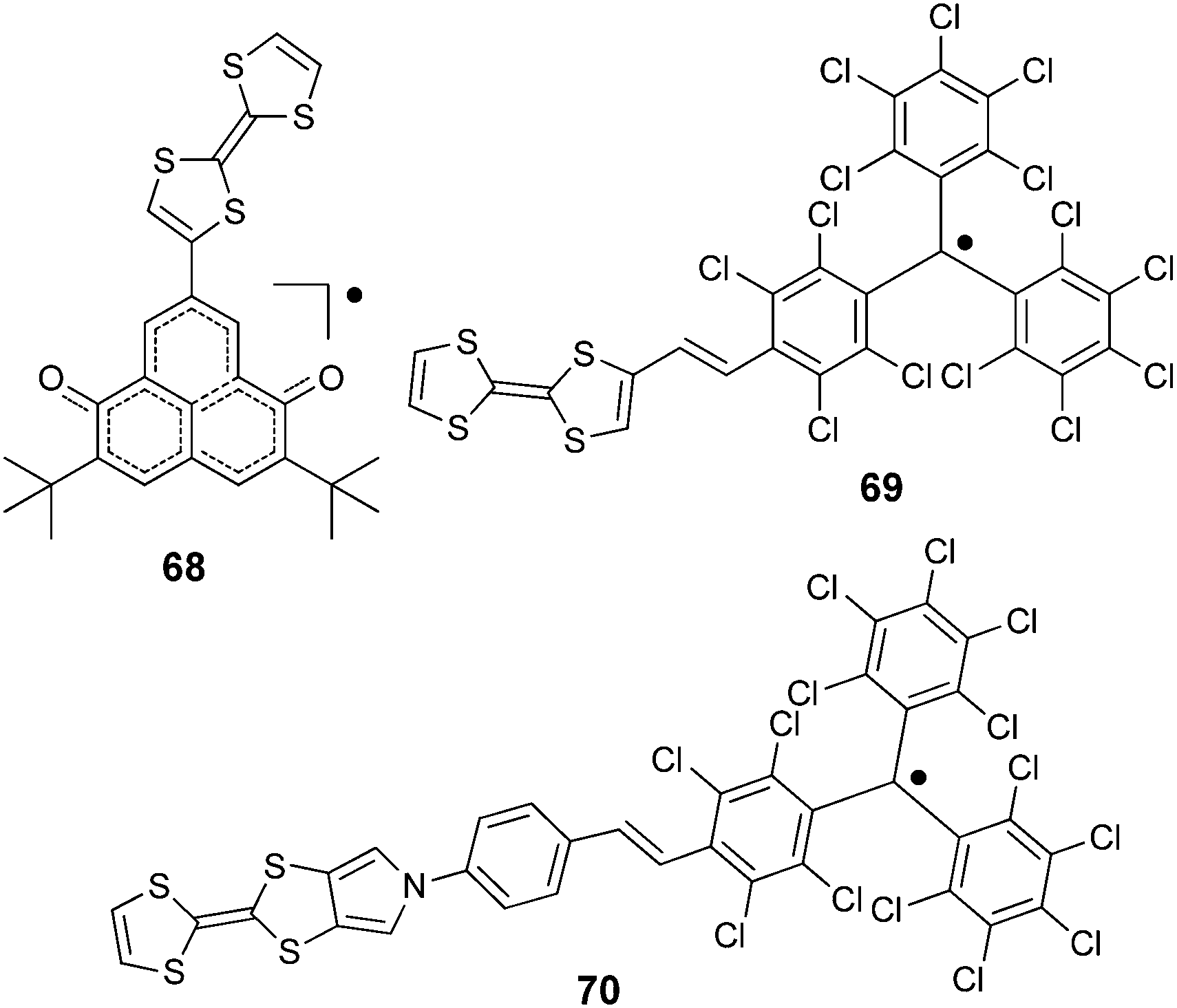 | ||
| Scheme 15 TTF–radicals 68–70. | ||
Cyclic voltammetry measurements showed the pair of TTF oxidation peaks together with the reversible reduction of the 6OP radical at a relatively high potential value (Table 9). Interestingly, this reduction was further anodically shifted upon the addition of 1,1,1-trifluoroethanol (TFE), thereby showing the stabilization of the 6OP anion by intermolecular hydrogen bonding with TFE molecules, which interact with the carbonyl group of 6OP.
| Compound | E 11/2,ox (V) | E 21/2,ox (V) | E 1red (V) | λ max (nm) ICT | ΔENHOMO–SOMO (ev) |
|---|---|---|---|---|---|
| a In PhCN vs. SCE, (TBA)ClO4. b In CH2Cl2. c DFT/ROBLYP/6-31G(d)//UBLYP/6-31G(d). d In CH2Cl2vs. Ag/AgCl, (TBA)PF6. e DFT/TD-UCAMB3LYP/6-31G*. f In THF. | |||||
| 68 | 0.39a | 0.78a | 0.11a | 1315b | 1.5c |
| 69 | 0.48d | 0.98d | −0.15d | 900b | 3.6e |
| 70 | 0.45d | 0.95d | −0.19d | 800f | 4.7e |
In the absorption spectrum in CH2Cl2 solution, the weak band centred at 1315 nm was assigned to ICT, while the sharp peak at 414 nm was assigned to the 6OP radical. In TFE solution, the UV-vis spectrum of 68 was completely different, showing a change of the solution colour from orange to green, with the appearance of a band at 605 nm, indicating the presence of the TTF+˙–6OP− zwitterionic type structure b (Fig. 23). The solvent-controlled equilibrium between the neutral radical a and zwitterion b species was also evidenced by solution EPR measurements. According to the extremely well-resolved hyperfine structure at 290 K with a g value of 2.0047, the radical form was observed in toluene, whereas in TFE, the zwitterion was favoured, as shown by the downfield shift of the absorption at g = 2.0082, with the signal containing nearly three equivalent 1H couplings, indicative of TTF spin bearing (Fig. 24).
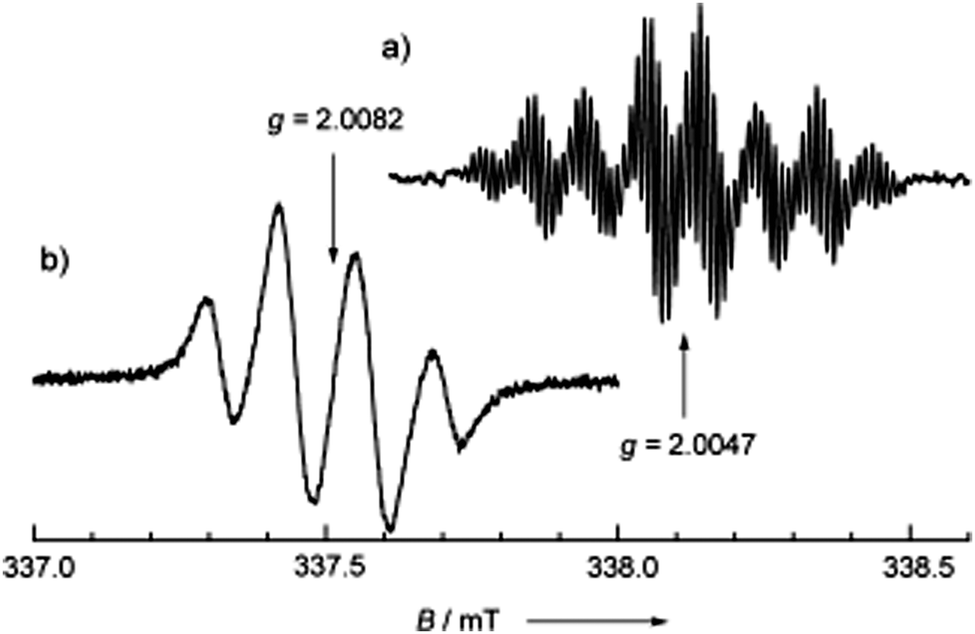 | ||
| Fig. 24 Liquid-phase ESR spectra of 68 in (a) toluene (1.0 × 10−4 M) and (b) CF3CH2OH (3.0 × 10−5 M) at 290 K. The microwave frequency used was 9.487717 GHz. Reproduced with permission from ref. 108; Copyright 2005, John Wiley and Sons. | ||
Interestingly, the equilibrium could also be controlled by temperature, as seen with a solution of 68 in a mixture CH2Cl2:TFE 199:1, which showed the pattern of 68a at room temperature, the one of 68b at 243 K and an intermediate state consisting of the superposition of the two EPR spectra at 263 K. Thus, the general picture for this system is spin distribution and IET control accompanied by thermo- and solvatochromism (Fig. 25).109
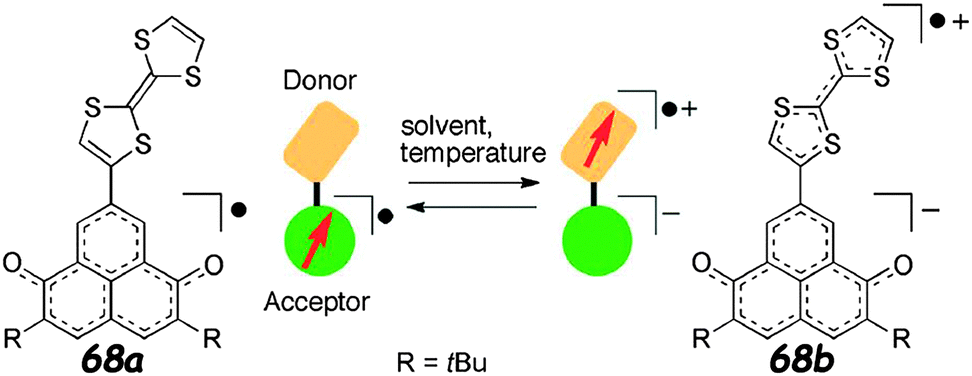 | ||
| Fig. 25 Solvent- and temperature-dependent spin-centre transfer of 68. Adapted with permission from ref. 109; Copyright 2014, John Wiley and Sons. | ||
The stabilization of the zwitterion species 68b was further supported by temperature variable cyclic voltammetry measurements and theoretical calculations.109 The latter suggested that the establishment of hydrogen bonding between TFE and 68 through the OH and CO groups, respectively, is a determining factor to control the spin distribution in the dyad (Fig. 26).
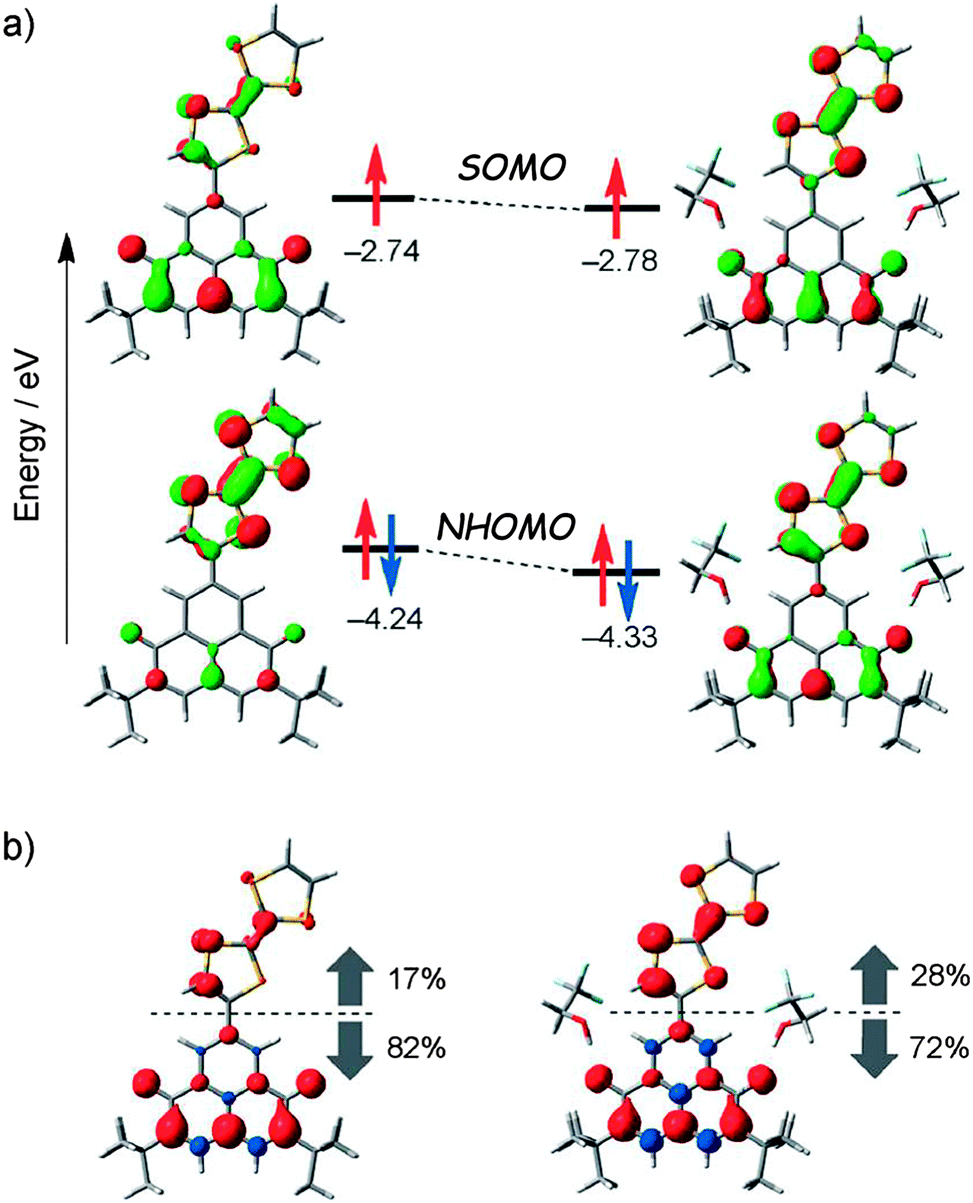 | ||
| Fig. 26 (a) MO energy levels of SOMOs and NHOMOs for 68 (left) and 68·(TFE)2 (right) calculated at the ROBLYP/6-31G(d)//UBLYP/6-31G(d) level of theory. (b) Spin-density distributions for 68 (left) and 68·(TFE)2 (right) calculated at the UBLYP/6-31G(d)//UBLYP/6-31G(d) level of theory. Red and blue regions denote positive and negative spin densities, respectively. The percentages denote spin densities located on the TTF and the 6OPO moieties against total spin densities. Reproduced with permission from ref. 109; Copyright 2014, John Wiley and Sons. | ||
TTF–PTM dyads (Scheme 15) 69104 and 70,110 reported by Rovira, Veciana et al., were synthesized by a Wittig–Horner–Wadsworth–Emmons reaction between TTF aldehyde (69) and TTF-pyrrolo aldehyde (70) and the methyl-phosphonate of the protonated form of PTM, followed by deprotonation and subsequent oxidation of the anion with silver nitrate. Cyclic voltammetry measurements showed a reversible oxidation for TTF into a radical cation and dication species with a slight anodic shift for 69 compared to 70, this very likely being a consequence of the longer bridge in the latter, together with the reversible reduction of PTM into a radical anion (Table 9). In the UV-vis spectra, both compounds showed a low energy band at around 900 nm for 69 and 800 nm for 70, which could be assigned to the intramolecular electron transfer (IET). This process is finely tuned by the nature of the solvent, as nicely shown by the solvatochromic effect and naked-eye-visible colour change for 69 in CH2Cl2, acetone and DMF (Fig. 27). In CH2Cl2, the spectrum is typical for a neutral PTM radical, whereby in DMF the compound mainly exists as a PTM anion, while in acetone both forms are present.
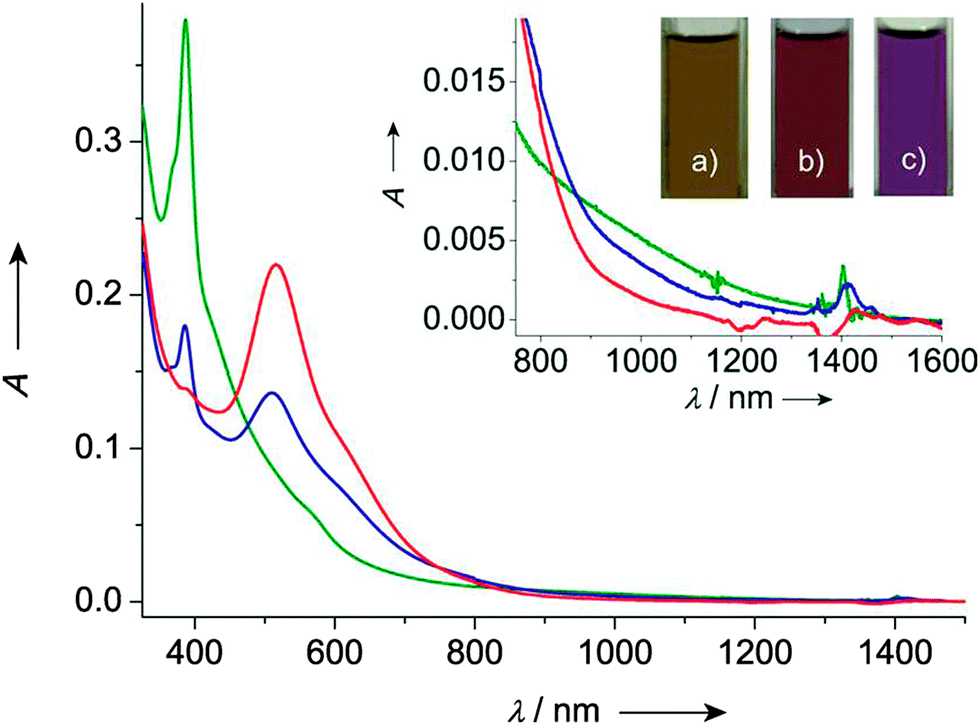 | ||
| Fig. 27 UV/vis/NIR spectra of dyad 69 in CH2Cl2 (green line), acetone (blue line) and DMF (red line). Inset: Low energy range of the absorption spectra and colours of 69 exhibited in (a) CH2Cl2, (b) acetone and (c) DMF solutions. Reproduced with permission from ref. 104; Copyright 2012, John Wiley and Sons. | ||
Variable temperature EPR measurements in different solvents indicated that in DMF solutions at room temperature and below, compound 69 exists as a π-dimer (form b in Fig. 23) due to TTF+˙ dimerization into diamagnetic pairs, as suggested by the EPR silent spectrum at 293 K. However, upon heating up to 375 K the dimer dissociates into separated radicals, according to the process illustrated in Fig. 28.111
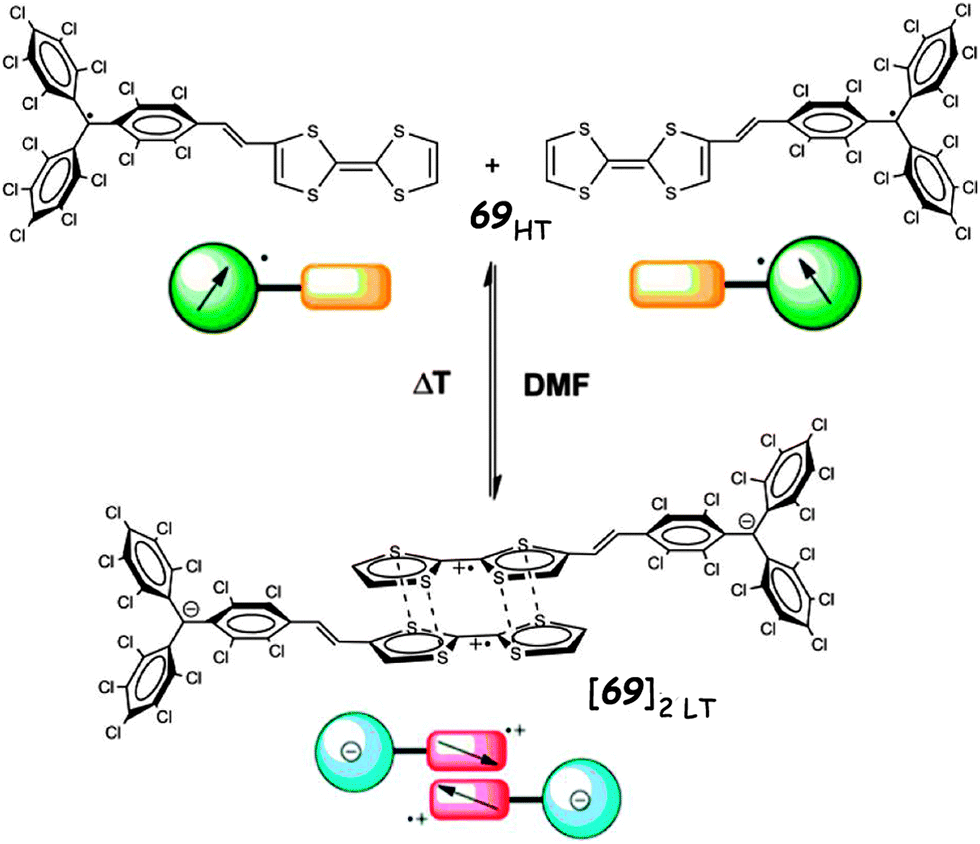 | ||
| Fig. 28 Reversible temperature-induced supramolecular switching of 69 in DMF solution. Adapted with permission from ref. 111; Copyright 2013, American Chemical Society. | ||
Further EPR and UV-vis spectroscopy studies focused on the oxidized and reduced species of 69 as well as on their intermolecular aggregates.112 Dyad 70, characterized by single crystal X-ray diffraction, shows segregation in the solid state of its D and A parts with a regular stacking of pyrrolo-TTF units.110 Contrary to 69, the presence of the zwitterionic state 70b was observed only partially in DMF, probably due to the weaker electron donor ability of TTF in 70 compared to 69.
In conclusion, TTF–radical dyads with strong coupling between the two units represent valuable precursors for new multifunctional materials, showing bistability and control of the spin distribution, and, possibly, orientation.
8. Conclusions and outlook
In this feature article, we reviewed the main families of non-fused TTF–acceptors reported in the literature since 2004, when the previous extensive review on this topic was published,11 with a special focus on their specific properties, such as electronic communication between the units, intramolecular charge transfer and its modulation by protonation, coordination, solvent, modulation of the emission properties and quenching mechanisms, photo-activated conductivity, control of the spin distribution, etc. Much progress has been achieved since the seminal theoretical work of Aviram and Ratner on TTF–σ–TCNQ,17 especially regarding the diversity of TTF–acceptor families proposed to date in the literature. Depending on the nature and specific properties of each acceptor, such as its electronic character, coordination ability to metal centres, basicity, emission, spin localization, etc., diverse properties and their modulation have been targeted by the research groups working in this promising and stimulating area at the interface between organic synthesis, supramolecular chemistry, coordination chemistry, photophysics and condensed matter physics. The families of compounds discussed herein have been classified according to the type of acceptor, which also determines the focus of the investigations. TTF–pyridines are excellent electroactive ligands and show modulation of the ICT by protonation and coordination. In TTF-triazines and -tetrazines, the donor–acceptor character has been combined with coordination ability through the introduction of chelating ligands. TTF-heteroazoles are interesting precursors for molecular photoconductors. When excellent fluorophores such as BODIPY and PDI are associated to TTF, partial to total emission quenching is observed due to photoinduced electron transfer (PET). However, the luminescence can be partially restored upon the oxidation of TTF, thus providing redox switchable systems. Unsaturated or aromatic hydrocarbons functionalized with electron withdrawing groups, such as CO, NO2, CN, constitute suitable acceptors in combination with TTF units and derivatives, which have proven to be of interest in nonlinear optics. Electron poor stable radicals, such as 6OP and PTM, have been attached to TTF within neutral radical dyads, which have demonstrated a rich solvato- and thermochromic behaviour combined with spin distribution control. Although many of the results reported on the compounds discussed herein will remain at an academic research level, their study allows acquiring new knowledge and a greater understanding about the fundamental processes occurring in molecular systems, such as electron and energy transfer, the mechanism of fluorescence quenching, the role of D–A interactions and crystal packing in photo-activated conductors and spin transfer. This new information will certainly strongly contribute to the development of molecular electronics and spintronics, actuation and sensing, molecular conductors and magnets. In molecular electronics, for example, donor–acceptor-based materials for organic field effect transistors (OFET) with ambipolar charge transport properties113 have proven their efficiency for electron and hole type mobilities upon the appropriate choice of donor and acceptor units.114 Comparatively, only few TTF–acceptor systems, to date, exclusively involving fused derivatives,115 have been investigated as ambipolar FET active materials, although there is clearly room for the development of more families of TTF–acceptors with different linkers between the units.116 Non-fused extended-TTF13 and TTF–acceptors14 have shown potential as sensitizers in DSSC, with a reported power conversion efficiency of 6.47% for a quinoxaline-fused TTF derivative,117 whereas a diketopyrrolopyrrole (DPP)–thienoTTF based copolymer has shown promising potential as a single material organic solar cell (SMOC).116b These recent examples highlight the interest and possibilities for the development of TTF–acceptors for organic solar cells. Photoconductivity12 is another property which seems underexploited to date, in spite of the several examples we have mentioned throughout the present manuscript. The potential of TTF–acceptor based materials as photoconductors, either as single crystals, thin films or supramolecular aggregates, possibly combined with other properties, is worth deeper investigation.Acknowledgements
This work was supported by the CNRS, University of Angers and the ANR. The authors thank all the co-workers appearing in the papers cited in the references.References
- I. Bertini, H. B. Gray, E. I. Stiefel and J. S. Valentine, Biological Inorganic Chemistry, University Science Books, Sausalito, CA, 2007 Search PubMed.
- M. Cordes and B. Giese, Chem. Soc. Rev., 2009, 38, 892–901 RSC.
- O. S. Wenger, Acc. Chem. Res., 2011, 44, 25–35 CrossRef CAS PubMed.
- E. Espíldora, J. L. Delgado and N. Martín, Isr. J. Chem., 2014, 54, 429–439 CrossRef.
- (a) J. L. Segura and N. Martin, Angew. Chem., Int. Ed., 2001, 40, 1372–1409 CrossRef CAS; (b) D. Canevet, M. Sallé, G. X. Zhang, D. Q. Zhang and D. B. Zhu, Chem. Commun., 2009, 2245–2269 RSC.
- (a) T. Ishiguro, K. Yamaji and G. Saito, Organic Superconductors, Spinger-Verlag, Heidelberg, Germany, 1998 Search PubMed; (b) T. Mori, Chem. Rev., 2004, 104, 4947–4969 CrossRef CAS PubMed.
- (a) E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García and V. Laukhin, Nature, 2000, 408, 447–449 CrossRef CAS PubMed; (b) E. Coronado and P. Day, Chem. Rev., 2004, 104, 5419–5448 CrossRef CAS PubMed.
- (a) N. Avarvari and J. D. Wallis, J. Mater. Chem., 2009, 19, 4061–4076 RSC; (b) F. Pop, P. Auban-Senzier, A. Frckowiak, K. Ptaszyński, I. Olejniczak, J. D. Wallis, E. Canadell and N. Avarvari, J. Am. Chem. Soc., 2013, 135, 17176–17186 CrossRef CAS PubMed; (c) F. Pop, P. Auban-Senzier, E. Canadell, G. L. J. A. Rikken and N. Avarvari, Nat. Commun., 2014, 5, 3757 Search PubMed.
- F. Pointillart, B. Le Guennic, O. Cador, O. Maury and L. Ouahab, Acc. Chem. Res., 2015, 48, 2834–2842 CrossRef CAS PubMed.
- (a) D. Lorcy, N. Bellec, M. Fourmigué and N. Avarvari, Coord. Chem. Rev., 2009, 253, 1398–1438 CrossRef CAS; (b) F. Riobé and N. Avarvari, Coord. Chem. Rev., 2010, 254, 1523–1533 CrossRef.
- M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891–4945 CrossRef CAS PubMed.
- (a) L. Zang, Acc. Chem. Res., 2015, 48, 2705–2714 CrossRef CAS PubMed; (b) K. P. Goetz, D. Vermeulen, M. E. Payne, C. Kloc, L. E. McNeil and O. D. Jurchescu, J. Mater. Chem. C, 2014, 2, 3065–3076 RSC.
- S. Wenger, P. A. Bouit, Q. L. Chen, J. Teuscher, D. Di Censo, R. Humphry-Baker, J. E. Moser, J. L. Delgado, N. Martín, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2010, 132, 5164–5169 CrossRef CAS PubMed.
- Y. Geng, F. Pop, C. Yi, N. Avarvari, M. Grätzel, S. Decurtins and S.-X. Liu, New J. Chem., 2014, 38, 3269–3274 RSC.
- M. R. Bryce, Adv. Mater., 1999, 11, 11–23 CrossRef CAS.
- (a) R. M. Metzger, Chem. Phys., 2006, 326, 176–187 CrossRef CAS; (b) C. Van Dyck and M. A. Ratner, Nano Lett., 2015, 15, 1577–1584 CrossRef CAS PubMed.
- A. Aviram and M. A. Ratner, Chem. Phys. Lett., 1974, 29, 277–283 CrossRef CAS.
- (a) P. de Miguel, M. R. Bryce, L. M. Goldenberg, A. Beeby, V. Khodorkovsky, L. Shapiro, A. Niemz, A. O. Cuello and V. Rotello, J. Mater. Chem., 1998, 8, 71–76 RSC; (b) D. F. Perepichka, M. R. Bryce, A. S. Batsanov, J. A. K. Howard, A. O. Cuello, M. Gray and V. M. Rotello, J. Org. Chem., 2001, 66, 4517–4524 CrossRef CAS PubMed.
- J. Wu, S.-X. Liu, A. Neels, F. Le Derf, M. Sallé and S. Decurtins, Tetrahedron, 2007, 63, 11282–11286 CrossRef CAS.
- J. Santos, B. M. Illescas, N. Martín, J. Adrio, J. C. Carretero, R. Viruela, E. Ortí, F. Spänig and D. M. Guldi, Chem. – Eur. J., 2011, 17, 2957–2964 CrossRef CAS PubMed.
- (a) D. F. Perepichka, M. R. Bryce, C. Pearson, M. C. Petty, E. J. L. McInnes and J. P. Zhao, Angew. Chem., Int. Ed., 2003, 42, 4636–4639 CrossRef CAS PubMed; (b) D. F. Perepichka and M. R. Bryce, Angew. Chem., Int. Ed., 2005, 44, 5370–5373 CrossRef CAS PubMed.
- E. Tsiperman, J. Y. Becker, V. Khodorkovsky, A. Shames and L. Shapiro, Angew. Chem., Int. Ed., 2005, 44, 4015–4018 CrossRef CAS PubMed.
- F. Otón, V. Lloveras, M. Mas-Torrent, J. Vidal-Gancedo, J. Veciana and C. Rovira, Angew. Chem., Int. Ed., 2011, 50, 10902–10906 CrossRef PubMed.
- J. P. Bergfield and M. A. Ratner, Phys. Status Solidi B, 2013, 250, 2249–2266 CrossRef CAS.
- F. Dumur, X. Guégano, N. Gautier, S.-X. Liu, A. Neels, S. Decurtins and P. Hudhomme, Eur. J. Org. Chem., 2009, 6341–6354 CrossRef CAS.
- M. R. Bryce, J. Mater. Chem., 2000, 10, 589–598 RSC.
- F. G. Brunetti, J. L. López, C. Atienza and N. Martín, J. Mater. Chem., 2012, 22, 4188–4205 RSC.
- J. J. Bergkamp, S. Decurtins and S.-X. Liu, Chem. Soc. Rev., 2015, 44, 863–874 RSC.
- D. F. Perepichka, M. R. Bryce, A. S. Batsanov, E. J. L. McInnes, J. P. Zhao and R. D. Farley, Chem. – Eur. J., 2002, 8, 4656–4669 CrossRef CAS.
- G. Ho, J. R. Heath, M. Kondratenko, D. F. Perepichka, K. Arseneault, M. Pézolet and M. R. Bryce, Chem. – Eur. J., 2005, 11, 2914–2922 CrossRef CAS PubMed.
- (a) S. C. Lee, A. Ueda, H. Kamo, K. Takahashi, M. Uruichi, K. Yamamoto, K. Yakushi, A. Nakao, R. Kumai, K. Kobayashi, H. Nakao, Y. Murakamie and H. Mori, Chem. Commun., 2012, 48, 8673–8675 RSC; (b) S. C. Lee, A. Ueda, A. Nakao, R. Kumai, H. Nakao, Y. Murakami and H. Mori, Chem. – Eur. J., 2014, 20, 1909–1917 CrossRef CAS PubMed.
- Q.-Y. Zhu, Y. Liu, W. Lu, Y. Zhang, G.-Q. Bian, G.-Y. Niu and J. Dai, Inorg. Chem., 2007, 46, 10065–10070 CrossRef CAS PubMed.
- Q.-Y. Zhu, L.-B. Huo, Y.-R. Qin, Y.-P. Zhang, Z.-J. Lu, J.-P. Wang and J. Dai, J. Phys. Chem. B, 2010, 114, 361–367 CrossRef CAS PubMed.
- R. Andreu, I. Malfant, P. G. Lacroix and P. Cassoux, Eur. J. Org. Chem., 2000, 737–741 CrossRef CAS.
- H. Xue, X.-J. Tang, L.-Z. Wu, L.-P. Zhang and C.-H. Tung, J. Org. Chem., 2005, 70, 9727–9734 CrossRef CAS PubMed.
- Y.-P. Zhao, L.-Z. Wu, G. Si, Y. Liu, H. Xue, L.-P. Zhang and C.-H. Tung, J. Org. Chem., 2007, 72, 3632–3639 CrossRef CAS PubMed.
- A. Zitouni, A. Hamel, S. Bouguessa, A. Gouasmia, A. El-Ghayoury, P. Frère and M. Sallé, Synth. Met., 2015, 204, 84–89 CrossRef CAS.
- T. Devic, N. Avarvari and P. Batail, Chem. – Eur. J., 2004, 10, 3697–3707 CrossRef CAS PubMed.
- F. Pointillart, T. Cauchy, Y. Le Gal, S. Golhen, O. Cador and L. Ouahab, Inorg. Chem., 2010, 49, 1947–1960 CrossRef CAS PubMed.
- J.-Y. Balandier, A. Belyasmine and M. Sallé, Eur. J. Org. Chem., 2008, 269–276 CrossRef CAS.
- S. Bakhta, M. Guerro, B. Kolli, F. Barrière, T. Roisnel and D. Lorcy, Tetrahedron Lett., 2010, 51, 4497–4500 CrossRef CAS.
- F. Pointillart, T. Cauchy, O. Maury, Y. Le Gal, S. Golhen, O. Cador and L. Ouahab, Chem. – Eur. J., 2010, 16, 11926–11941 CrossRef CAS PubMed.
- T. Devic, P. Batail and N. Avarvari, Chem. Commun., 2004, 1538–1539 RSC.
- M. Behl and R. Zentel, Macromol. Chem. Phys., 2004, 205, 1633–1643 CrossRef CAS.
- (a) J. Bonvoisin, J.-P. Launay, M. Van der Auweraer and F. C. De Schryver, J. Phys. Chem., 1994, 98, 5052–5057 CrossRef CAS; (b) R. Fink, C. Frenz, M. Thelakkat and H.-W. Schmidt, Macromolecules, 1997, 30, 8177–8181 CrossRef CAS; (c) J. Pang, Y. Tao, S. Freiberg, X.-P. Yang, M. D'Iorio and S. Wang, J. Mater. Chem., 2002, 12, 206–212 RSC; (d) W. Wang, H. Sun and A. E. Kaifer, Org. Lett., 2007, 9, 2657–2660 CrossRef CAS PubMed.
- M.-X. Wang and H.-B. Yang, J. Am. Chem. Soc., 2004, 126, 15412–15422 CrossRef CAS PubMed.
- F. Riobé, P. Grosshans, H. Sidorenkova, M. Geoffroy and N. Avarvari, Chem. – Eur. J., 2009, 15, 380–387 CrossRef PubMed.
- F. Pop, F. Riobé, S. Seifert, T. Cauchy, J. Ding, N. Dupont, A. Hauser, M. Koch and N. Avarvari, Inorg. Chem., 2013, 52, 5023–5034 CrossRef CAS PubMed.
- A. García, B. Insuasty, M. Ángeles Herranz, R. Martin-Álvarez and N. Martín, Org. Lett., 2009, 11, 5398–5401 CrossRef PubMed.
- (a) P. Gamez, P. de Hoog, O. Roubeau, M. Lutz, W. L. Driessen, A. L. Spek and J. Reedijk, Chem. Commun., 2002, 1488–1489 RSC; (b) P. U. Maheswari, B. Modec, A. Pevec, B. Kozlevčar, C. Massera, P. Gamez and J. Reedijk, Inorg. Chem., 2006, 45, 6637–6645 CrossRef CAS PubMed; (c) C. Yuste, L. Canadillas-Delgado, A. Labrador, F. S. Delgado, C. Ruiz-Pérez, F. Lloret and M. Julve, Inorg. Chem., 2009, 48, 6630–6640 CrossRef CAS PubMed.
- D. G. Branzea, A. Fihey, T. Cauchy, A. El-Ghayoury and N. Avarvari, Inorg. Chem., 2012, 51, 8545–8556 CrossRef CAS PubMed.
- S. V. Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2007, 129, 828–838 CrossRef CAS PubMed.
- (a) C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC; (b) B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Chem. Soc. Rev., 2008, 37, 68–83 RSC; (c) A. Frontera, P. Gamez, M. Mascal, T. J. Mooibroek and J. Reedijk, Angew. Chem., Int. Ed., 2011, 50, 9564–9583 CrossRef CAS PubMed; (d) P. Ballester, Acc. Chem. Res., 2013, 46, 874–884 CrossRef CAS PubMed; (e) I. Nazarenko, F. Pop, Q. Sun, A. Hauser, F. Lloret, M. Julve, A. El-Ghayoury and N. Avarvari, Dalton Trans., 2015, 44, 8855–8866 RSC.
- (a) H. T. Chifotides and K. R. Dunbar, Acc. Chem. Res., 2013, 46, 894–906 CrossRef CAS PubMed; (b) A. Frontera, Coord. Chem. Rev., 2013, 257, 1716–1727 CrossRef CAS.
- (a) N. Saracoglu, Tetrahedron, 2007, 63, 4199–4236 CrossRef CAS; (b) G. Clavier and P. Audebert, Chem. Rev., 2010, 110, 3299–3314 CrossRef CAS PubMed.
- Q. Zhou, P. Audebert, G. Clavier, R. Méallet-Renault, F. Miomandre, Z. Shaukat, T.-T. Vu and J. Tang, J. Phys. Chem. C, 2011, 115, 21899–21906 CAS.
- (a) P. Audebert, F. Miomandre, G. Clavier, M.-C. Vernières, S. Badré and R. Méallet-Renault, Chem. – Eur. J., 2005, 11, 5667–5673 CrossRef CAS PubMed; (b) Y.-H. Gong, F. Miomandre, R. Méallet-Renault, S. Badré, L. Galmiche, J. Tang, P. Audebert and G. Clavier, Eur. J. Org. Chem., 2009, 6121–6128 CrossRef CAS; (c) C. Quinton, V. Alain-Rizzo, C. Dumas-Verdes, G. Clavier, F. Miomandre and P. Audebert, Eur. J. Org. Chem., 2012, 1394–1403 CrossRef CAS.
- F. Pop, J. Ding, L. M. Lawson Daku, A. Hauser and N. Avarvari, RSC Adv., 2013, 3, 3218–3221 RSC.
- C. Quinton, V. Alain-Rizzo, C. Dumas-Verdes, F. Miomandre and P. Audebert, Electrochim. Acta, 2013, 110, 693–701 CrossRef CAS.
- Y. Morita, T. Murata, H. Yamochi, G. Saito and K. Nakasuji, Synth. Met., 2003, 135–136, 579–580 CrossRef CAS.
- (a) Y. Morita, S. Maki, M. Ohmoto, H. Kitagawa, T. Okubo, T. Mitani and K. Nakasuji, Synth. Met., 2003, 135–136, 541–542 CrossRef CAS; (b) T. Murata, E. Miyazaki, S. Maki, Y. Umemoto, M. Ohmoto, K. Nakasuji and Y. Morita, Bull. Chem. Soc. Jpn., 2012, 85, 995–1006 CrossRef CAS.
- (a) T. Murata, Y. Morita, K. Fukui, K. Sato, D. Shiomi, T. Takui, M. Maesato, H. Yamochi, G. Saito and K. Nakasuji, Angew. Chem., Int. Ed., 2004, 43, 6343–6346 CrossRef CAS PubMed; (b) T. Murata, Y. Morita, Y. Yakiyama, K. Fukui, H. Yamochi, G. Saito and K. Nakasuji, J. Am. Chem. Soc., 2007, 129, 10837–10846 CrossRef CAS PubMed.
- T. Biet, T. Cauchy and N. Avarvari, Chem. – Eur. J., 2012, 18, 16097–16103 CrossRef CAS PubMed.
- T. Biet and N. Avarvari, Org. Biomol. Chem., 2014, 12, 3167–3174 CAS.
- T. Biet and N. Avarvari, CrystEngComm, 2014, 16, 6612–6620 RSC.
- H. Fujiwara, S. Yokota, S. Hayashi, S. Takemoto and H. Matsuzaka, Physica B, 2010, 405, S15–S18 CrossRef CAS.
- F. Pop, A. Amacher, N. Avarvari, J. Ding, L. M. Lawson Daku, A. Hauser, M. Koch, J. Hauser, S.-X. Liu and S. Decurtins, Chem. – Eur. J., 2013, 19, 2504–2514 CrossRef CAS PubMed.
- F. Pop, S. Seifert, J. Hankache, J. Ding, A. Hauser and N. Avarvari, Org. Biomol. Chem., 2015, 13, 1040–1047 CAS.
- A. F. Cozzolino, I. Vargas-Baca, S. Mansour and A. H. Mahmoudkhani, J. Am. Chem. Soc., 2005, 127, 3184–3190 CrossRef CAS PubMed.
- S. Yokota, K. Tsujimoto, S. Hayashi, F. Pointillart, L. Ouahab and H. Fujiwara, Inorg. Chem., 2013, 52, 6543–6550 CrossRef CAS PubMed.
- V. P. Rao, A. K. Y. Jen and J. B. Caldwell, Tetrahedron Lett., 1994, 35, 3849–3852 CrossRef CAS.
- A. Insuasty, A. Ortiz, A. Tigreros, E. Solarte, B. Insuasty and N. Martín, Dyes Pigm., 2011, 88, 385–390 CrossRef CAS.
- (a) U. Mitschke and P. Bäuerle, J. Mater. Chem., 2000, 10, 1471–1507 RSC; (b) G. Hughes and M. R. Bryce, J. Mater. Chem., 2005, 15, 94–107 RSC.
- (a) H. Fujiwara, Y. Sugishima and K. Tsujimoto, Tetrahedron Lett., 2008, 49, 7200–7203 CrossRef CAS; (b) H. Fujiwara, Y. Sugishima and K. Tsujimoto, J. Phys.: Conf. Ser., 2008, 132, 012025 CrossRef.
- K. Furukawa, Y. Sugishima, H. Fujiwara and T. Nakamura, Chem. Lett., 2011, 40, 292–294 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- J. Xiong, L. Sun, Y. Liao, G.-N. Li, J.-L. Zuo and X.-Z. You, Tetrahedron Lett., 2011, 52, 6157–6161 CrossRef CAS.
- K. L. Huang, N. Bellec, M. Guerro, F. Camerel, T. Roisnel and D. Lorcy, Tetrahedron, 2011, 67, 8740–8746 CrossRef CAS.
- (a) K. Tsujimoto, R. Ogasawara and H. Fujiwara, Tetrahedron Lett., 2013, 54, 1251–1255 CrossRef CAS; (b) K. Tsujimoto, R. Ogasawara, T. Nakagawa and H. Fujiwara, Eur. J. Inorg. Chem., 2014, 3960–3972 CrossRef CAS.
- T. Weil, T. Vosch, J. Hofkens, K. Peneva and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 9068–9093 CrossRef CAS PubMed.
- (a) X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed; (b) F. Würthner and M. Stolte, Chem. Commun., 2011, 47, 5109–5115 RSC.
- (a) S. W. Eaton, L. E. Shoer, S. D. Karlen, S. M. Dyar, E. A. Margulies, B. S. Veldkamp, C. Ramanan, D. A. Hartzler, S. Savikhin, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2013, 135, 14701–14712 CrossRef CAS PubMed; (b) M. G. Debije and P. P. C. Verbunt, Adv. Energy Mater., 2012, 2, 12–35 CrossRef CAS.
- (a) M. P. O'Neil, M. P. Niemczyk, W. A. Svec, D. Gosztola, G. L. Gaines and M. R. Wasielewski, Science, 1992, 257, 63–65 Search PubMed; (b) V. M. Blas-Ferrando, J. Ortiz, K. Ohkubo, S. Fukuzumi, F. Fernández-Lázaro and Á. Sastre-Santos, Chem. Sci., 2014, 5, 4785–4793 RSC.
- (a) F. Würthner, Chem. Commun., 2004, 1564–1579 RSC; (b) F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- M. Jaggi, C. Blum, N. Dupont, J. Grilj, S.-X. Liu, J. Hauser, A. Hauser and S. Decurtins, Org. Lett., 2009, 11, 3096–3099 CrossRef CAS PubMed.
- M. Jaggi, C. Blum, B. S. Marti, S.-X. Liu, S. Leutwyler and S. Decurtins, Org. Lett., 2010, 12, 1344–1347 CrossRef CAS PubMed.
- H. Qiu, C. Wang, J. Xu, G. Lai and Y. Shen, Monatsh. Chem., 2008, 139, 1357–1362 CrossRef CAS.
- Y. Zhang, Z. Xu, H. Qiu, G. Lai and Y. Shen, J. Photochem. Photobiol., A, 2009, 204, 32–38 CrossRef CAS.
- X. Guo, D. Zhang, H. Zhang, Q. Fan, W. Xu, X. Ai, L. Fan and D. Zhu, Tetrahedron, 2003, 59, 4843–4850 CrossRef CAS.
- (a) S. Leroy-Lhez, J. Baffreau, L. Perrin, E. Levillain, M. Allain, M.-J. Blesa and P. Hudhomme, J. Org. Chem., 2005, 70, 6313–6320 CrossRef CAS PubMed; (b) S. Leroy-Lhez, L. Perrin, J. Baffreau and P. Hudhomme, C. R. Chim., 2006, 9, 240–246 CrossRef CAS.
- X. Zheng, D. Zhang and D. Zhu, Tetrahedron Lett., 2006, 47, 9083–9087 CrossRef CAS.
- K. Qvortrup, M. Å. Petersen, T. Hassenkam and M. B. Nielsen, Tetrahedron Lett., 2009, 50, 5613–5616 CrossRef CAS.
- Y. Zhang, L. Cai, C.-Y. Wang, G. Lai and Y. Shen, New J. Chem., 2008, 32, 1968–1973 RSC.
- Y. Zhang, Z. Xu, L. Cai, G. Lai, H. Qiua and Y. Shen, J. Photochem. Photobiol., A, 2009, 200, 334–345 CrossRef.
- S. Castellanos, A. A. Vieira, B. M. Illescas, V. Sacchetti, C. Schubert, J. Moreno, D. M. Guldi, S. Hecht and N. Martín, Angew. Chem., Int. Ed., 2013, 52, 13985–13990 CrossRef CAS PubMed.
- M. V. Solano, E. A. Della Pia, M. Jevric, C. Schubert, X. Wang, C. van der Pol, A. Kadziola, K. Norgaard, D. M. Guldi, M. B. Nielsen and J. O. Jeppesen, Chem. – Eur. J., 2014, 20, 9918–9929 CrossRef CAS PubMed.
- (a) R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. L. Bredas, M. Logdlund and W. R. Salaneck, Nature, 1999, 397, 121–128 CrossRef CAS; (b) N. A. D. Yamamoto, L. L. Lavery, B. F. Nowacki, I. R. Grova, G. L. Whiting, B. Krusor, E. R. de Azevedo, L. Akcelrud, A. C. Arias and L. S. Roman, J. Phys. Chem. C, 2012, 116, 18641–18648 CrossRef CAS.
- K. Tsujimoto, R. Ogasawara, Y. Kishi and H. Fujiwara, New J. Chem., 2014, 38, 406–418 RSC.
- A. Miyazaki and T. Enoki, New J. Chem., 2009, 33, 1249–1254 RSC.
- (a) M. Å. Petersen, A. S. Andersson, K. Kilså and M. B. Nielsen, Eur. J. Org. Chem., 2009, 1855–1858 CrossRef CAS; (b) M. B. Nielsen, S. L. Broman, M. Å. Petersen, A. S. Andersson, T. S. Jensen, K. Kilså and A. Kadziola, Pure Appl. Chem., 2010, 82, 843–852 CrossRef CAS.
- S. Kato, M. Kivala, W. B. Schweizer, C. Boudon, J.-P. Gisselbrecht and F. Diederich, Chem. – Eur. J., 2009, 15, 8687–8691 CrossRef CAS PubMed.
- A. S. Andersson, F. Diederich and M. B. Nielsen, Org. Biomol. Chem., 2009, 7, 3474–3480 CAS.
- A. S. Andersson, L. Kerndrup, A. Ø. Madsen, K. Kilså and M. B. Nielsen, J. Org. Chem., 2009, 74, 375–382 CrossRef CAS PubMed.
- J. Guasch, L. Grisanti, V. Lloveras, J. Vidal-Gancedo, M. Souto, D. C. Morales, M. Vilaseca, C. Sissa, A. Painelli, I. Ratera, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2012, 51, 11024–11028 CrossRef CAS PubMed.
- (a) Y. Morita, T. Ohba, N. Haneda, S. Maki, J. Kawai, K. Hatanaka, K. Sato, D. Shiomi, T. Takui and K. Nakasuji, J. Am. Chem. Soc., 2000, 122, 4825–4826 CrossRef CAS; (b) S. Nishida, J. Kawai, M. Moriguchi, T. Ohba, N. Haneda, K. Fukui, A. Fuyuhiro, D. Shiomi, K. Sato, T. Takui, K. Nakasuji and Y. Morita, Chem. – Eur. J., 2013, 19, 11904–11915 CrossRef CAS PubMed.
- (a) O. Elsner, D. Ruiz-Molina, J. Vidal-Gancedo, C. Rovira and J. Veciana, Chem. Commun., 1999, 579–580 RSC; (b) C. Sporer, I. Ratera, D. Ruiz-Molina, Y. Zhao, J. Vidal-Gancedo, K. Wurst, P. Jaitner, K. Clays, A. Persoons, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2004, 43, 5266–5268 CrossRef CAS PubMed; (c) I. Ratera, C. Sporer, D. Ruiz-Molina, N. Ventosa, J. Baggerman, A. M. Brouwer, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2007, 129, 6117–6129 CrossRef CAS PubMed; (d) M. Souto, D. C. Morales, J. Guasch, I. Ratera, C. Rovira, A. Painelli and J. Veciana, J. Phys. Org. Chem., 2014, 27, 465–469 CrossRef CAS.
- (a) M. Chahma, X. Wang, A. van der Est and M. Pilkington, J. Org. Chem., 2006, 71, 2750–2755 CrossRef CAS PubMed; (b) M. Chahma, K. Macnamara, A. van der Est, A. Alberola, V. Polo and M. Pilkington, New J. Chem., 2007, 31, 1973–1978 RSC.
- S. Nishida, Y. Morita, K. Fukui, K. Sato, D. Shiomi, T. Takui and K. Nakasuji, Angew. Chem., Int. Ed., 2005, 44, 7277–7280 CrossRef CAS PubMed.
- S. Nishida, K. Fukui and Y. Morita, Chem. – Asian J., 2014, 9, 500–505 CrossRef CAS PubMed.
- M. Souto, M. V. Solano, M. Jensen, D. Bendixen, F. Delchiaro, A. Girlando, A. Painelli, J. O. Jeppesen, C. Rovira, I. Ratera and J. Veciana, Chem. – Eur. J., 2015, 21, 8816–8825 CrossRef CAS PubMed.
- M. Souto, J. Guasch, V. Lloveras, P. Mayorga, J. T. López Navarrete, J. Casado, I. Ratera, C. Rovira, A. Painelli and J. Veciana, J. Phys. Chem. Lett., 2013, 4, 2721–2726 CrossRef CAS.
- J. Guasch, L. Grisanti, M. Souto, V. Lloveras, J. Vidal-Gancedo, I. Ratera, A. Painelli, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2013, 135, 6958–6967 CrossRef CAS PubMed.
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296–1323 CrossRef CAS PubMed.
- (a) M. Wang, M. Ford, H. Phan, J. Coughlin, T.-Q. Nguyen and G. C. Bazan, Chem. Commun., 2016, 52, 3207–3210 RSC; (b) R. Stalder, S. R. Puniredd, M. R. Hansen, U. Koldemir, C. Grand, W. Zajaczkowski, K. Müllen, W. Pisula and J. R. Reynolds, Chem. Mater., 2016, 28, 1286–1297 CrossRef CAS.
- (a) F. Oton, R. Pfattner, E. Pavlica, Y. Olivier, G. Bratina, J. Cornil, J. Puigdollers, R. Alcubilla, X. Fontrodona, M. Mas-Torrent, J. Veciana and C. Rovira, CrystEngComm, 2011, 13, 6597–6600 RSC; (b) L. Tan, Y. Guo, Y. Yang, G. Zhang, D. Zhang, G. Yu, W. Xu and Y. Liu, Chem. Sci., 2012, 3, 2530–2541 RSC; (c) R. Pfattner, E. Pavlica, M. Jaggi, S.-X. Liu, S. Decurtins, G. Bratina, J. Veciana, M. Mas-Torrent and C. Rovira, J. Mater. Chem. C, 2013, 1, 3985–3988 RSC; (d) A. Amacher, H. Luo, Z. Liu, M. Bircher, M. Cascella, J. Hauser, S. Decurtins, D. Zhang and S.-X. Liu, RSC Adv., 2014, 4, 2873–2878 RSC.
- (a) D. Cortizo-Lacalle, S. Arumugam, S. E. T. Elmasly, A. L. Kanibolotsky, N. J. Findlay, A. R. Inigo and P. J. Skabara, J. Mater. Chem., 2012, 22, 11310–11315 RSC; (b) S. Arumugam, D. Cortizo-Lacalle, S. Rossbauer, S. Hunter, A. L. Kanibolotsky, A. R. Inigo, P. A. Lane, T. D. Anthopoulos and P. J. Skabara, ACS Appl. Mater. Interfaces, 2015, 7, 27999–28005 CrossRef PubMed.
- A. Amacher, C. Yi, J. Yang, M. P. Bircher, Y. Fu, M. Cascella, M. Grätzel, S. Decurtins and S.-X. Liu, Chem. Commun., 2014, 50, 6540–6542 RSC.
Footnote |
| † Present address: School of Chemistry, The University of Nottingham, NG72RD Nottingham, UK. E-mail: Flavia.Pop@nottingham.ac.uk |
| This journal is © The Royal Society of Chemistry 2016 |