
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA switchable dual organocatalytic system and the enantioselective total synthesis of the quadrane sesquiterpene suberosanone†
Yajun
Ren
a,
Marc
Presset‡
a,
Jeremy
Godemert
a,
Nicolas
Vanthuyne
a,
Jean-Valère
Naubron
b,
Michel
Giorgi
b,
Jean
Rodriguez
*a and
Yoann
Coquerel
*a
aAix Marseille Université, Centrale Marseille, CNRS, iSm2 UMR7313, F-13397 Marseille, France. E-mail: jean.rodriguez@univ-amu.fr; yoann.coquerel@univ-amu.fr; Fax: +33 491 289 187; Tel: +33 491 289 088
bAix Marseille Université, CNRS, Spectropole FR1739, F-13397 Marseille, France
First published on 15th April 2016
Abstract
The combination of aminocatalysis with N-heterocyclic carbene catalysis has been extended to a switchable dual catalytic system, which allowed a direct enantioselective entry to bridged bicyclo[3.n.1] ring systems and the total synthesis of the natural product (1R)-suberosanone.
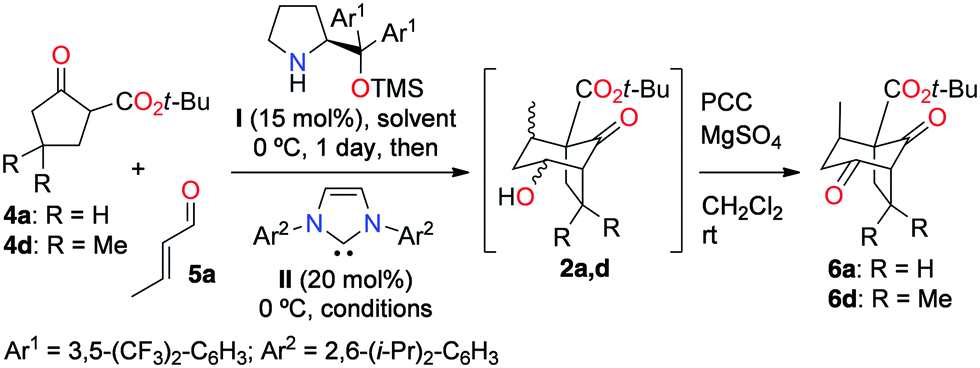
Quadranes form a small class of cytotoxic sesquiterpene natural products, originated either from terrestrial or marine organisms, and belonging to the larger group of polyquinanes.1 It is worth mentioning that natural products have in the past and continue to play a crucial role in the discovery of drugs for the benefit of mankind.2 Although the first known naturally occurring quadrane, namely quadrone, was isolated back in 1978, despite considerable synthetic efforts, no enantioselective total synthesis of a quadrane natural product has been reported.1,3 Actually, most quadranes are of unknown absolute configuration even though they are all presumed to exist naturally as their (1R) enantiomers.1,4 The absolute configuration of the quadrane suberosanone (1) was recently assigned as (1S) by chemical synthesis.5 Intrigued by this oddity, and motivated by the development of an original enantioselective organocatalytic method for the synthesis of functionalized bridged bicyclo[3.2.1]octane compounds6 applicable to the synthesis of most, if not all, quadranes, we embarked on the total synthesis of (1R)-suberosanone (1). It was devised that (1R)-1 would be accessible in a few steps starting from the bicyclo[3.2.1]octane (1R)-2, which we planned to prepare using a yet-to-discover enantioselective dual organocatalytic Michael/aldolization sequence from the β-ketoester 4 and crotonaldehyde (5a, Scheme 1).7
 | ||
| Scheme 1 Retrosynthetic analysis for suberosanone, and planned enantioselective dual organocatalytic Michael/aldol sequence. BB = Brønsted base activation. IM = iminium activation. | ||
Based on the accumulated knowledge on enantioselective organocatalytic Michael additions with 1,3-dicarbonyl compounds, we were confident that highly enantio-enriched Michael adduct intermediates of type 3 could be obtained from pronucleophiles of type 4 under iminium activation.8 An additional Brønsted base would then be required to operate the ring-closing aldolization step. Although enticing, this idea raised some challenging issues: (i) iminium activation is a reversible process;8 (ii) previous work on the synthesis of bridged ring systems of type 2via Brønsted base-promoted Michael/aldolization sequences in the racemic series established that these reactions are fully reversible when performed with common Brønsted bases (e.g. DBU);9 (iii) Michael adducts of type 3 readily racemize, precluding their isolation in high enantiopurity under standard conditions;10a and (iv) intermediates of type 3 amenable to applications to the total synthesis of quadranes exhibit a gem-dimethyl group in the vicinity of the aldolization site, which should both notably slow down the aldolization step and favor Brønsted base-catalyzed retro-Michael/Michael processes leading to racemization. Recently, it was discovered that hindered imidazolylidene N-heterocyclic carbenes (NHCs) are potent Brønsted bases capable of catalyzing Michael/aldolization sequences, but with little tendency for reversibility.11 It was hypothesized that the planned enantioselective Michael/aldolization sequence leading to (1R)-2 could be achieved using a sequential combination of the aminocatalyst I and the NHC catalyst II in a one-pot dual catalytic process.12
In order to test this idea, the prototypical reactions of the β-ketoesters 4a and 4d with crotonaldehyde (5a) were examined under various dual catalytic conditions (Table 1). Because all the four possible diastereomers of 2a,d can be expected in different proportions, the crude reaction products were directly oxidized into the corresponding diketones 6a,d to simplify analysis. The benchmark result was obtained by the sequential combination of catalyst I and DBU, which afforded product 6a with low enantioselectivity due to DBU-catalyzed racemization processes (entry 1). In contrast, it was found that the sequential addition of catalysts I and II to solutions of 4a and 5a produced, after oxidation, the diketone 6a in generally good yields and stereoselectivities (entries 2–5). The reaction in dichloromethane (entry 2) gave the best enantioselectivity but a low yield, which could be improved in toluene (entry 3). Alternatively, methanol afforded comparable results in a somewhat accelerated reaction (entry 4), while substituting catalyst I by the so-called MacMillan's second generation catalyst13 gave 6a in lower yield and enantioselectivity (entry 5). Other dual organocatalytic systems combining an aminocatalyst with a NHC have been reported in the past few years.14 Among these, Lathrop and Rovis reported an elegant enantioselective Michael/crossed benzoin sequence from the methyl ester analog of 4a (the precursor of 6c, see Fig. 1) and aldehyde 5a using a combination of catalyst I and an achiral triazolylidene NHC catalyst to give a fused bicyclo[3.3.0]octane product.10,15 Remarkably, and in sharp contrast to the Lathrop and Rovis study, no fused bicyclo[3.3.0]octane products could be detected in the intermediate crude products 2, illustrating the perfectly switchable nature of the overall dual organocatalytic transformation as a function of the NHC catalyst employed (triazolylidene vs. hindered imidazolylidene).16 More generally, this represents the first described switchable dual catalytic enantioselective transformation. Stimulated by these results, we then moved to the more challenging substrate 4d (entries 6–9). In that case, the reaction was only found possible in methanol and afforded product 6d with moderate enantioselectivity under the conditions previously used for 6a (compare entries 4 and 7). It was hypothesized that reducing the amount of free NHC II in solution would limit the undesired competitive NHC-catalyzed Michael addition leading to racemization. Indeed, a reaction conducted with only 3 mol% of II allowed the isolation of product 6d with a significantly better enantioselectivity (entry 8). Following the same idea, masking the NHC as its azolium salt in situ should allow maintaining of only a tiny amount of the free NHC II in solution. In practice, it was found that adding acetic acid together with II after the Michael addition step afforded the product 6d in good yield and improved enantioselectivity, at the expense of prolonged reaction time (entry 9).17 The reaction was then examined using other cyclic α-activated ketones and α,β-unsaturated aldehydes, which afforded the bridged bicyclic products 6a–m with good to excellent yields and stereoselectivities (Fig. 1). The enhancement of diastereoselectivity observed in many cases was expected and ensued from the decomposition of the minor diastereomers of 2 having equatorial substituents by methanol-mediated retro-Dieckmann fragmentation processes (see the ESI†).9,18 In a few cases, the MacMillan's second generation catalyst13 performed better than its counterpart I (e.g. for 6i and 6j, see the ESI†). The determination of the absolute configurations in products 6a–m is based on previous work,8,10 and was confirmed by derivatization, X-ray diffraction analysis, and full chiroptical characterization of the targeted natural product (see below and the ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Conditions | Yielda | drb | erc |
| a For the isolated pure product (0.5 mmol scale reactions). b Determined by NMR analysis of the crude product. c For the major diastereomer, determined by HPLC. d Reaction performed with DBU instead of II. e Reaction performed with so-called MacMillan's second generation catalyst instead of I. f Reaction performed with only 3 mol% of II. | |||||
| 1d | MeOH | 2 days | 6a, 77% | 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
5:1 |
| 2 | CH2Cl2 | 2 days | 6a, 28% | 4:1 |
27:1 |
| 3 | Toluene | 2 days | 6a, 71% | 4:1 |
26:1 |
| 4 | MeOH | 1 day | 6a, 75% | 4:1 |
18:1 |
| 5e | MeOH | 1 day | 6a, 60% | 4:1 |
10:1 |
| 6 | Toluene | 2 days | 6d, <5% | — | — |
| 7 | MeOH | 1 day | 6d, 80% | 2:1 |
8:1 |
| 8f | MeOH | 3 days | 6d, 90% | 2:1 |
12:1 |
| 9 | MeOH | AcOH (20 mol%), 4 days | 6d, 90% | 2:1 |
16:1 |
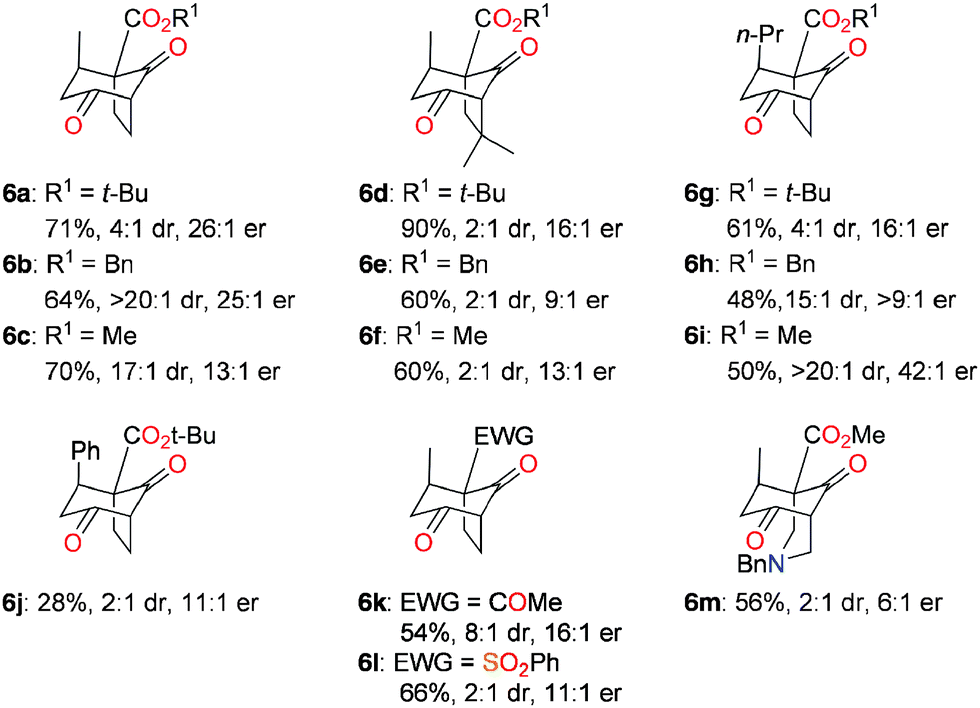 | ||
| Fig. 1 Scope of the dual organocatalytic reaction. See the ESI† for full experimental details. | ||
With the new enantioselective methodological tool described above in hand, the total synthesis started with a scale up study, which afforded 2d as a mixture of four diastereomers in 90% yield on a 12 gram scale (Scheme 2). The oxidation of an analytic sample of 2d delivered the diketone 6d with similar yield and stereoselectivities when compared to the methodological scale series (2:1 dr, 8:1 er, compare with Table 1, entry 7).19 The Barton–McCombie deoxygenation of 2d afforded the key intermediate 7 in 85% yield (2:1 dr, 9.2 g batch), which was later in the synthesis confirmed to maintain the 8:1 er of the major diastereomer. The initially planned strategy to convert 7 into the natural product 1 and its congeners was relied on the early olefination of the ketone carbonyl group at C2. Regrettably, we soon had to face the problem that the C2 carbonyl group and the corresponding olefins were extremely difficult to react, and when possible, with the desired diastereoselectivity. These difficulties were attributed to severe cumulative steric effects at C2 caused by its neopentylic position, a 1,3-diaxial interaction on the β-face, a gem-dimethyl group on the α-face, and a conformational rigidity. Overall, the chemical transformation of the key intermediate 7 revealed to be much more difficult than anticipated, but also more exciting. Ultimately, we had to proceed through the protection of the ketone group at C2 in two steps for the reasons given above, which actually turned out beneficial for the issue of the total synthesis. The diastereoselective reduction of the ketone group in 7 (2:1 dr) with sodium borohydride afforded the corresponding mixture of diastereomers 8a and 8b (77%, 2:1 dr). Following the serendipitous observation that 8a and 8b could be kinetically differentiated in etherification reactions, they were separated by the selective benzylation of the minor diastereomer 8b having an equatorial methyl group to give 9b using NaH/BrBn. A single recrystallization of the remaining diastereomerically pure alcohol 8a (>50:1 dr, 8:1 er) afforded enantiopure crystalline 8a (52%, >199:1 er) suitable for X-ray diffraction analysis, which confirmed both its relative and absolute configurations to be as depicted (see the ESI†).20 The alcohol 8a was then converted into its benzyl ether 9a using KH/BrBn, and the subsequent Arndt–Eistert homologation21 afforded the carboxylic acid 11via the methyldiazoketone 10. The carboxylic moiety in 11 was transformed into the corresponding methyl ketone with methyl lithium, and the C2 carbonyl group was restored to afford the desired diketone 12. From 12, the endgame strategy was largely inspired by previous work on the total syntheses of quadranes.1 It was found that t-BuOK promoted an efficient ring-closing aldolization/dehydration sequence from 12 to give the corresponding cyclopentenone 13, the diastereoselective α-methylation of which under standard conditions furnished the tricyclic late intermediate 14. Finally, the diastereoselective hydrogenation of the double bond in 14 afforded quantitatively the target product (1R)-suberosanone (1). The enantioselective synthesis of 12 also constitutes a formal enantioselective total synthesis of the related quadrane natural product (1R)-suberosenone.5
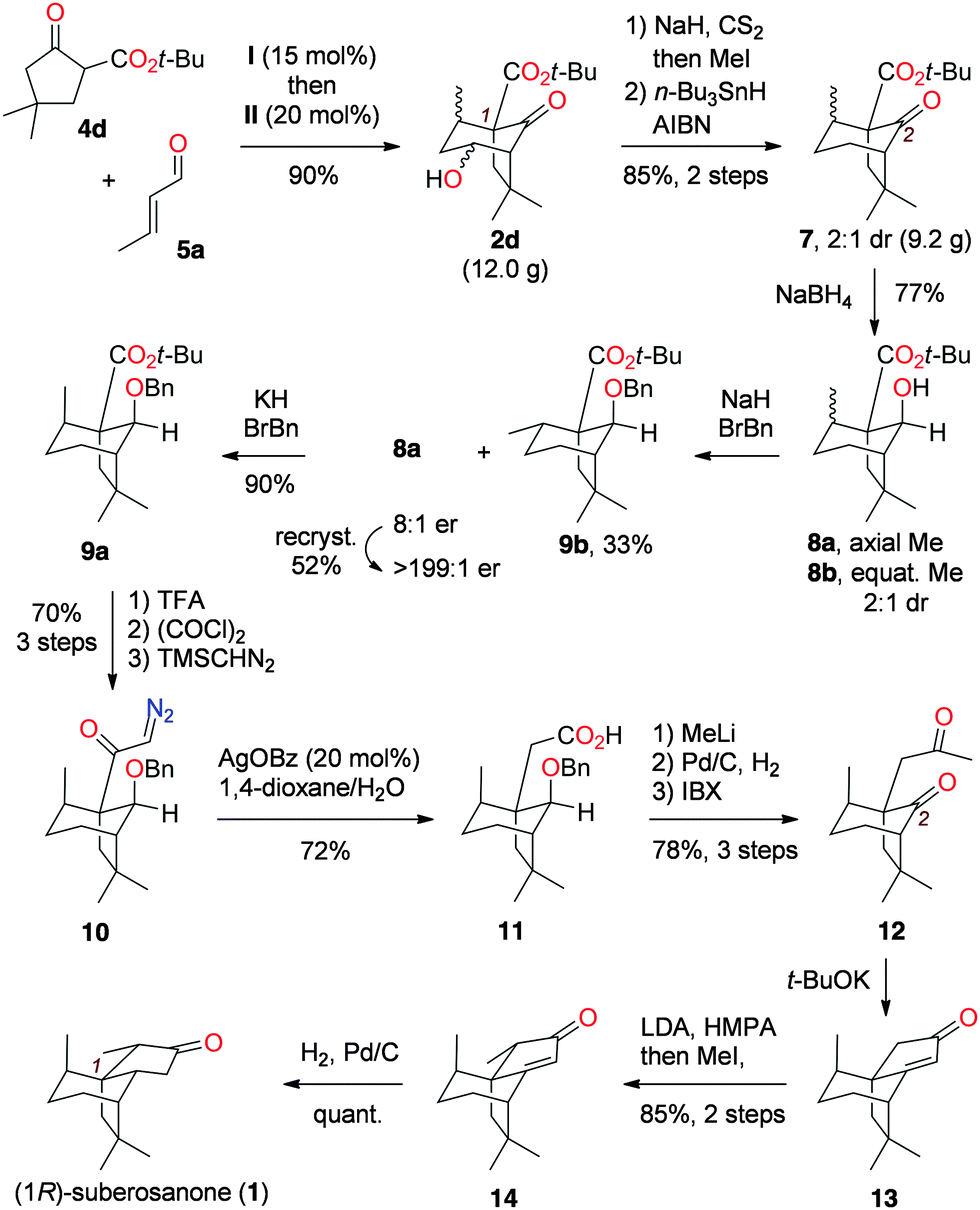 | ||
| Scheme 2 Total synthesis of (1R)-suberosanone (1). | ||
With synthetic samples of (1R)-1 in hand, we tackled the issue of the determination of the absolute configuration of naturally occurring 1. To do so, we performed a full chiroptical characterization of (1R)-1, including optical rotatory dispersion (ORD), electronic circular dichroism (ECD) and vibrational circular dichroism (VCD) studies (see the ESI†). The [α]D of the natural sample of 1 extracted from the gorgonian Isis hippuris was measured at [α]25D = −60 (c = 0.1, CHCl3),22 while the synthetic sample of (1R)-1 prepared by Dumas and co-workers showed [α]19D = +53.4 (c = 0.1, CHCl3).5 On that basis, the absolute configuration of naturally occurring 1 was assigned as (1S). The measurement of the optical rotation of our synthetic samples of (1R)-1 (er > 199:1) afforded the following values: [α]25D = +20 (c = 0.1, CHCl3), [α]15D = +20 (c = 0.1, CHCl3), and [α]25D = +11 (c = 0.1, EtOH).23 Additionally, Stephens and co-workers predicted [α]D = −20.6 (gas phase) for (1R)-1 by DFT calculations.4 A puzzling situation! Although we have no sound explanation for these discrepancies in the [α]D values,24 some clues came from the comparison of the ORD of our synthetic (1R)-1 with the predicted ORD (DFT) for (1R)-1, as well as with the experimental and theoretical ORDs of (1R)-quadrone (Fig. 2). On the basis of similar shapes of ORDs, and in line with Stephens and co-workers’ intuition on the occurrence of a single absolute stereogenicity in quadranes biosynthesis, the absolute configuration of naturally occurring 1 is likely to be (1R). However, a clear-cut answer to this question can only arise from additional chiroptical characterization of the naturally occurring material.
 | ||
| Fig. 2 Experimental and calculated ORDs of (1R)-suberosanone (1) and (1R)-quadrone. Data from ref. 4 and our own measurements. | ||
In summary, a remarkable divergence was identified in dual organocatalytic transformations combining a secondary amine and a NHC catalyst, the NHC operating as the reactivity switch. A practical enantioselective method for the synthesis of functionalized bicyclo[3.n.1] derivatives resulted. This original switchable bicatalytic transformation opens up avenues for the conceptualization of switchable multicatalytic transformations. The dual catalytic method described herein was initially elaborated to provide the first enantioselective route to the cytotoxic quadrane natural products, which allowed the total synthesis of (1R)-suberosanone and the formal synthesis of (1R)-suberosenone. This work provides all the necessary chiroptical information for the determination of the absolute configuration of naturally occurring suberosanone when more data are available.
Financial support from the China Scholarship Council (YR fellowship), the Ecole Normale Supérieure Cachan (MP fellowship), Aix-Marseille Université, Centrale Marseille, and the Centre National de la Recherche Scientifique (CNRS) is gratefully acknowledged. The CRCMM is acknowledged for support computing facilities.
Notes and references
- Review: M. Presset, Y. Coquerel and J. Rodriguez, Eur. J. Org. Chem., 2010, 2247 CrossRef CAS.
- G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 3670 CrossRef CAS PubMed and previous reviews from the same authors.
- A couple reports on non-racemic diastereoselective total syntheses of quadranes, from the chiral pool and using a chiral auxiliary, respectively, have erroneously been entitled “Enantioselective total synthesis of…”. See M. Torihata, T. Nakahata and S. Kuwahara, Org. Lett., 2007, 9, 2557 CrossRef CAS PubMed , and ref. 5.
- P. J. Stephens, D. M. McCann, F. J. Devlin and A. B. Smith, III, J. Nat. Prod., 2006, 69, 1055 CrossRef CAS PubMed.
- M. Kousara, A. Ferry, F. Le Bideau, K. L. Serré, I. Chataigner, E. Morvan, J. Dubois, M. Chéron and F. Dumas, Chem. Commun., 2015, 51, 3458 RSC.
- Highlight: (a) M. Presset, Y. Coquerel and J. Rodriguez, ChemCatChem, 2012, 4, 172 CrossRef CAS . Review: ; (b) M. Presset, Y. Coquerel and J. Rodriguez, Chem. Rev., 2013, 113, 525 CrossRef CAS PubMed . Recent contributions: ; (c) M. Tsakos, M. R. J. Elsegood and C. G. Kokotos, Chem. Commun., 2013, 49, 2219 RSC; (d) A. Lefranc, L. Gunée, S. Goncalves-Contal and A. Alexakis, Synlett, 2014, 2947 CAS; (e) A. Lefranc, L. Gremaud and A. Alexakis, Org. Lett., 2014, 16, 5242 CrossRef CAS PubMed; (f) A. R. Burns, A. G. E. Madec, D. W. Low, I. D. Roya and H. W. Lam, Chem. Sci., 2015, 6, 3550 RSC.
- Selected reviews on multicatalysis: (a) L. M. Ambrosini and T. H. Lambert, ChemCatChem, 2010, 2, 1373 CrossRef CAS; (b) S. Piovesana, D. M. Scarpino Schietroma and M. Bella, Angew. Chem., Int. Ed., 2011, 50, 6216 CrossRef CAS PubMed; (c) R. C. Wende and P. R. Schreiner, Green Chem., 2012, 14, 1821 RSC; (d) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633 RSC; (e) Z. Du and Z. Shao, Chem. Soc. Rev., 2013, 42, 1337 RSC; (f) G. Jürjens, A. Kirschning and D. A. Candito, Nat. Prod. Rep., 2015, 32, 723 RSC.
- Selected reviews: (a) K. L. Jensen, G. Dickmeiss, H. Jiang, L. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248 CrossRef CAS PubMed; (b) S. Meninno and A. Lattanzi, Chem. Commun., 2013, 49, 3821 RSC.
- Y. Coquerel, M.-H. Filippini, D. Bensa and J. Rodriguez, Chem. – Eur. J., 2008, 14, 3078 CrossRef CAS PubMed.
- (a) S. P. Lathrop and T. Rovis, J. Am. Chem. Soc., 2009, 131, 13628 CrossRef CAS PubMed . With β-ketosulfones: ; (b) D. Enders, A. Grossmann, H. Huang and G. Raabe, Eur. J. Org. Chem., 2011, 4298 CrossRef CAS.
- (a) T. Boddaert, Y. Coquerel and J. Rodriguez, Chem. – Eur. J., 2011, 17, 2266 CrossRef CAS PubMed . For the reversibility issue, see also: ; (b) E. M. Phillips, M. Riedrich and K. A. Scheidt, J. Am. Chem. Soc., 2010, 132, 13179 CrossRef CAS PubMed . For other applications from our laboratory, see: ; (c) M. Hans, L. Delaude, J. Rodriguez and Y. Coquerel, J. Org. Chem., 2014, 79, 2758 CrossRef CAS PubMed; (d) F. Perez, Y. Ren, T. Boddaert, J. Rodriguez and Y. Coquerel, J. Org. Chem., 2015, 80, 1092 CrossRef CAS PubMed; (e) F. Nawaz, M. Zaghouani, D. Bonne, O. Chuzel, J. Rodriguez and Y. Coquerel, Eur. J. Org. Chem., 2013, 8253 CrossRef CAS . See also: J. Chen and Y. Huang, Nat. Commun., 2014, 5, 3437 Search PubMed.
- Hindered NHCs such as II react reversibly with aldehydes to produce shielded Breslow intermediates of reduced nucleophilicity, limiting their applications in the benzoin reaction. For a discussion, see: C. Burstein, S. Tschan, X. Xie and F. Glorius, Synthesis, 2006, 2418 CAS.
- G. Lelais and D. W. C. MacMillan, Aldrichimica Acta, 2006, 39, 79 CAS.
- Review: J. Gu, W. Du and Y.-C. Chen, Synthesis, 2015, 3451 CAS.
- The triazolylidene catalyst employed in ref. 10 is ca. 106 times less basic than the NHC catalyst II. (a) E. M. Higgins, J. A. Sherwood, A. G. Lindsay, J. Armstrong, R. S. Massey, R. W. Alder and A. C. O'Donoghue, Chem. Commun., 2011, 47, 1559 RSC; (b) R. S. Massey, C. J. Collett, A. G. Lindsay, A. D. Smith and A. C. O'Donoghue, J. Am. Chem. Soc., 2012, 134, 20421 CrossRef CAS PubMed.
- For switchable monocatalytic transformations with NHCs, see: (a) C. Guo, B. Sahoo, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17402 CrossRef CAS PubMed; (b) C. Guo, M. Fleige, D. Janssen-Müller, C. G. Daniliuc and F. Glorius, Nat. Chem., 2015, 7, 842 CrossRef CAS PubMed.
- A control experiment using sodium acetate (20 mol%) as the Brønsted base left, as expected, the Michael adduct intermediate 3 unchanged after prolonged reaction time.
- This diastereomeric enrichment is regrettably not possible with the t-Bu ester derivatives and the products incorporating a gem-dimethyl group.
- The scale up study was realized before the conditions in Table 1, entries 8 and 9 were identified.
- CCDC 1449766 contains the supplementary crystallographic data (CIF) for compound 8a.
- For reviews: (a) W. Kirmse, Eur. J. Org. Chem., 2002, 2193 CrossRef CAS; (b) Y. Coquerel and J. Rodriguez, in Molecular Rearrangements in Organic Synthesis, ed. C. Rojas, John Wiley & Sons, Hoboken, NJ, 2015, ch. 3, pp. 59–84 Search PubMed.
- Isolation and characterization: J.-H. Sheu, K.-C. Hung, G.-H. Wang and C.-Y. Duh, J. Nat. Prod., 2000, 63, 1603 CrossRef CAS.
- Mean values of 5 or more independent measurements on different synthetic samples performed by different operators.
- We can remember that, as early as 1960, in the introduction of his pioneer book on the subject, Carl Djerassi wrote: “The chief message of this book can actually be constructed as a complaint against this unfortunate temporary victory of monochromatic polarimetry over rotatory dispersion.” And later on: “…rotations in the ultraviolet region are (for colorless compounds) invariably greater than for the conventional sodium D line. Comparisons of compounds with small [α]Dare therefore better carried out at a lower wavelength, which can be selected from the dispersion curve.” C. Djerassi, in Optical Rotatory Dispersion, McGraw-Hill series in Advanced Chemistry, ed. W. C. Fernelius, L. P. Hammett, D. N. Hume, J. A. Pole, J. D. Roberts and H. H. Williams, McGraw-Hill Book Company, New York, 1960 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedures, characterization data and copies of NMR spectra and HPLC chromatograms for all compounds; CIF for compound 8a. CCDC 1449766. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc01689h |
| ‡ Present address: Université Paris-Est, ICMPE (UMR 7182), CNRS, UPEC, F-94320 Thiais, France. |
| This journal is © The Royal Society of Chemistry 2016 |