
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
‘Clickable’ ZnO nanocrystals: the superiority of a novel organometallic approach over the inorganic sol–gel procedure†
Agnieszka
Grala
ab,
Małgorzata
Wolska-Pietkiewicz
a,
Wojciech
Danowski
a,
Zbigniew
Wróbel
b,
Justyna
Grzonka
bcd and
Janusz
Lewiński
*ab
aWarsaw University of Technology, Faculty of Chemistry, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: lewin@ch.pw.edu.pl
bPolish Academy of Sciences, Institute of Physical Chemistry, Kasprzaka 44/52, 01-224 Warsaw, Poland
cWarsaw University of Technology, Faculty of Materials Science and Engineering, Wołoska 141, 02-507 Warsaw, Poland
dInstitute of Electronic Materials Technology, Wolczynska 133, 01-919 Warsaw, Poland
First published on 3rd May 2016
Abstract
We demonstrate for the first time a highly efficient Cu(I)-catalyzed alkyne–azide cycloaddition reaction on the surface of ZnO nanocrystals with retention of their photoluminescence properties. Our comparative studies highlight the superiority of a novel self-supporting organometallic method for the preparation of brightly luminescent and well-passivated ZnO nanocrystals over the traditional sol–gel procedure.
The crucial features of inorganic nanocrystals (NCs), such as unique physicochemical properties and stability, are strongly affected by the organic shell coating their inorganic core.1 Therefore, an appropriate choice of the functionalizing ligands grafted onto the surface is a key factor in the design of functional NCs. Moreover, the external organic coating significantly influences both interactions with the environment and further application possibilities of nanomaterials. The most valuable surface transformations refer to the conjugation of NCs with other molecules that provide desired functionalities. In this regard, one of the most powerful tools enabling numerous modifications is the so-called click chemistry, defined by Sharpless as a group of selective, fast and highly efficient reactions.2 Copper(I)-catalyzed azide–alkyne cycloaddition (CuACC), being highly specific and utilizing mild reaction conditions, has become the most widely investigated click reaction to achieve an efficient and rapid surface modification of nanostructures.3 Despite many advantages, the application of Cu as a catalyst imposes numerous impediments, particularly copper ions are known to quench the photoluminescence (PL) of semiconductor NCs.4 The same behaviour is commonly assumed for ZnO nanostructures. However, according to our literature survey, this issue has been scarcely discussed, and essentially the reported observations are not supported by the corresponding experimental data.5
The interest in ZnO-based nanomaterials as heavy metal-free substituents of commonly used CdX NCs has been recently booming in various directions.6 The use of ZnO NCs as fluorescent agents in biological imaging is an especially promising application.7 For this purpose, there is a special need for elaboration of effective surface modification methods providing nanocrystals conjugated with biomolecules. To realize such a demand, the most appropriate tool is the CuACC reaction; however, the NCs' external ligand shell should be impermeable to Cu ions. To the best of our knowledge, there are no reports on successful strategies allowing ZnO NC functionalization using classic copper(I)-catalyzed click chemistry with preservation of their PL properties. Recently, our group developed a novel self-supporting organometallic approach for the synthesis of liquid crystalline carboxylate ligand-coated ZnO NCs which were effectively used for the preparation of highly-ordered hybrid nanomaterials at the air/water interface.8 Simultaneously, we also demonstrated that ZnO NCs obtained by this organometallic procedure exhibit essentially a very low negative impact on mammalian cell lines.9 We anticipated that the observed low cytotoxicity stems from the NC effective surface passivation and the impermeable organic shell. Thus, we wondered if this type of well-passivated ZnO NC could be prone to the ligand shell functionalization using CuACC while retaining the PL properties of the nanoparticles.
In order to verify our hypothesis that the self-supporting organometallic procedure provides ZnO NCs with the ligand shell sufficiently shielding against Cu ions during the CuACC process, we selected ZnO NCs stabilized by deprotonated 10-undecynoic acid (una-H) for Cu(I)-mediated click chemistry. Initially, the una ligand-passivated ZnO NCs (hereafter denoted as una-ZnO NCs) were synthesized according to the self-supporting method,8 which involves: (i) the equimolar reaction between Et2Zn and una-H in a donor solvent and (ii) the exposure of the in situ synthesized [EtZn(una)] precursor10 to air resulting in its subsequent transformation to una-ZnO NCs, forming stable colloidal solutions in THF, CHCl3 and DMSO (Scheme 1). Prior to further processing and after the purification (see the ESI†), the una-ZnO NCs were characterized using a wide library of analytical techniques. The resulting NCs are coated by the monoanionic carboxylate ligand as proven by thermogravimetric analysis (TGA). The thermal decomposition of una-ZnO NCs shows one maximum at ca. 450 °C corresponding to the coating monoanionic una ligands, while pure una-H decomposes at ca. 238 °C (Fig. 1). Inspection of the una-ZnO NCs using high-resolution transmission electron microscopy (HRTEM) and dynamic light scattering (DLS) gave inorganic core average diameters of 4.8 ± 0.5 nm (Fig. 1; for the corresponding powder X-ray diffraction data see Fig. S8, ESI†) and the average hydrodynamic diameter of 8.7 nm (Fig. S9, ESI†),11 respectively. In the IR spectrum of una-ZnO NCs, the presence of two strong or medium intensity bands characteristic of the asymmetric νas(COO) and symmetric νs(COO) stretching at 1530 and 1359 cm−1, (Δν = 171 cm−1) indicated the bridging bidentate μ2-coordination mode of the coating carboxylate moieties (Fig. 4). The 1H NMR spectrum of una-ZnO NCs exhibited broader signals corresponding to that of the parent una-H molecules (Fig. 3a and Fig. S6, ESI†). The most essential information indicating that the una ligand binding to the surface of NCs provided a broadened triplet at 2.33 ppm assigned to the resonance of O2CCH2 protons which are the closer to the NC surface. The corresponding UV-Vis absorption and photoluminescence (PL) spectra are shown in Fig. 5.
 | ||
| Scheme 1 Preparation of ZnO NCs: (i) the precursor synthesis, (ii) its transformation to una-ZnO NCs, and (iii) the functionalization of brightly luminescent una-ZnO NCs by the CuACC reaction affording tr-NCs with preserved PL. | ||
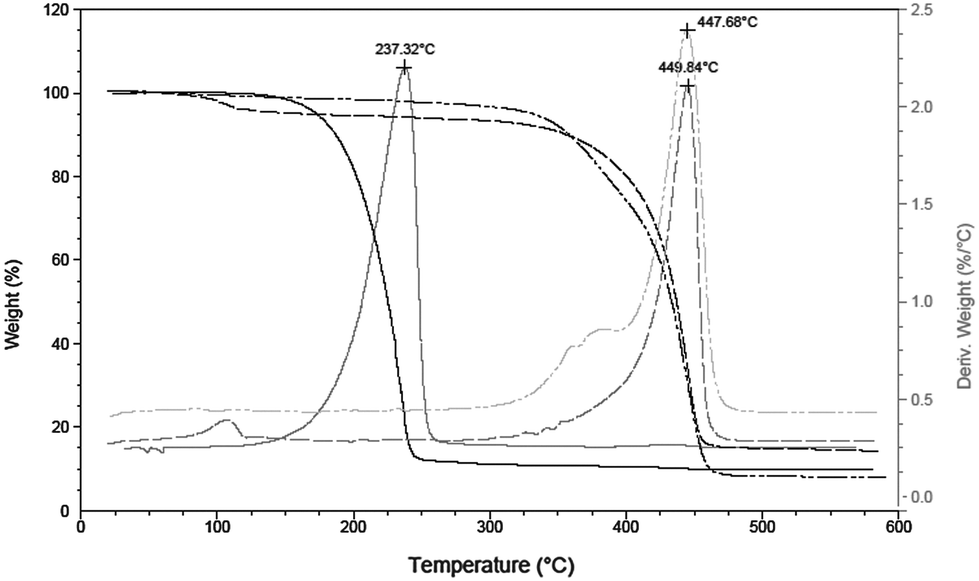 | ||
| Fig. 1 TGA (black) and derivative thermogravimetric (grey) analysis of una-H (solid line), una-ZnO NCs (dashed line) and tr-ZnO NCs (dashed-dotted line). | ||
The surface modification of una-ZnO NCs is illustrated in Scheme 1, path (iii). The purified alkyne-terminated NCs were dried in vacuo and redispersed in oxygen-free THF. Then, the mixture of S-(3-azidopropyl)thioacetate and a catalytic amount of ([Cu(C12H8N2)[P(C6H5)3]2]NO3) was added and the reaction was carried out for 1 h in an inert gas atmosphere to yield the functionalized tr-ZnO NCs. Then, the processed NCs were washed (see the ESI†) in order to remove impurities and the catalyst.
The 1H NMR spectra of (a) the parent una-ZnO NCs before the click reaction and (b) modified tr-ZnO NCs are shown in Fig. 3. The most relevant signal for monitoring the reaction progress is the resonance at 1.93 ppm associated with the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH group. In Fig. 3b, this signal is residual, which means the reaction between the alkyne group and the azide (S-(3-azidopropyl)thioacetate) proceeded almost quantitatively. The appearance of a new set of resonances, including the signal at 7.34 ppm from the CH–N proton, confirms the formation of triazole. These findings are in agreement with the IR spectra. For una-ZnO NCs, two characteristic bands at 3281 cm−1 and 2120 cm−1 are present, which correspond to the CC and CH stretching vibrations, respectively (Fig. 4). After the click reaction, these two resonances are no longer observed. The HRTEM micrographs indicate that tr-ZnO NCs are well-separated and spherically-shaped (Fig. 2c and d), which means that surface modification did not induce the aggregation of NCs. According to our expectations the average hydrodynamic diameter of tr-NCs slightly increased to 10.1 nm (ESI†).
CH group. In Fig. 3b, this signal is residual, which means the reaction between the alkyne group and the azide (S-(3-azidopropyl)thioacetate) proceeded almost quantitatively. The appearance of a new set of resonances, including the signal at 7.34 ppm from the CH–N proton, confirms the formation of triazole. These findings are in agreement with the IR spectra. For una-ZnO NCs, two characteristic bands at 3281 cm−1 and 2120 cm−1 are present, which correspond to the CC and CH stretching vibrations, respectively (Fig. 4). After the click reaction, these two resonances are no longer observed. The HRTEM micrographs indicate that tr-ZnO NCs are well-separated and spherically-shaped (Fig. 2c and d), which means that surface modification did not induce the aggregation of NCs. According to our expectations the average hydrodynamic diameter of tr-NCs slightly increased to 10.1 nm (ESI†).
 | ||
| Fig. 2 The HR TEM micrographs of (a and b) una-ZnO NCs, (c and d) tr-ZnO NCs. | ||
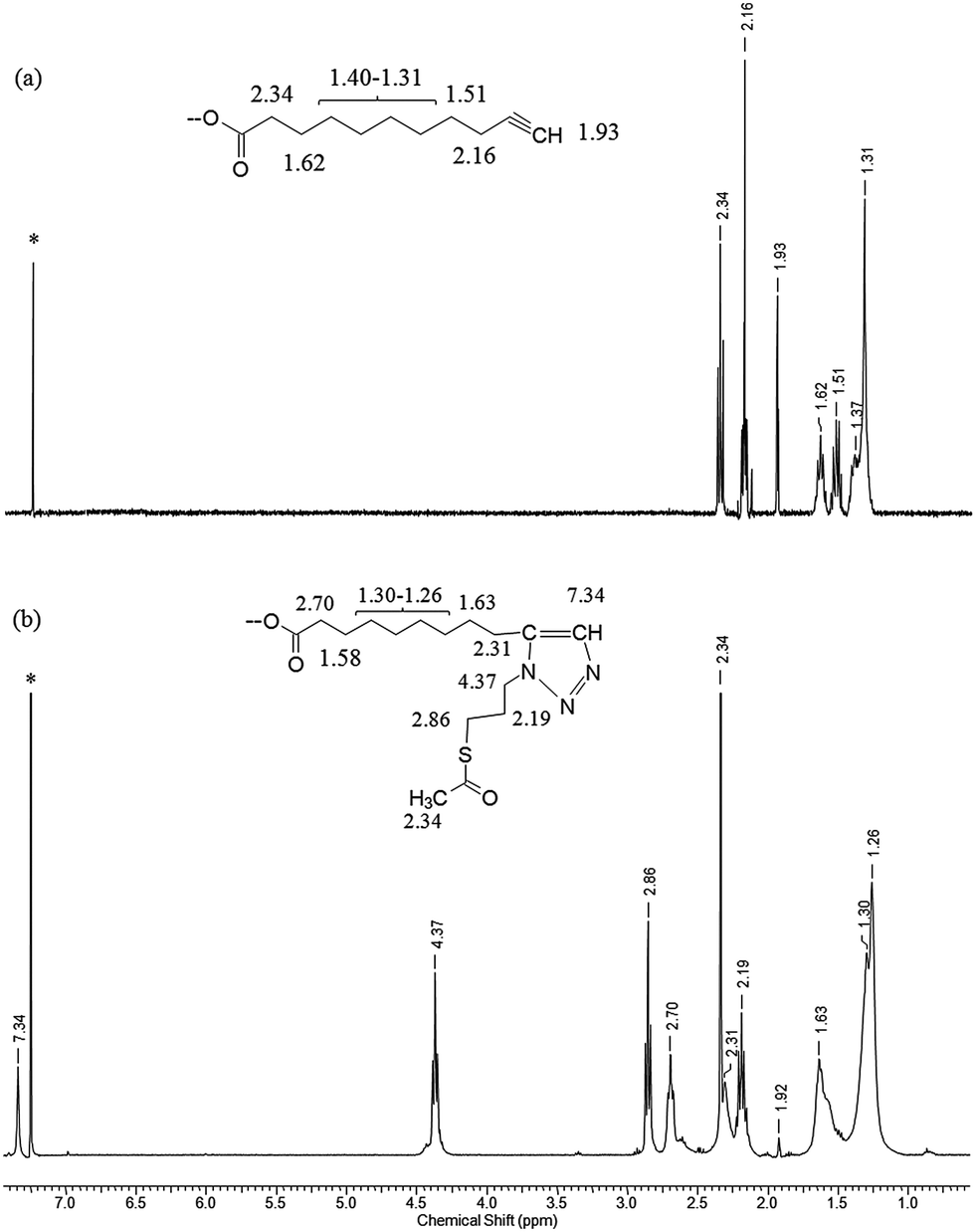 | ||
| Fig. 3 1H NMR spectra of (a) the parent una-ZnO NCs and (b) tr-ZnO NCs after the CuACC reaction (*CDCl3, 400 MHz, RT). | ||
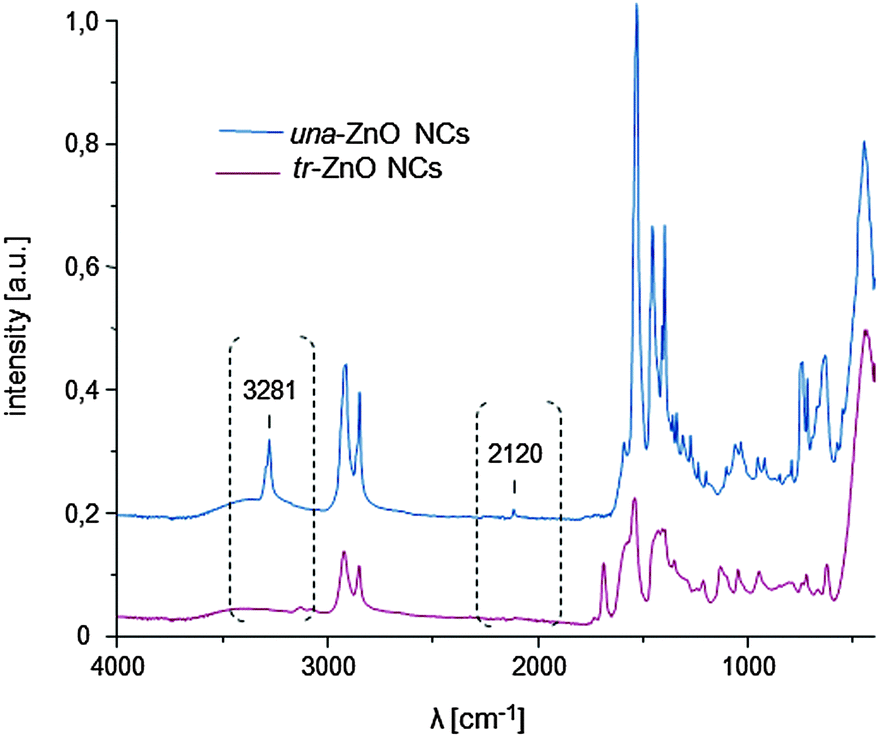 | ||
| Fig. 4 The IR spectra of ZnO NCs before and after the click reaction. | ||
The photoluminescence spectra of both una-ZnO NCs before and after their functionalization using CuACC are shown in Fig. 5. In both cases, the emission peaks are centered at ca. 520 nm. After the CuACC process, the luminescence of the NCs is only negligibly suppressed. Thus, the data clearly indicate that the Cu(I) catalyst used in the CuACC reaction did not influence the PL properties of the resulting tr-ZnO NCs. The quenching commonly found for other types of semiconductor NCs is rationalized by the permeable nature of the used ligand systems for copper ions.12 In our case, the observed retention of PL indicates that the self-supporting organometallic approach provides ZnO NCs with well passivated and impermeable organic shell relevant to the CuACC reaction. The anticipated impermeability perfectly correlates with the above-mentioned very low negative impact of the analogously synthesized ZnO NCs on mammalian cell lines.9
 | ||
| Fig. 5 (a) PL emission spectra of ZnO NCs before (black) and after the CuACC reaction (red); (b) PL spectra of: inorgA-ZnO NCs obtained via an inorganic method with 10-undecynoic acid on the surface (black); after the click reaction with an azide in the presence of a Cu(I) catalyst (red); corresponding to the bar graphs representing the suppression of the PL. | ||
To highlight the superiority of our synthetic method over most common inorganic sol–gel processes,13 we performed the analogous functionalization mediated by CuACC for una-coated ZnO NCs obtained by the traditional inorganic method (hereafter denoted as inorgA-ZnO). Briefly, the inorgA-ZnO NCs were prepared in ethanolic solution in the reaction of zinc acetate and lithium hydroxide, followed by the ligand exchange with 10-undecynoic acid (for details see the ESI†). Next, the inorgA-ZnO NCs were subjected to the CuACC reaction under the same conditions as previously described for una-ZnO NCs. As it is shown in Fig. 5, the click reaction resulted in immediate and irreversible quenching of the NC PL (similar observations were noted by others in the case of the CuACC functionalization of ZnO NCs prepared by the inorganic sol–gel method, however no experimental data were presented).5 However, taking into account that in the traditional inorganic sol–gel process the rate of nucleation and particle growth could hardly be controlled for a regime of quantum dot sizes,13b,14 it is reasonable to expect the spontaneous formation of various surface defects and permeable coating organic shell of the resulting NCs. Under these circumstances, Cu ions can easily approach the surface and subsequently quench the PL.4a,12
In conclusion, the presented data clearly indicate the advantage of the elaborated self-supporting organometallic procedure for the synthesis of ZnO NCs in a regime of quantum dot sizes over the traditional sol–gel inorganic method widely reported in the literature. The transformation of the designed alkylzinc [RZn-X] precursor in the presence of air provides high-quality ZnO NCs coated by a impermeable shell composed of X-type ligands which are appropriate for the classic Cu(I)-catalyzed click chemistry with retention of their PL properties. Conversely, the inorganic method affords the NCs with insufficiently capped surfaces to retain the PL properties after the analogous surface modification. It is also noteworthy that unlike the sol–gel method,5 this new organometallic approach directly affords nanomaterials adequate for post-synthetic modifications, i.e. ZnO NCs do not require pre-processing, such as ligand exchange, before further functionalization. Our strategy opens a new route to the direct and efficient surface functionalization of ZnO NCs, including coupling with biomolecules for the use as biolabels and other applications. Further studies on both the surface structure of ZnO NCs as well as click reactions between ZnO NCs and biologically active molecules are in progress.
The authors gratefully acknowledge the National Science Centre (Grant No. 2014/13/B/ST5/04420) and the Foundation for Polish Science Team Programme co-financed by the “European Regional Development Fund (Grant TEAM/2011-7/8) for financial support.
Notes and references
- R. A. Sperling and W. J. Parak, Philos. Trans. R. Soc., A, 2010, 368, 1333 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- (a) R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620 CrossRef CAS PubMed; (b) N. Li and W. H. Binder, J. Mater. Chem., 2011, 21, 16717 RSC; (c) K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904 CrossRef CAS PubMed; (d) W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572 CrossRef CAS.
- (a) W. H. Binder, R. Sachsenhofer, C. J. Straif and R. Zirbs, J. Mater. Chem., 2007, 17, 2125–2132 RSC; (b) H.-S. Han, N. K. Devaraj, J. Lee, S. A. Hilderbrand, R. Weissleder and M. G. Bawendi, J. Am. Chem. Soc., 2010, 132, 7838 CrossRef CAS PubMed; (c) A. Bernardin, A. Cazet, L. Guyon, P. Delannoy, F. Vinet, D. Bonnaffe and I. Texier, Bioconjugate Chem., 2010, 21, 583 CrossRef CAS PubMed; (d) G. Beaune, S. Tamang, A. Bernardin, P. Bayle-Guillemaud, D. Fenel, G. Schoehn, F. Vinet, P. Reiss and I. Texier, ChemPhysChem, 2011, 12, 2247 CrossRef CAS PubMed.
- (a) M. Sato, K. Shimatani, Y. Iwasaki, S. Morito, H. Tanaka, Y. Fujita and M. Nakamura, Appl. Surf. Sci., 2011, 258, 786 CrossRef CAS; (b) L. M. Bishop, J. C. Yeager, X. Chen, J. N. Wheeler, M. D. Torelli, M. C. Benson, S. D. Burke, J. A. Pedersen and R. J. Hamers, Langmuir, 2012, 28, 1322 CrossRef CAS PubMed.
- (a) H. Morkoç and U. Özgür, Zinc Oxide: Fundamentals, Materials and Device Technology, Willey-VCH, Weinheim, 2009 Search PubMed; (b) C. Klingshirn, ChemPhysChem, 2007, 8, 782 CrossRef CAS PubMed; (c) A. B. Djurisic and Y. H. Leung, Small, 2006, 2, 944 CrossRef CAS PubMed; (d) Q. Zhang, C. S. Dandeneau, X. Zhou and G. Cao, Adv. Mater., 2009, 21, 4087 CrossRef CAS.
- (a) H.-M. Xiong, Adv. Mater., 2013, 25, 5329 CrossRef CAS PubMed; (b) Y. Wang, S. Song, J. Liu, D. Liu and H. Zhang, Angew. Chem., Int. Ed., 2015, 54(2), 536 CAS; (c) J. Zhou, Y. Yang and C.-Y. Zhang, Chem. Rev., 2015, 115, 11669 CrossRef CAS PubMed.
- J. Paczesny, M. Wolska-Pietkiewicz, I. Binkiewicz, Z. Wróbel, M. Wadowska, K. Matuła, I. Dzicielewski, D. Pociecha, J. Smalc-Koziorowska, J. Lewiński and R. Hołyst, Chem. – Eur. J., 2015, 21, 16941 CrossRef CAS PubMed.
- M. Wolska-Pietkiewicz, K. Tokarska, A. Grala, A. Wojewódzka, K. Wójcik, Z. Wróbel, J. Grzonka, K.J. Kurzydłowski, P. J. Cywiński, M. Chudy and J. Lewiński, under review.
- A. Grala, M. Wolska-Pietkiewicz, A. Wojewódzka, M. Dabergut, I. Justyniak and J. Lewiński, Organometallics, 2015, 34, 4959 CrossRef CAS.
- Likely the size of solvated ZnO NCs include the dimensions of the oxide core and the coating carboxylate, and a solvation shell, which is consistent with the current view on a colloidal surface structure, see: (a) T. Schindler, M. Schmiele, T. Schmutzler, T. Kassar, D. Segets, W. Peukert, A. Radulescu, A. Kriele, R. Gilles and T. Unruh, Langmuir, 2015, 31, 10130 CrossRef CAS PubMed; (b) M. Zobel, R. B. Neder and S. A. J. Kimber, Science, 2015, 347, 292 CrossRef CAS PubMed.
- A. V. Isarov and J. Chrysochoos, Langmuir, 1997, 13, 3142 CrossRef CAS.
- (a) L. Spanhel and M. A. Anderson, J. Am. Chem. Soc., 1991, 113, 2826 CrossRef CAS; (b) E. A. Meulenkamp, J. Phys. Chem. B, 1998, 102, 5566 CrossRef CAS.
- (a) M. Voigt, M. Klaumunzer, H. Thiem and W. Peukert, J. Phys. Chem. C, 2010, 114, 6243 CrossRef CAS; (b) B. L. Caetano, C. V. Santilli, F. Meneau, V. Briois and S. H. Pulcinelli, J. Phys. Chem. C, 2011, 115, 4404 CrossRef CAS; (c) A. Layek, G. Mishra, A. Sharma, M. Spasova, S. Dhar, A. Chowdhury and R. Bandyopadhyaya, J. Phys. Chem. C, 2012, 116, 24757 CrossRef CAS; (d) T. Schindler, J. Walter, W. Peukert, D. Segets and T. Unruh, J. Phys. Chem. B, 2015, 119, 15370 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc01430e |
| This journal is © The Royal Society of Chemistry 2016 |