
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Redox-active tetraruthenium metallacycles: reversible release of up to eight electrons resulting in strong electrochromism†
Daniel
Fink
,
Bernhard
Weibert
and
Rainer F.
Winter
*
Fachbereich Chemie, Universität Konstanz, Universitätsstraße 10, D-78453 Konstanz, Germany. E-mail: rainer.winter@uni-konstanz.de
First published on 12th April 2016
Abstract
Tetraruthenium macrocycles with 1,4-divinylphenylene and diarylamine-substituted isophthalic acids as the sides display up to eight one-electron redox steps and rich electrochromic behaviour with strong absorptions of the dications in the near infrared and of the tetra- and hexacations at low energies in the visible.
Metallacycles are typically constructed from metal coligand fragments as nodes and two kinds of ditopic bridging ligands, so-called linkers as the sides. The shapes and sizes of such structures are determined by the preferred coordination geometries of the metal ions and the topologies of the linkers. Hence, a great number of metallacycles with vastly different architectures and astounding levels of complexity have been realized.1–6 Considering that many of these structures contain redox-active metal ions or linkers, or even both, only relatively few studies were specifically devoted to investigating or capitalizing on that property. Inherent perspectives such as triggering changes in guest-binding inside the cavities of metallacycles, manipulating their optical and magnetic properties, or studying intracage charge transfer phenomena have only sporadically been tackled. The elegant work of Hupp and coworkers, who explored charge transfer between diimine or porphyrinic linkers in mixed-valent tetrarhenium rectangles with ligand-based redox activity,7–10 and of Kaim, Stang and Therrien on tetraplatinum, -rhenium or -ruthenium metallacycles with oxidizable or reducible linkers stand as instructive examples.11–16
We here report on tetraruthenium metallacycles which are constructed from two pairs of diruthenium 1,4-divinylphenylene- and triarylamine-derived ditopic linkers as two different kinds of redox-active entities that allow for the pairwise release of up to eight electrons per macrocycle resulting in strong polyelectrochromism.17–19 Discrete divinylphenylene-bridged diruthenium complexes {Ru(CO)(L)(PiPr3)2}2(μ-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–C6H4–CHCH-1,4) (L = mono- or bidentate monoanionic ligand) are generally oxidized in two consecutive, reversible one-electron steps. Their mixed-valent radical cations exhibit complete charge and spin delocalization as is indicated by the observation of just one Ru(CO) IR-band (the observed small band splitting arises from the non-degeneracy of the symmetrical and antisymmetrical Ru(CO) stretching vibrations) and resolved EPR hyperfine splitting of the unpaired spin to four equivalent 31P nuclei of the PiPr3 coligands.20,21 Triarylamines constitute paradigmatic organic redox-systems and have found great use as selective oxidants and in redox catalysis.22–26 More electron-rich representatives can be further oxidized to persistent dications.26 A macrocycle constructed from two of these entities each may thus reversibly release six or even eight electrons per macrocycle.27,28
CH–C6H4–CHCH-1,4) (L = mono- or bidentate monoanionic ligand) are generally oxidized in two consecutive, reversible one-electron steps. Their mixed-valent radical cations exhibit complete charge and spin delocalization as is indicated by the observation of just one Ru(CO) IR-band (the observed small band splitting arises from the non-degeneracy of the symmetrical and antisymmetrical Ru(CO) stretching vibrations) and resolved EPR hyperfine splitting of the unpaired spin to four equivalent 31P nuclei of the PiPr3 coligands.20,21 Triarylamines constitute paradigmatic organic redox-systems and have found great use as selective oxidants and in redox catalysis.22–26 More electron-rich representatives can be further oxidized to persistent dications.26 A macrocycle constructed from two of these entities each may thus reversibly release six or even eight electrons per macrocycle.27,28
When the 1,4-divinylphenylene-bridged diruthenium complex 1-Cl as 180° building block was combined with diphenyl- or (di-4-anisylamino)benzene-3,5-dicarboxylic acid (2-H or 2-OMe) as a 0° building block4 with K2CO3 base in MeOH/CH2Cl 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, a complex mixture of products was initially formed as indicated by the presence of several sets of vinyl proton and 31P NMR signals.
:1, a complex mixture of products was initially formed as indicated by the presence of several sets of vinyl proton and 31P NMR signals.
On further warming to 50 °C for 48 h that mixture became uniform, showing just one set of proton NMR signals for the divinylphenylene and dicarboxylate building blocks and a single 31P NMR resonance. Target macrocycles 3-H and 3-OMe (Scheme 1) were isolated in yields of ca. 60% as pale yellow microcrystalline solids and were characterized by 1H, 13C{1H} and 31P{1H} NMR spectroscopy, ESI-MS and combustion analysis (ESI†).
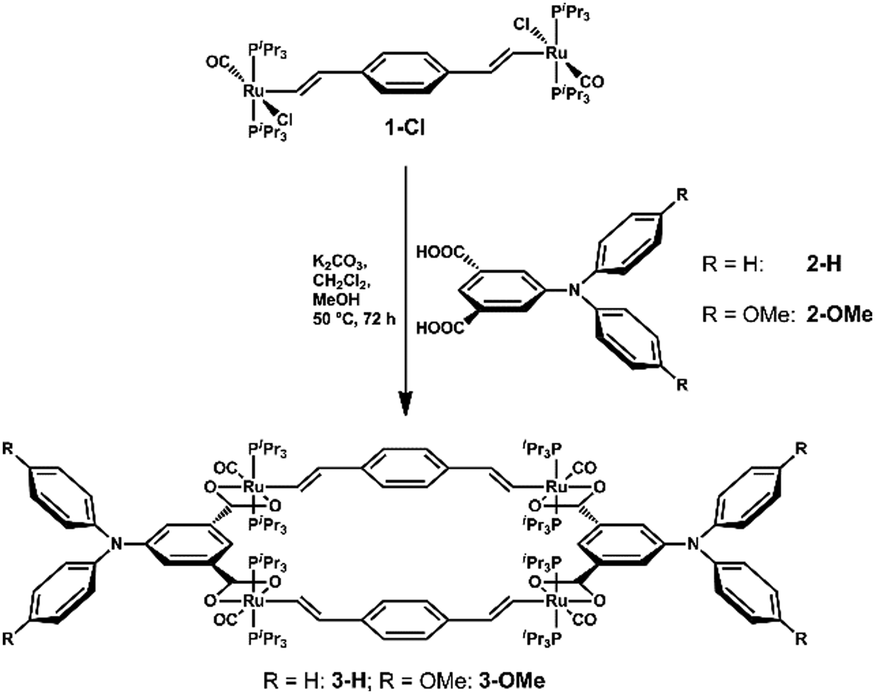 | ||
| Scheme 1 Synthesis of the tetraruthenium metallamacrocycles. | ||
Crystals of 3-OMe that lent themselves for X-ray crystallography were grown from benzene/MeOH and CH2Cl2/MeOH, respectively, and the results are shown in Fig. 1. In both structures, the divinylphenylene diruthenium moieties adopt a cisoid conformation and both vinyl groups point outward the inner cavity of the metallamacrocycle. This generates an oval cavity of approximate dimensions of 7.9 Å × 12.4 Å or 7.8 Å × 12.4 Å as measured between opposing hydrogen atoms. Forcing the divinylphenylene units into a metallamacrocyclic ring causes some minor torsional strain. In the benzene solvate 3-OMe·7C6H6 this strain is evident from the torsion of one ruthenium alkenyl moiety of each divinylphenylene linker by ϕ = 27.3° with respect to the adjacent phenylene ring whereas the other remains in a more coplanar arrangement with ϕ = 6.6°. Torsional angles O–C–C–C at the benzene-3,5-dicarboxylates of 7.6 and 4.5° and at the Ru–CC–C linkages of 3.5 and 4.8° are nearly ideal and the P–Ru–P vectors of opposing nodes are almost parallel as indicated by P–Ru–Ru–P torsion angles of 3.5 and 4.1°. In the dichloromethane solvate 3-OMe·8CH2Cl2 the divinylphenylene moieties achieve a somewhat higher degree of coplanarity with the central phenylene rings with torsion angles ϕ of 10.8 and 13.3° while the torsional angles O–C–C–C and Ru–CC–C are larger at 7.2° and 14.9° or 7.2° and 8.4°. The overall result is a twist of the P–Ru–P vectors as measured by P–Ru–Ru–P torsions of 19.0° and 21.9°, which in turn induces a bending of one divinylphenylene moiety to above and the other one to below the mean plane of the macrocycle (Fig. 1c). The two structures can thus be viewed as snapshots demonstrating the conformational degrees of freedom inherent to these macrocycles.
 | ||
| Fig. 1 Structures of 3-OMe as the benzene solvate (a) top view, (b) side view and (c) side view of the CH2Cl2 solvate. Solvent molecules other than the benzene molecule in the cavity and hydrogen atoms are removed for reasons of clarity. The ellipsoids are displayed at a 50% probability level. | ||
In 3-OMe·7C6H6, one molecule of benzene stacks in a coplanar, but laterally offset fashion with respect to the divinylphenylene sides, whereas the cavity of the CH2Cl2 solvate accommodates two solvent molecules that show no H-bonding interactions or short contacts with the cavity walls (see ESI†). This indicates that the tetraruthenium macrocycles, like other structures of this type, might also exhibit interesting host–guest chemistry.29,30
The electrochemical properties of macrocycles 3-H and 3-OMe are best compared to those of precursor 1-Cl and its bis(benzoato) substitution product, {Ru(CO)(PiPr3)2(κ2O,O′-OOCPh)}2(μ-CHCH–C6H4–CHCH-1,4) (1-OOCPh). Both complexes display two consecutive, reversible one-electron waves at half-wave potentials E1/2 of −0.09 and +0.17 V20 or −0.25 and +0.02 V, respectively.31 The cathodic shift of ∼150 mV on substitution of the chloro by the benzoato ligands follows the increase in valence electron count at the Ru atoms from 16 to 18 and the concomitant raise in energy of the redox-orbital(s), which receive major contributions from the π-conjugated bridging ligand.20,21,32–37 For 3-H, three consecutive waves are apparent in the cyclic voltammograms (CH2Cl2/NBu4PF6, Fig. 2). On close inspection one notes a small splitting of the first wave into two closely spaced one-electron processes. No such effect is however seen for the second and third waves. E1/2 values under these conditions are summarized in Table 1. Nearly identical results were obtained with the even less ion-pairing B{C6H3(CF3)2-3,5}4− counterion (ESI†). Since all waves are associated with similar peak currents and based on the overall composition and potentials we can safely assign the first two composite waves as the oxidations of the divinylphenylene sides and the third wave as the concurrent oxidations of the amine appendices. Thus, all three waves correspond to two closely spaced or coincident one-electron processes.
 | ||
| Fig. 2 Cyclic voltammograms (CH2Cl2/0.1 M NBu4PF6, r.t. at v = 0.1 V s−1) (left) and square wave voltammograms (right) of 3-H and 3-OMe along with deconvolutions. | ||
| E 0/+1/2 | E +/2+1/2 | E 2+/4+1/2 | E 4+/6+1/2 | E 6+/8+1/2 | |
|---|---|---|---|---|---|
| 3-H | −272 | −188 | 33 | 739 | — |
| 3-OMe | −275 | −194 | 34 | 431 | 1026 |
In the case of 3-OMe, a fourth such wave is seen close to the anodic discharge limit of the electrolyte, which obviously corresponds to the second oxidations of the peripheral triarylamine sites (Fig. 2). The above assignments are reconfirmed by the fact that substitution of the phenyl by anisyl substituents only affects the redox potentials past the 4+ states (Table 1).
Bis(alkenylarylene)-bridged diruthenium complexes with extended conjugated bridging ligands and [2.2]paracyclophanyl-bridged analogs with cofacial and coplanar arranged styryl decks were found to exhibit rather substantial degrees of ground-state delocalization in the mixed-valent states despite potential splittings ΔE1/2 near the purely statistical limit.32,38 This is, however, not the case here as shown by the results of IR and UV/Vis/NIR (NIR = near infrared) spectroelectrochemistry. Spectroscopic changes on the first composite two-electron oxidation, where one electron is removed from each divinylphenylene diruthenium entity, are essentially identical to the first oxidation of 1-Cl or 1-OOCPh. Thus, the Ru(CO) IR band blue-shifts by 34 cm−1 with some discernible splitting into a less intense band at higher and a more intense one at lower energy (Fig. 3 and ESI†). That splitting is due to a non-degeneracy of the symmetrical and antisymmetrical combinations of Ru(CO) stretches. Further spectroscopic changes include the growth of bands in the region of ring stretching and bending vibrations and of an intense absorption in the NIR at ca. 7700 cm−1 (ESI†). That band also shows up as part of an intense, structured NIR absorption in the UV/Vis/NIR experiments along with an equally intense one in the Vis (see Table 2, Fig. 3 and ESI†). Characteristic spectroscopic data are collected in Table 2.
 | ||
| Fig. 3 Changes in the UV/Vis/NIR (left) and in the ν(CO) region of the IR spectra (right) of macrocyle 3-OMe upon oxidation to 3-OMe2+ (top), 3-OMe4+ (middle) and 3-OMe6+ (bottom) (CH2Cl2/NBu4PF6, r.t.). | ||
| ν(CO) (cm−1) | λ (nm) (ε × 10−3 (M−1 cm−1)) | |
|---|---|---|
| 3-H | 1901 | 306 (60.3), 357 (75.6) |
| 3-H2+ | 1943, 1927 | 293 (49.8), 538 (42.4), 590 (60.4), 1093 (36.0), 1267 (59.9) |
| 3-H4+ | 1975 | 276 (53.1), 605 (101.9) |
| 3-OMe | 1900 | 304 (67.4), 355 (87.8) |
| 3-OMe2+ | 1942, 1925 | 282 (70.3), 537 (57.8), 590 (76.9), 1093 (52.0), 1267 (79.4) |
| 3-OMe4+ | 1973 | 276 (60.2), 606 (104.3) |
| 3-OMe6+ | 1975 | 275 (54.5), 354 (30.4), 610 (107.0), 787 (42.1) |
The relation between ΔE1/2 (in Volts) and Kc, the equilibrium constant of the comproportionation reaction 3-H2+ + 3-H ⇄ 2 3-H+, Kc = exp[n·F·ΔE1/2/(R·T)] = exp(39.33·ΔE1/2) at T = 295 K dictates that, after release of one equivalent of charge, the mixed-valent (MV) forms 3-H+ or 3-OMe+ are the dominant species in the equilibrated solutions, even when ΔE1/2 is vanishingly small. Still, no spectrum recorded during electrolysis has features other than those corresponding to the respective dications and the neutral precursors and, in particular, none that could be ascribed to charge transfer from the remaining reduced divinylphenylene side to the already oxidized one. This means that the radical cations are mixed-valent species of Class I with non-interacting redox sites.39,40 Obviously the dicarboxylic linkers, which do not contribute to the redox orbitals HOMO and HOMO−1,31,41 and the lateral positioning of the divinylphenylene moieties of the macrocycles mutually insulate the two divinylphenylene sides from each other. As a consequence, 3-H2+ and 3-OMe2+ are EPR active and display a somewhat broad isotropic signal at g = 2.039 (ESI†).
The same overall behaviour repeats on the second composite oxidation encompassing the concurrent 2+/3+ and the 3+/4+ waves, corresponding to the second oxidation of the divinylphenylene sides (Fig. 3 and ESI†). Thus, the overall spectroscopic changes with a further blue-shift of the Ru(CO) band by ca. 40 cm−1, the collapse of the NIR band and the further growth of the Vis absorption are very similar or virtually identical to those observed on the second oxidation of diruthenium complexes 1-Cl and 1-OOCPh. Again, no absorption bands other than those of the di- and tetracations are seen at intermediate stages of the electrolysis where 3-H3+ or 3-OMe3+ constitute the major species in solution. Hence, the trications are also valence localized MV species of Class I. The divinylphenylene diruthenium entities thus behave as two independent electrophoric units and the only consequence of forging two of them together in a metallacyclic structure is a predictable doubling of molar extinction coefficients to rather impressive values of ca. 80000 for the prominent NIR absorption of 3-OMe2+ and >100000 for the Vis band of 3-OMe4+ (neutral 3-OMe is transparent in these regimes).
Owing to the inherent instability of 3-H6+, the spectral consequences of further oxidizing the triarylamine appendices could only be monitored for 3-OMe. They include a very modest shift of the Ru(CO) IR band from 1973 to 1975 cm−1 and of the prominent Vis band from 606 nm in 3-OMe4+ to 610 nm in 3-OMe6+ (see Table 2). Additional bands at 354 nm and at 787 nm with extinction coefficients of 30400 and 42100 M−1 cm−1 closely resemble those of the radical cation of 2-OMe (λmax = 365, 793 nm).42 Moreover, chemically generated 3-OMe6+ displays a non-binomial triplet at g = 2.014 and a 14N hyperfine splitting of 9.6 G in its EPR spectrum as it is typical of aminium-type radicals,43 thereby reconfirming the above assignments of the individual redox processes (ESI†). The only negligible impact of amine oxidation on the position of the π → π* absorption band of the bis(oxidized) divinylphenylene diruthenium entities and the Ru(CO) band shifts further emphasizes the lack of electronic interaction between the two different kinds of redox sites. Further oxidation to 3-OMe8+ was, however, not possible under the conditions of spectroelectrochemistry, probably due to the proximity of that wave to the solvent background and a poor solubility of this polycation.
In conclusion, we have prepared and investigated amine-functionalized tetraruthenium metallamacrocycles in good yields from divinylphenylene–diruthenium complexes and amine functionalized isophthalic acids. These compounds can be oxidized by up to eight electrons with stepwise charge loss from the divinylphenylene linkers and the triarylamine appendices. Spectroscopic consequences of oxidation up to 3-OMe6+ were monitored by IR and UV/Vis/NIR spectroelectrochemistry and revealed strongly electrochromic behaviour.44 The lateral placement of the redox-active divinylphenylene entities and the insulating nature of the dicarboxylate linkers shut down intramolecular charge transfer between the individual redox sites and lead to paramagnetic behaviour.
The authors thank Maximilian Dürr and Ivana Ivanović-Burmazović from the Friedrich-Alexander Universität Erlangen-Nürnberg for the acquisition of the ESI-MS spectra and Stefan Scheerer for the recording and simulation of the EPR spectra.
Notes and references
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed.
- C. G. Oliveri, P. A. Ulmann, M. J. Wiester and C. A. Mirkin, Acc. Chem. Res., 2008, 41, 1618–1629 CrossRef CAS PubMed.
- Y.-F. Han, W.-G. Jia, W.-B. Yu and G.-X. Jin, Chem. Soc. Rev., 2009, 38, 3419–3434 RSC.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC.
- M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC.
- P. H. Dinolfo, M. E. Williams, C. L. Stern and J. T. Hupp, J. Am. Chem. Soc., 2004, 126, 12989–13001 CrossRef CAS PubMed.
- P. H. Dinolfo and J. T. Hupp, J. Am. Chem. Soc., 2004, 126, 16814–16819 CrossRef CAS PubMed.
- P. H. Dinolfo, S. J. Lee, V. Coropceanu, J.-L. Brédas and J. T. Hupp, Inorg. Chem., 2005, 44, 5789–5797 CrossRef CAS PubMed.
- P. H. Dinolfo, V. Coropceanu, J.-L. Brédas and J. T. Hupp, J. Am. Chem. Soc., 2006, 128, 12592–12593 CrossRef CAS PubMed.
- H. Hartmann, S. Berger, R. Winter, J. Fiedler and W. Kaim, Inorg. Chem., 2000, 39, 4977–4980 CrossRef CAS PubMed.
- W. Kaim, B. Schwederski, A. Dogan, J. Fiedler, C. J. Kuehl and P. J. Stang, Inorg. Chem., 2002, 41, 4025–4028 CrossRef CAS PubMed.
- N. Das, A. M. Arif, P. J. Stang, M. Sieger, B. Sarkar, W. Kaim and J. Fiedler, Inorg. Chem., 2005, 44, 5798–5804 CrossRef CAS PubMed.
- J. Mattsson, P. Govindaswamy, A. K. Renfrew, P. J. Dyson, P. Štěpnička, G. Süss-Fink and B. Therrien, Organometallics, 2009, 28, 4350–4357 CrossRef CAS.
- V. Vajpayee, S. Bivaud, S. Goeb, V. Croué, M. Allain, B. V. Popp, A. Garci, B. Therrien and M. Sallé, Organometallics, 2014, 33, 1651–1658 CrossRef CAS.
- M. Yuan, F. Weisser, B. Sarkar, A. Garci, P. Braunstein, L. Routaboul and B. Therrien, Organometallics, 2014, 33, 5043–5045 CrossRef CAS.
- R. J. Mortimer, Chem. Soc. Rev., 1997, 26, 147–156 RSC.
- M. D. Ward, J. Solid State Electrochem., 2005, 9, 778–787 CrossRef CAS.
- R. J. Mortimer, Annu. Rev. Mater. Res., 2011, 41, 241–268 CrossRef CAS.
- J. Maurer, B. Sarkar, B. Schwederski, W. Kaim, R. F. Winter and S. Záliš, Organometallics, 2006, 25, 3701–3712 CrossRef CAS.
- S. Záliš, R. F. Winter and W. Kaim, Coord. Chem. Rev., 2010, 254, 1383–1396 CrossRef.
- E. Steckhan, Angew. Chem., Int. Ed. Engl., 1986, 25, 683–701 CrossRef.
- S. Dapperheld, E. Steckhan, K.-H. G. Brinkhaus and T. Esch, Chem. Ber., 1991, 124, 2557–2567 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- T. P. Bender, J. F. Graham and J. M. Duff, Chem. Mater., 2001, 13, 4105–4111 CrossRef CAS.
- S. Amthor, B. Noller and C. Lambert, Chem. Phys., 2005, 316, 141–152 CrossRef CAS.
- C.-Y. Yao, J. Yao and Y.-W. Zhong, Inorg. Chem., 2011, 50, 6847–6849 CrossRef CAS PubMed.
- J.-H. Tang, S.-H. Wu, J.-Y. Shao, H.-J. Nie and Y.-W. Zhong, Organometallics, 2013, 32, 4564–4570 CrossRef CAS.
- B. Therrien, Eur. J. Inorg. Chem., 2009, 2445–2453 CrossRef CAS.
- N. P. E. Barry, O. Zava, P. J. Dyson and B. Therrien, J. Organomet. Chem., 2012, 705, 1–6 CrossRef CAS.
- E. Wuttke, Y.-M. Hervault, W. Polit, M. Linseis, P. Erler, S. Rigaut and R. F. Winter, Organometallics, 2014, 33, 4672–4686 CrossRef CAS.
- M. Linseis, S. Záliš, M. Zabel and R. F. Winter, J. Am. Chem. Soc., 2012, 134, 16671–16692 CrossRef CAS PubMed.
- P. Mücke, M. Linseis, S. Záliš and R. F. Winter, Inorg. Chim. Acta, 2011, 374, 36–50 CrossRef.
- W. Y. Man, J.-L. Xia, N. J. Brown, J. D. Farmer, D. S. Yufit, J. A. K. Howard, S. H. Liu and P. J. Low, Organometallics, 2011, 30, 1852–1858 CrossRef CAS.
- U. Pfaff, A. Hildebrandt, M. Korb, S. Oßwald, M. Linseis, K. Schreiter, S. Spange, R. F. Winter and H. Lang, Chem. – Eur. J., 2016, 22, 783–801 CrossRef CAS PubMed.
- S. Scheerer, N. Rotthowe, O. S. Abdel-Rahman, X. He, S. Rigaut, H. Kvapilová, S. Záliš and R. F. Winter, Inorg. Chem., 2015, 54, 3387–3402 CrossRef CAS PubMed.
- Y.-P. Ou, J. Zhang, M. Xu, J. Xia, F. Hartl, J. Yin, G.-A. Yu and S. H. Liu, Chem. – Asian J., 2014, 9, 1152–1160 CrossRef CAS PubMed.
- P. Mücke, M. Zabel, R. Edge, D. Collison, S. Clément, S. Záliš and R. F. Winter, J. Organomet. Chem., 2011, 696, 3186–3197 CrossRef.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247–422 CrossRef CAS.
- B. Brunschwig, C. Creutz and N. Sutin, Chem. Soc. Rev., 2002, 31, 168–184 RSC.
- F. Pevny, R. F. Winter, B. Sarkar and S. Záliš, Dalton Trans., 2010, 8000–8011 RSC.
- H. Murata and P. M. Lahti, J. Org. Chem., 2007, 72, 4974–4977 CrossRef CAS PubMed.
- G. A. Pearson, M. Rocek and R. I. Walter, J. Phys. Chem., 1978, 82, 1185–1192 CrossRef CAS.
- N. Deibel, M. G. Sommer, S. Hohloch, J. Schwann, D. Schweinfurth, F. Ehret and B. Sarkar, Organometallics, 2014, 33, 4756–4765 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization, NMR and mass spectra, X-ray crystallography, voltammograms, spectroelectrochemistry. CCDC 1450420 and 1450421. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc00936k |
| This journal is © The Royal Society of Chemistry 2016 |