
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New synthesis route for ternary transition metal amides as well as ultrafast amide–hydride hydrogen storage materials†
Hujun
Cao
*a,
Antonio
Santoru
a,
Claudio
Pistidda
a,
Theresia M. M.
Richter
b,
Anna-Lisa
Chaudhary
a,
Gökhan
Gizer
a,
Rainer
Niewa
b,
Ping
Chen
c,
Thomas
Klassen
a and
Martin
Dornheim
a
aInstitute of Materials Research, Materials Technology, Helmholtz-Zentrum Geesthacht GmbH, Max-Planck-Straße 1, D-21502 Geesthacht, Schleswig-Holstein, Germany. E-mail: hujun.cao@hzg.de; Fax: +49 04152/87-2625; Tel: +49 04152/87-2643
bInstitute of Inorganic Chemistry, University Stuttgart, Pfaffenwaldring 55, Stuttgart 70569, Germany
cDalian National Laboratory for Clean Energy Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P. R. China
First published on 1st March 2016
Abstract
K2[Mn(NH2)4] and K2[Zn(NH2)4] were successfully synthesized via a mechanochemical method. The mixture of K2[Mn(NH2)4] and LiH showed excellent rehydrogenation properties. In fact, after dehydrogenation K2[Mn(NH2)4]-8LiH fully rehydrogenates within 60 seconds at ca. 230 °C and 5 MPa of H2. This is one of the fastest rehydrogenation rates in amide–hydride systems known to date. This work also shows a strategy for the synthesis of transition metal nitrides by decomposition of the mixtures of M[M′(NH2)n] (where M is an alkali or alkaline earth metal and M′ is a transition metal) and metal hydrides.
Transition metal nitrides attract increasing attention owing to their interesting physical and chemical properties (e.g. optical, magnetic, electrical, mechanical and catalytic properties).1 Zn3N2 powder was first synthesized by Juza and Hahn in 1940.2a This material is a promising candidate for optoelectronic applications because of its high electron mobility and high electrical conductivity.2 Manganese nitrides (i.e. Mn4N, Mn3N2, Mn6N5, and Mn2N), are well known for their phase-dependent magnetic properties.3 Apart from the binary transition metal nitrides, the development of ternary transition metal nitrides have attracted considerable attention due to their various properties.4 In the past decades, a lot of ternary transition metal nitrides have been explored such as Ca[NiN], Sr[NiN], Li7[MnN4], Li7[VN4] and Li[ZnN].4 Owing to N2 having extremely strong covalent bonds and therefore high stability, it is very difficult to employ it to synthesize nitrogen containing materials (i.e. transition metal nitrides). Usually the synthesis conditions for transition metal nitrides vary remarkably.5 The lighter 3d-transition metals are thermodynamically more stable than the heavier ones. To synthesize nitrides of Ti to Ni from the elements, often high temperatures and high pressures are favorable, for example, Ni3N was synthesized by use of supercritical nitrogen at ∼1800 K and 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MPa.6 However, such reaction conditions are challenging for large-scale synthesis and practical applications. The use of NH3 instead of N2 allows a considerable reduction of reaction times, pressures and temperatures. Ammonothermal synthesis, using supercritical NH3, gives access to synthesize some transition metal nitrides like Mn4N, Mn3N2, Fe4−xNixN, Ni3N and Cu3N.7 Using this method very well crystallized powder, or even single crystals of transition metal nitrides like EuN and Mn3N2 can be obtained, the formation process still needs high pressures, high temperatures and long reaction times.8 Usually, ternary transition metal amides (M[M′(NH2)n]) are synthesized by ammonothermal method, and they are important intermediates for synthesis of transition metal nitrides. For example, Mn3N2 was obtained by decomposition of Na2[Mn(NH2)4] (reaction (1)). However, Na2[Mn(NH2)4] was synthesized by reaction of MnI2 and NaNH2 at 673–873 K and 600 MPa of NH3 for more than 30 days (reaction (2)).9
000 MPa.6 However, such reaction conditions are challenging for large-scale synthesis and practical applications. The use of NH3 instead of N2 allows a considerable reduction of reaction times, pressures and temperatures. Ammonothermal synthesis, using supercritical NH3, gives access to synthesize some transition metal nitrides like Mn4N, Mn3N2, Fe4−xNixN, Ni3N and Cu3N.7 Using this method very well crystallized powder, or even single crystals of transition metal nitrides like EuN and Mn3N2 can be obtained, the formation process still needs high pressures, high temperatures and long reaction times.8 Usually, ternary transition metal amides (M[M′(NH2)n]) are synthesized by ammonothermal method, and they are important intermediates for synthesis of transition metal nitrides. For example, Mn3N2 was obtained by decomposition of Na2[Mn(NH2)4] (reaction (1)). However, Na2[Mn(NH2)4] was synthesized by reaction of MnI2 and NaNH2 at 673–873 K and 600 MPa of NH3 for more than 30 days (reaction (2)).9| 3Na2[Mn(NH2)4] → Mn3N2 + 6NaNH2 + 4NH3 | (1) |
| 4NH3 + MnI2 + 2NaNH2 → Na2[Mn(NH2)4] + 2NH4I | (2) |
M[M′(NH2)n] compounds contain NH2− group like alkali/alkaline earth metal amides which are excellent candidates for hydrogen storage.10 M[M′(NH2)n] compounds also contain transition metals and most of transition metals have some “catalytic effect” on hydrogen dissociation.11 Using M[M′(NH2)n] as a hydrogen storage media might be an important step towards the development of new hydrogen storage systems with novel hydrogen sorption properties. In addition, some metal nitrides are easily obtained by dehydrogenation of amide–hydride mixtures. For example, LiNH2-2LiH converts to Li3N after dehydrogenation.10c This could be a viable method to rapidly and conveniently obtain ternary and/or quaternary transition metal nitrides by decomposing mixtures of M[M′(NH2)n] and metal hydrides under mild temperature and gas pressure conditions.
Various mechanochemical reactions have been developed for synthesis of intermetallics, alloy compounds, nanocrystalline powders, magnetic materials, ceramic powders, hydrogen storage materials and catalytic materials.12 That is because mechanochemical synthesis methods offer several advantages over traditional processing routes. These advantages include low-temperature, solid state reactions, fewer processing steps, ease to scale-up and consequently, lower costs for material production.
In this work, for the first time K2[Mn(NH2)4] and K2[Zn(NH2)4] were synthesized by mechanochemical reaction. K2[Mn(NH2)4]-8LiH mixture showed excellent hydrogen absorption properties. In addition, an excellent material for electrode application (i.e. Li7[MnN4]) was obtained by dehydrogenation of K2[Mn(NH2)4]-8LiH.
The formula of K2[Zn(NH2)4] was proposed for the product of the reaction between a zinc salt and KNH2 in liquid NH3 by Juza in 1937.13 Prior to this, it was first synthesized by Fitzgerald in 1907, when crystals were grown from Zn/Zn(NH2)2 in a solution of KNH2 in liquid NH3 at ambient temperature.14 In 1969, Brisseau and Rouxel found that K2[Zn(NH2)4] is a triclinic unit cell. More detailed unit cell parameters with a = 6.730(1) Å, b = 7.438(1) Å, c = 8.019(2) Å, α = 72.03(2)°, β = 84.45(2)°, and γ = 63.82(1)° were determined until 1997.15 Monoclinic K2[Mn(NH2)4] was first synthesized in 1975 by Drew et al. with the same method as for K2[Zn(NH2)4].16 In 2013, a new monoclinic K2[Zn(NH2)4], which is isotypic to K2[Mn(NH2)4] was obtained by Richter et al. using an ammonothermal synthesis method under 720 K and 249 MPa pressures of ammonia for ca. 7 days.17 For this work, K2[Zn(NH2)4] (triclinic) and K2[Mn(NH2)4] were synthesized under moderate conditions by ball milling K and Zn/Mn in a molar ratio of 2:1 at 0.7 MPa of NH3 for 12 h. The characterization was carried out by high resolution X-ray diffraction experiments (Hard X-ray diffraction beamline – P02, PETRA III, Hamburg). Based on the Rietveld analysis results, K2[Zn(NH2)4] (triclinic) and K2[Mn(NH2)4] were obtained with high purities (Fig. 1). According to the Rietveld refinements, ca. 4.6 ± 0.2 wt% of unreacted Mn was found in the K2[Mn(NH2)4] sample. This is most likely due to the fact that a fraction of metallic manganese was adhered on areas of the walls of the milling vial were only few collisions took place. However the results indicate that K2[Zn(NH2)4] (triclinic) and K2[Mn(NH2)4] were synthesized by ball milling metallic manganese/zinc with potassium under moderate conditions without use of high pressures, high temperatures and long reaction times. However, the unit cell parameters of the mechanochemically synthesized K2[Zn(NH2)4] (triclinic) and K2[Mn(NH2)4] differ slightly from the values reported in literatures (Table S1, ESI†).15,16 These differences maybe attribute to the different synthesis methods and measurement uncertainties.
 | ||
| Fig. 1 High resolution powder X-ray diffraction patterns (black dots) and Rietveld refinements (red line) of the as-synthesized triclinic K2[Zn(NH2)4] – (a) and K2[Mn(NH2)4] – (b). Bragg reflections for each phase are indicated by the tick marks. The bottom line represents the difference curve (Iobs − Icalc). Weighted R-factors as low as Rw (%) = 6.83 and Rw (%) = 6.35 were obtained for (a) and (b) respectively. | ||
Thermogravimetry (TG) combined with differential thermal analysis (DTA) was employed to investigate the thermal decomposition process of the as-prepared triclinic K2[Zn(NH2)4] and K2[Mn(NH2)4] (Fig. 2). After heating to ca. 500 °C, the weight losses of both K2[Zn(NH2)4] and K2[Mn(NH2)4] were about 35 wt% in the TG curves. However, the thermal decomposition process of K2[Mn(NH2)4] is very different from that of K2[Zn(NH2)4]. Based on the weight losses, ca. 2 equiv. NH3 were released from K2[Mn(NH2)4] and K2[Zn(NH2)4] at ca. 360 and 420 °C, respectively. In addition, there was no evidence of weight losses for K2[Mn(NH2)4] in the temperature range between 200 and 300 °C, meaning that a relatively stable compound could be formed at this point. DTA measurements show that both K2[Mn(NH2)4] and K2[Zn(NH2)4] decompose in a multi-steps reaction. The K2[Mn(NH2)4] decomposition process can be clearly divided into three steps upon heating from 30 to 500 °C. For K2[Zn(NH2)4] three separate thermal events at 135, 186 and 220 °C can be observed clearly, which correlate well with the results of Richter et al.17 The first signal at 135 °C can be attributed to the phase transition from triclinic to monoclinic. After heating to ca. 420 °C, mechanochemically synthesized K2[Zn(NH2)4] and ammonothermally synthesized K2[Zn(NH2)4] undergo similar weight losses (16.5 wt% and 16.4 wt% respectively), meaning that the purities of these two materials are extremely close to each other's (Fig. S1, ESI†).
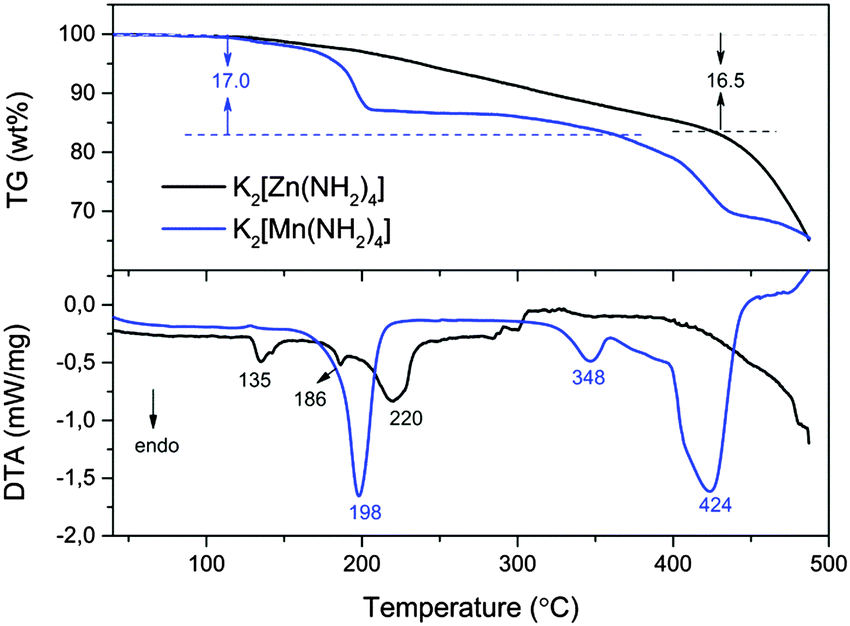 | ||
| Fig. 2 TG-DTA curves of the as-synthesized triclinic K2[Zn(NH2)4] and K2[Mn(NH2)4], heating from 30 °C to 500 °C with a heating rate of 5 °C min−1. | ||
Recently, we found that the mixture of K2[Zn(NH2)4] and 8LiH is a good hydrogen storage material due to its excellent hydrogen absorption properties.18 Here the hydrogen sorption properties of K2[Mn(NH2)4]-8LiH were studied and compared with the reference material LiNH2-2LiH. The results of this investigation are summarized in Fig. S2 (ESI†) and Fig. 3. Fig. S2 (ESI†) shows that K2[Mn(NH2)4]-8LiH decomposes in the temperature range between room temperature to 500 °C in 3 steps. Most of the hydrogen together with an almost undetectable amount of ammonia were released during the first two decomposition steps. Fig. 3 summarizes the volumetric de/re-hydrogenation properties (HERA, Quebec, Canada) of K2[Mn(NH2)4]-8LiH and the reference material LiNH2-2LiH. The results show that after heating to 400 °C ca. 4.2 and 4.3 wt% of hydrogen were released from K2[Mn(NH2)4]-8LiH and LiNH2-2LiH, respectively. Interestingly, the onset decomposition temperature of K2[Mn(NH2)4]-8LiH is about 60 °C lower than that of the LiNH2-2LiH sample. Noteworthy, from the inset in Fig. 3, it is known that most of hydrogen (∼60% of the total capacity) can be hydrogenated by K2[Mn(NH2)4]-8LiH sample during the time from 3990 to 4053 s (at which time the temperature is close to ∼230 °C), however, less than 2% of hydrogen was absorbed by the reference LiNH2-2LiH during the same time. The rehydrogenation rate of K2[Mn(NH2)4]-8LiH is one of the fastest absorption reaction rates known for amide–hydride systems.
 | ||
| Fig. 3 De/re-hydrogenation curves of the as ball milled K2[Mn(NH2)4]-8LiH sample, heating from room temperature to 400 °C at a ramping rate of 3 °C min−1 under vacuum for dehydrogenation, heating from room temperature to 300 °C at a heating rate of 3 °C min−1 and 5 MPa of H2 for re-hydrogenation. Inset is the re-hydrogenation curve of as ball milled K2[Mn(NH2)4]-8LiH and LiNH2-2LiH samples under different absorption time. | ||
It is known that LiNH2-2LiH converts into Li3N upon dehydrogenation to 500 °C, which shows a high reactivity towards most nitrides.10c Thus; the decomposition of such amide–hydride systems is a strategy to obtain transition metal nitrides. Li7[MnN4], due to its high specific gravimetric capacity, in excess of 300 mAh g−1 and excellent retention is a good candidate as a cathode material.19 Usually, Li7[MnN4] is synthesized by heating the mixture of Li3N and Mn under N2 at high temperature (>700 °C) for long time (>24 h).4c,20 In this work, Li7[MnN4] is synthesized by dehydrogenation of K2Mn(NH2)4-8LiH below 500 °C within 100 min. However, Li7[MnN4] is formed together with K and Mn4N and unknown compounds (Fig. 4).
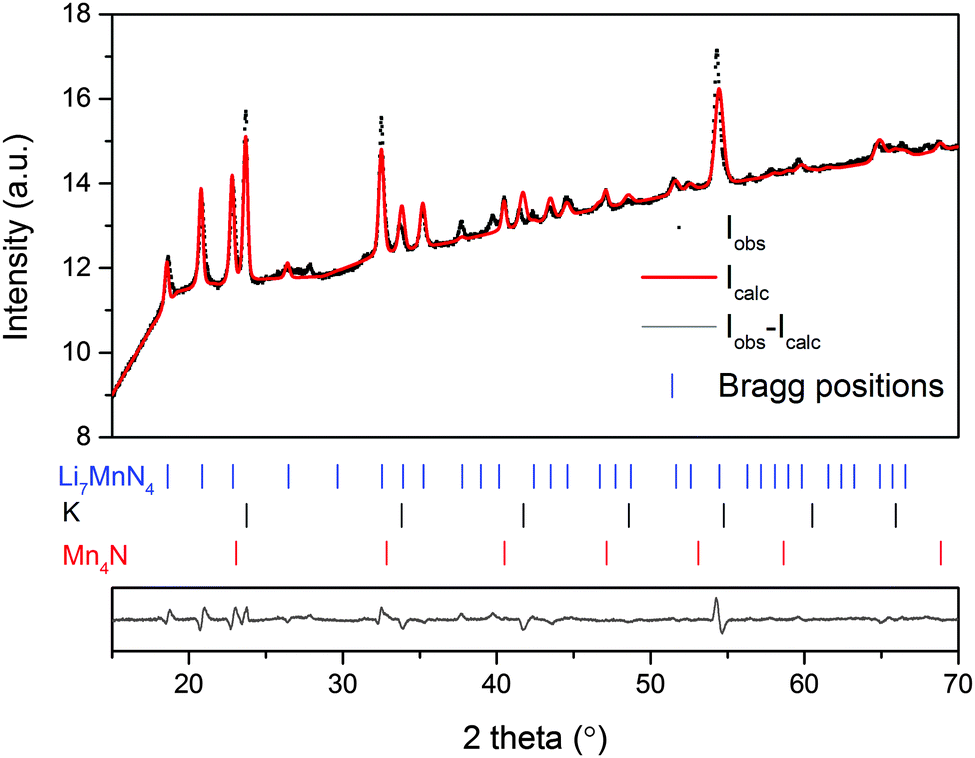 | ||
| Fig. 4 Rietveld analysis for the X-ray powder diffraction pattern of the as ball milled K2[Mn(NH2)4]-8LiH sample after thermal desorption to 500 °C in TG, (small black dots), Rw (%) = 1.26322. | ||
In summary, K2[Mn(NH2)4] and K2[Zn(NH2)4] were synthesized for the first time by mechanochemical reaction. These phases are extremely promising both as hydrogen storage materials and as intermediates toward the formation of transition metal nitrides. With the method described in this work, M[M′(NH2)n] can be synthesized in large-scale under mild temperatures, without the need of applying high gas pressures and long reaction times. After mixing K2[Mn(NH2)4] with 8LiH, it released 4.2 wt% of H2 below 400 °C. Compared to the reference LiNH2-2LiH, K2[Mn(NH2)4]-8LiH showed improved de/re-hydrogenation performance. The dehydrogenated K2[Mn(NH2)4]-8LiH sample can be rehydrogenated within 60 seconds at ca. 230 °C and 5 MPa of H2 with a rehydrogenation rate of ca. 3 wt% per min, which is one of the fastest rehydrogenation rates measured in amide–hydride systems. Moreover, this study has shown evidence of binary and ternary transition metal nitrides formation upon dehydrogenation of the mixtures of M[M′(NH2)n] and metal hydrides.
Financial support by the Helmholtz-Chinese Academy of Science Joint Research Group “RevHy”-“Study on the synthesis, structures and performance of complex hydrides systems for Reversible high capacity Hydrogen storage at low temperatures”, Marie Curie Initial Training Network “ECOSTORE – Novel Complex Metal Hydrides for Efficient and Compact Storage of Renewable Energy as Hydrogen and Electricity” (grant 607040) is thankfully acknowledged.
We thank Dr Bednarcik Jozef and Dr Martin Etter for their kind helps on the high resolution X-ray diffraction experiments.
Notes and references
- (a) D. S. Bem and H. C. Z. Loye, J. Solid State Chem., 1993, 104, 467 CrossRef CAS; (b) M. Nishijima, N. Tadokoro, Y. Takeda, N. Imanishi and O. Yamamoto, J. Electrochem. Soc., 1994, 141, 2966 CrossRef CAS; (c) M. Nishijima, T. Kagohashi, M. Imanishi, Y. Takeda, O. Yamamoto and S. Kondo, Solid State Ionics, 1996, 83, 107 CrossRef CAS; (d) M. Nishijima, T. Kagohashi, Y. Takeda, M. Imanishi and O. Yamamoto, J. Power Sources, 1997, 68, 510 CrossRef CAS; (e) G. Hitoki, A. Ishikawa, T. Takata, J. N. Kondo, M. Hara and K. Domen, Chem. Lett., 2002, 736 CrossRef CAS; (f) J. L. Tirado, Mater. Sci. Eng., R, 2003, 40, 103 CrossRef; (g) D. J. Ham and J. S. Lee, Energies, 2009, 2, 873 CrossRef CAS; (h) B. Cao, G. M. Veith, J. C. Neuefeind, R. R. Adzic and P. G. Khalifah, J. Am. Chem. Soc., 2013, 135, 19186 CrossRef CAS PubMed; (i) M.-S. Balogun, W. Qiu, W. Wang, P. Fang, X. Lu and Y. Tong, J. Mater. Chem. A, 2015, 3, 1364 RSC; (j) S. Dong, X. Chen, X. Zhang and G. Cui, Coord. Chem. Rev., 2013, 257, 1946 CrossRef CAS; (k) J. Guo, F. Chang, P. Wang, D. Hu, P. Yu, G. Wu, Z. Xiong and P. Chen, ACS Catal., 2015, 5, 2708 CrossRef CAS; (l) R. S. Ningthoujam and N. S. Gajbhiye, Prog. Mater. Sci., 2015, 70, 50 CrossRef CAS.
- (a) R. Juza and H. Hahn, Z. Anorg. Allg. Chem., 1940, 244, 125 CrossRef CAS; (b) M. Futsuhara, K. Yoshioka and O. Takai, Thin Solid Films, 1998, 322, 274 CrossRef CAS; (c) R. Long, Y. Dai, L. Yu, M. Guo and B. Huang, J. Phys. Chem. B, 2007, 111, 3379 CrossRef CAS PubMed; (d) S.-H. Yoo, A. Walsh, D. O. Scanlon and A. Soon, RSC Adv., 2014, 4, 3306 RSC.
- (a) W. R. L. Lambrecht, M. Prikhodko and M. S. Miao, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 174411 CrossRef; (b) A. Janotti, S.-H. Wei and L. Bellaiche, Appl. Phys. Lett., 2003, 82, 766 CrossRef CAS; (c) K. Suzuki, T. Kaneko, H. Yoshida, Y. Obi, H. Fujimori and H. Morita, J. Alloys Compd., 2000, 306, 66 CrossRef CAS; (d) H. Jacobs and C. Stüve, J. Less-Common Met., 1984, 96, 323 CrossRef CAS.
- (a) R. Juza, E. Anschütz and H. Puff, Angew. Chem., 1959, 71, 161 Search PubMed; (b) R. Niewa, F. J. DiSalvo, D. K. Yang, D. B. Zax, H. Luo and W. B. Yelon, J. Alloys Compd., 1998, 266, 32 CrossRef CAS; (c) R. Juza, K. Langer and K. Von Benda, Angew. Chem., Int. Ed. Engl., 1968, 7, 360 CrossRef CAS; (d) T. M. M. Richter, N. S. A. Alt, E. Schlücker and R. Niewa, Z. Anorg. Allg. Chem., 2015, 641, 1016 CrossRef CAS.
- (a) E. Gregoryanz, C. Sanloup, M. Somayazulu, J. Badro, G. Fiquet, H.-k. Mao and R. J. Hemley, Nat. Mater., 2004, 3, 294 CrossRef CAS PubMed; (b) T. Hashimoto, F. Wu, J. S. Speck and S. Nakamura, Nat. Mater., 2007, 6, 568 CrossRef CAS PubMed; (c) F. Rivadulla, M. Banobre-Lopez, C. X. Quintela, A. Pineiro, V. Pardo, D. Baldomir, M. A. Lopez-Quintela, J. Rivas, C. A. Ramos, H. Salva, J.-S. Zhou and J. B. Goodenough, Nat. Mater., 2009, 8, 947 CrossRef CAS PubMed; (d) S. T. Oyama, The Chemistry of Transition Metal Carbides and Nitrides, Springer, 1995 Search PubMed.
- M. Hasegawa and T. Yagi, J. Alloys Compd., 2005, 403, 131 CrossRef CAS.
- (a) M. Zając, J. Gosk, E. Grzanka, S. Stelmakh, M. Palczewska, A. Wysmołek, K. Korona, M. Kamińska and A. Twardowski, J. Alloys Compd., 2008, 456, 324 CrossRef; (b) H. Jacobs, D. Rechenbach and U. Zachwieja, J. Alloys Compd., 1995, 227, 10 CrossRef CAS; (c) A. Leineweber, H. Jacobs and S. Hull, Inorg. Chem., 2001, 40, 5818 CrossRef CAS PubMed; (d) T. M. M. Richter and R. Niewa, Inorganics, 2014, 2, 29 CrossRef CAS.
- H. Jacobs and U. Fink, Z. Anorg. Allg. Chem., 1978, 438, 151 CrossRef CAS.
- (a) G. Kreiner and H. Jacobs, J. Alloys Compd., 1992, 183, 345 CrossRef CAS; (b) B. Fröhling and H. Jacobs, Z. Anorg. Allg. Chem., 1997, 623, 1108 CrossRef.
- (a) H. Cao, Y. Zhang, J. Wang, Z. Xiong, G. Wu and P. Chen, Prog. Nat. Sci., 2012, 22, 550 CrossRef; (b) P. Chen and M. Zhu, Mater. Today, 2008, 11, 36 CrossRef CAS; (c) P. Chen, Z. Xiong, J. Luo, J. Lin and K. L. Tan, Nature, 2002, 420, 302 CrossRef CAS PubMed.
- M. Pozzo and D. Alfè, Int. J. Hydrogen Energy, 2009, 34, 1922 CrossRef CAS.
- (a) A.-L. Chaudhary, D. A. Sheppard, M. Paskevicius, M. Saunders and C. Buckley, RSC Adv., 2014, 42, 21979 RSC; (b) J. Huot, D. B. Ravnsbæk, J. Zhang, F. Cuevas, M. Latroche and T. R. Jensen, Prog. Mater. Sci., 2013, 58, 30 CrossRef CAS; (c) H. Y. Leng, T. Ichikawa, S. Hino, N. Hanada, S. Isobe and H. Fujii, J. Power Sources, 2006, 156, 166 CrossRef CAS; (d) C. Xu, S. De, A. M. Balu, M. Ojeda and R. Luque, Chem. Commun., 2015, 51, 6698 RSC; (e) P. G. McCormick, T. Tsuzuki, J. S. Robinson and J. Ding, Adv. Mater., 2001, 13, 1008 CrossRef CAS; (f) B. D. Stojanovic, J. Mater. Process. Technol., 2003, 143–144, 78 CrossRef CAS; (g) V. Sepelak, A. Duvel, M. Wilkening, K.-D. Becker and P. Heitjans, Chem. Soc. Rev., 2013, 42, 7507 RSC.
- R. Juza, Z. Anorg. Allg. Chem., 1937, 231, 121 CrossRef CAS.
- F. F. Fitzgerald, J. Am. Chem. Soc., 1907, 29, 656 CrossRef CAS.
- (a) L. Brisseau, J. Rouxel and C. R. Hebd, C. R. Acad. Sci., 1969, 268, 2308 CAS; (b) B. Fröhling and H. Jacobs, Z. Anorg. Allg. Chem., 1997, 623, 1103 CrossRef.
- M. Drew, L. Guémas, P. Chevalier, P. Palvadeau and J. Rouxel, Rev. Chim. Miner., 1975, 12, 419 CAS.
- T. M. M. Richter, S. Zhang and R. Niewa, Z. Kristallogr. - Cryst. Mater., 2013, 228, 351 CAS.
- H. Cao, T. M. M. Richter, C. Pistidda, A. L. Chaudhary, A. Santoru, G. Gizer, R. Niewa, P. Chen, T. Klassen and M. Dornheim, ChemSusChem, 2015, 22, 3777 CrossRef PubMed.
- (a) L. Ji, Z. Lin, M. Alcoutlabi and X. Zhang, Energy Environ. Sci., 2011, 4, 2682 RSC; (b) J. Cabana, C. M. Ionica-Bousquet, C. P. Grey and M. R. Palacín, Electrochem. Commun., 2010, 12, 315 CrossRef CAS.
- R. Niewa and F. J. DiSalvo, Chem. Mater., 1998, 10, 2733 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; TG-DTA curves of monoclinic K2[Zn(NH2)4]; TG-DTA-MS curves and de/re-hydrogenation curves of the as ball milled K2[Mn(NH2)4]-8LiH sample. See DOI: 10.1039/c6cc00719h |
| This journal is © The Royal Society of Chemistry 2016 |