
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unraveling a two-step oxidation mechanism in electrochemical Cu-MOF synthesis†
Philipp
Schäfer
a,
Monique A.
van der Veen
*b and
Katrin F.
Domke
*a
aMax Planck Institute for Polymer Research, Molecular Spectroscopy Department, Ackermannweg 10, D 55128 Mainz, Germany. E-mail: domke@mpip-mainz.mpg.de
bDelft University of Technology, Faculty of Applied Sciences, Chemical Engineering Department, Section of Catalysis Engineering, Julianalaan 136, NL 2628 BL Delft, Netherlands. E-mail: M.A.vanderVeen@tudelft.nl
First published on 1st March 2016
Abstract
To employ the full potential of electrochemical (ec) synthesis to grow metal–organic frameworks (MOFs) in more complex organizations at the mesoscale, it is vital to understand the underlying crystallization reaction pathway. For the MOF most typically grown electrochemically, CuBTC, we systematically investigated the role of oxygen species in the synthesis.
Metal–organic frameworks (MOFs) attract significant interest as versatile materials for gas separation1 and storage,2 and more recently as light harvesters for novel energy conversion schemes.3,4 Their topology (crystal structure, pore size, pore connectivity) and functionality (chemistry at reactive sites, luminescence/absorption properties of framework, guest donor/acceptor moieties) can, in principle, be tuned to desire by making use of the wealth of metal ion and organic linker combinations the chemist's tool kit provides. While classically, MOFs are synthesized solvothermally at elevated pressure and/or temperature,5,6 recently, novel anodic and cathodic7–12 electrochemical (ec) fabrication protocols are emerging as electrosynthesis offers milder conditions, greater energy efficiency and energetic control over the MOF growth.7,13,14
It has been shown that by applying an appropriate synthesis potential, the size of MOF crystals can be directly controlled in the sub- to 5 μm size range,7,8 or that different linkers can be built in on demand to fabricate multi-functionality MOFs in situ.10 These cases demonstrate the extraordinary possibilities electrosynthesis offers for controlled MOF growth. However, generalizing the ec approach requires a detailed understanding of the underlying reaction mechanisms which have to date only been speculated on.7,15 Questions regarding the chemical species involved, the role of the substrate surface and the energetics and kinetics of the overall reaction pathway of ec MOF formation need to be answered to mature ec synthesis into a readily available tool for rational MOF design.
In this work, we unravel the reaction mechanism of the ec formation of a showcase MOF, CuBTC (HKUST-1; Cu 1,3,5 tricarboxylic acid). Prepared according to the original protocol from 2005, electrosynthesized CuBTC, finds large-scale application e.g. for carbon dioxide/methane16 or propylene/propane17 gas mixture separation, or in novel sensors and electronic devices.18 Despite CuBTC being one of the most extensively studied MOFs,7,19 its ec formation mechanism has not yet been unraveled, hindering full exploitation of electrosynthetic MOF design possibilities. It has been postulated that, Cu is anodically oxidized in one step to Cu2+ followed by linker coordination in solution.7,15 Other groups, however, have observed MOF crystals intergrown with the surface that suggest on-surface growth.20
To solve the prevailing controversy and open a route for rational MOF topology and functionality design, we set out to identify the chemical species involved in CuBTC electro-synthesis and unravel the reaction mechanism. We systematically varied the experimental conditions such as the Cu source, the presence of O2 and the applied potential (Table 1), generally following the protocol found in literature:7,21 a Cu plate was immersed at open circuit in a mixture of 100 mL absolute EtOH, 3 g BTC linker and 1 g methyl tributyl ammonium methyl sulfate electrolyte. After electrode immersion, a potential of E = 1 V vs. Ag/AgCl was applied for 20 min. To prevent Cu2O formation on the Cu electrode from ambient aerobic corrosion in O2-free experiments, EtOH was degassed with Ar for 20 min prior to Cu immersion, and the ec cell was constantly kept under Ar. To remove the natural oxide layer (comprised mainly of Cu2O and a small amount of CuO and Cu(OH)222), the Cu electrode was etched with 25% HNO3 and 25% HCl23 before transfer to the synthesis cell.
| Sample type | Cu source | O2 presence | E /reaction time | CuBTC formation |
|---|---|---|---|---|
| a Potentials reported vs. Ag/AgCl/3 M KCl. b SEM/Raman characterization in ESI. | ||||
| A | Cu with natural oxide layer | Yes | 1 V/20 min | Yes |
| B | Cu, oxide free | No | 1 V/20 min | No |
| C | Cu with artificial oxide layer | No | 1 V/20 min | Yes |
| D | Cu2O | Yes | No/16 hours | Yes |
| E | Cu2O | Low amount | No/16 hours | Yes (truncated) |
| Fb | CuO | Yes | No/16 hours | No |
The resulting topography and chemical composition of each sample type was characterized to determine whether CuBTC grows under the respective conditions. CuBTC is known to form octahedral crystals that can be seen with scanning electron microscopy (SEM).24 Raman in-plane ring bending vibrations at 743 and 825 cm−1 of the BTC moiety in CuBTC confirm the presence of CuBTC (Fig. S2 and Table S1, ESI†).25 X-ray diffraction (XRD) was used for CuBTC identification where a sufficient amount of MOF crystals was obtained (see ESI,† for experimental details).
Fig. 1 shows the SEM and Raman results for the different sample types under investigation. Samples A were fabricated under synthesis conditions similar to the ec synthesis protocols found in literature,7,21i.e. starting from a Cu-electrode with a natural oxide layer and taking no precautions to avoid the presence of O2. As expected, octahedral crystals of CuBTC of <1 μm to 5 μm in diameter form (Fig. 1A). Electrode coverage is incomplete at ∼20 particles per 100 μm2, likely due to the low potential of 1 V. An increased synthesis time should improve surface coverage. The corresponding Raman spectrum in the inset of Fig. 1A displays the characteristic bending vibrations of CuBTC at 743 and 825 cm−1, and XRD diffractograms confirm the successful synthesis of CuBTC (Fig. S1A, ESI†). Note that the Raman spectrum also shows a significant contribution of Cu2O (see Fig. S3A, ESI,† broad peaks at 525 and 626 cm−1).
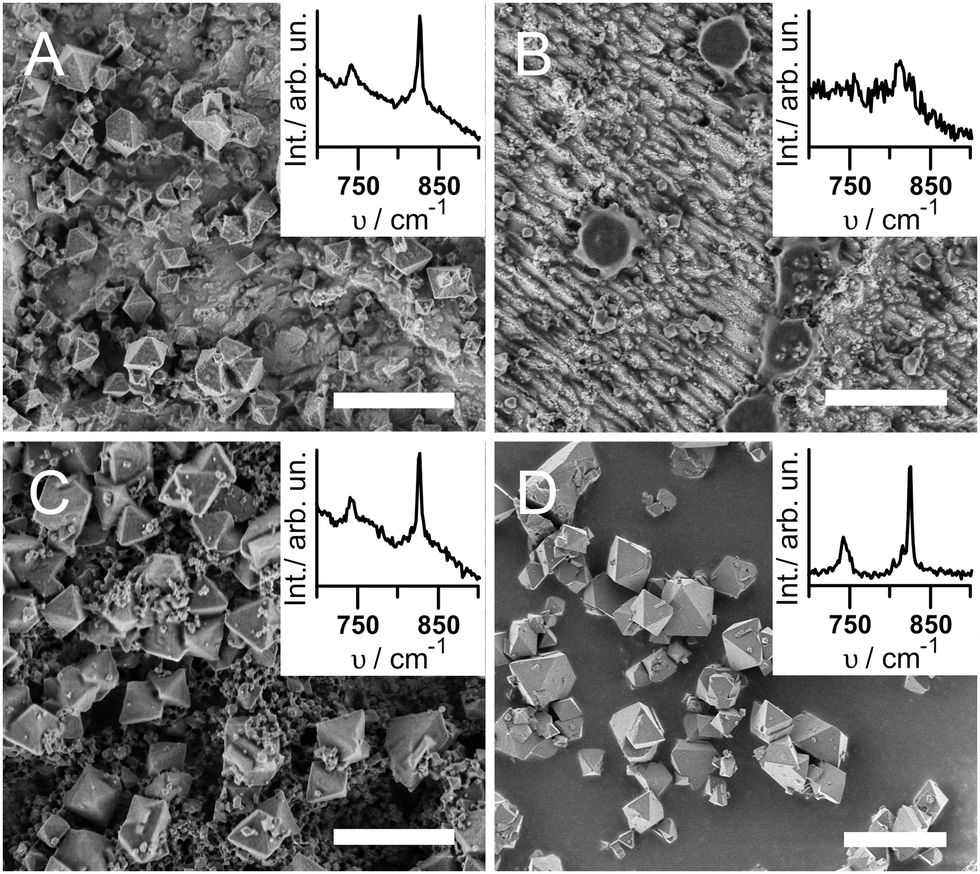 | ||
| Fig. 1 SEM micrographs and Raman spectra of Samples A to D. Scale bar: 10 μm. | ||
Samples B were produced under oxide- and O2-free conditions, thus void of all components unnecessary for the Cu2+ dissolution-coordination mechanism proposed in literature. The SEM image (Fig. 1B) shows parallel Cu tracks due to the acidic etching of the surface. With exception of only about 1 particle per 100 μm2 of ca. 0.5 to 1 μm diameter, no CuBTC crystals are visible. The CuBTC response in the Raman spectra can hardly be distinguished from the noise (Fig. 1B inset), and no XRD signal of CuBTC was obtained (Fig. S1B, ESI†).
To find the smallest set of components required to enable CuBTC synthesis, we electrochemically oxidized the surface of the Cu plate on purpose by immersing it in the electrolyte solution and applying a potential of 1 V vs. Ag/AgCl for 25 min prior to O2-free MOF synthesis (Samples C). SEM, Raman and XRD confirm the successful synthesis of CuBTC (Fig. 1C and Fig. S1C, ESI†); the Raman spectrum shows a significant Cu2O contribution (Fig. S3C, ESI†). Samples C are covered with a mixture of about 5 crystals per 100 μm2 of octahedral crystals of 3 to 5 μm diameter and ca. 50 particles per 100 μm2 in the sub-μm diameter range. At some spots, the smaller crystals cover the larger particles.
From experiments A to C, we learn that oxygen plays an important role in ec CuBTC synthesis as CuBTC crystals do not grow under O2-oxygen free conditions and Cu2O is always present on the electrodes. To further investigate the role of oxygen, we used pure oxides as precursors for CuBTC growth. For Samples D, we immersed Cu2O powder in an ethanolic solution of 0.15 M BTC without any other additions for 14 hrs under ambient conditions. No potential was applied. A blue powder was isolated by centrifugation and characterized. The SEM micrograph in Fig. 1D shows octahedral crystals of 1 to 5 μm diameter that are intergrown into larger agglomerates of 10 to 15 μm length. Raman spectra (Fig. 1D inset) and XRD (Fig. S1D, ESI†) confirm CuBTC synthesis. Interestingly, an analogous experiment with CuO powder as starting material did not produce any CuBTC (Samples F, ESI†).
To identify the necessary oxidation agent for the oxidation of Cu(I) to Cu(II) in absence of an applied potential, Samples E were synthesized like Samples D, but in an O2-deficient environment. The solution was degassed for 15 min, but during transfer into a glove box, some O2 could re-dissolve. The SEM micrographs show about 10% of the Cu2O surface covered with octahedral crystals of diameters between 0.3 and 0.7 μm, and partly with what seem to be incomplete CuBTC crystals with diameters of 100 to 200 nm (Fig. 2, red). This is the only Sample for which we observe incomplete CuBTC octahedrons. It is unlikely that these crystals deposited onto the Cu2O substrate during centrifugation; rather they must have grown at the surface, and their growth was halted prematurely due to lack of O2. Similarly, also the few octahedral crystals (Fig. 2, blue) are visibly attached to the Cu2O substrate. From these results, we conclude that O2 is the necessary oxidant for the oxidation of Cu2O to CuBTC without ec potential. Furthermore, the visible connection between the CuBTC and the Cu2O indicates that CuBTC nucleation likely takes place directly at the Cu2O solid/liquid interface. Summing up, the first step to ec CuBTC formation is the oxidation of Cu to Cu1+ achieved either by ec oxidation of the Cu anode or by providing O2 and/or H2O (H2O enables CuBTC synthesis even under inert conditions.20) as reactant. Cu2O is the predominant oxidation product of Cu in ethanol.26 Note that also Cu(OH)2 readily converts into CuBTC in presence of the linker.27 While Cu(OH)2 as an intermittent, short-lived reaction intermediate cannot be excluded, our spectroscopic data does not indicate stable Cu(OH)2 formation (i.e. no Raman band at 460 cm−1, Fig. S2b, ESI†). Furthermore, CuBTC forms directly from Cu1+2O powder while Cu2+O does not convert to CuBTC under the given experimental conditions.
 | ||
| Fig. 2 SEM of Samples E. Red: truncated crystals; blue: intergrowth with surface. Scale bar: 500 nm. | ||
As known from literature, Cu2O can be produced from Cu by the following oxidation reactions with H2O or O2:22,28
| 2Cu + H2O ↔ Cu2O + 2H+ + 2e− | (1) |
| 4Cu + O2 ↔ 2Cu2O | (2) |
Instead of using O2 or H2O as oxidant, Cu can be electro-chemically oxidized. In absence of O2 and H2O, the amount of available Cu+12O is expected to limit the CuBTC yield. Indeed, for Samples C, we did not observe continuous detachment of large MOF crystals from the electrode into the solution phase, indicating that CuBTC growth stopped after all provided Cu2O had been consumed.
For the second oxidation step of Cu1+2O to Cu2+BTC, we propose the following ec half reaction:
| 3Cu2O + 4H3BTC ↔ 2Cu3BTC2 + 3H2O + 6e− + 6H+ | (3) |
| O2 + 4H+ + 4e− ↔ 2H2O | (4) |
Direct oxidation of solid Cu2O to CuBTC at the ec solid/liquid interface to start crystal nucleation likely explains the tight attachment of CuBTC crystals to the electrode as previously observed7 as well as the incomplete CuBTC crystals visibly bound to Cu2O that we observe under oxygen-limited conditions; however, a dissolution process with Cu2O as intermittent cannot be completely excluded. It remains unclear whether further crystal growth happens at the Cu2O–CuBTC–electrolyte interface or continues through a solution-based process with Cu2+ ions attaching to the outer facets of the growing crystal. Campagnol et al. show that electrosynthesized Cu isonicotinate grows at the MOF–electrolyte interface.29 The significant amount of water (50%)30 that makes Cu2+ ions more soluble in the form of [Cu(H2O)6]2+ could explain a solution-based process. The lack of H2O in our system, however, makes a direct comparison difficult.
Let us summarize the reaction mechanism for the ec oxidation of Cu0 to CuBTC: in presence of O2 and/or H2O, Cu is oxidized in a one-electron step to Cu2O. A second oxidation step from Cu1+ to Cu2+ in the presence of BTC directly converts Cu2O to CuBTC. Involvement of an intermediate Cu(I) reaction step has not been proposed so far and opens up new pathways to increase control of the synthesis.
Our newly gained knowledge that CuBTC crystallization proceeds through Cu2O formation allows us to suggest a novel method to fabricate patterned MOF devices (Fig. 3A). As a proof of principle, we prepared a Cu substrate with both Cu and Cu2O present by ec oxidizing the surface in the presence of NaOH.31 Then, we selectively removed Cu2O in a spot of ∼1 cm diameter in the centre of the sample by pipetting a drop of 10% HCl. After removal of that drop and subsequent rinsing with EtOH, the Cu plate was covered with Cu2O except for the acid-etched blank Cu area. Under synthesis conditions B (O2 exclusion, 1 V), CuBTC was grown solely on the oxide-covered part of the sample as discernible in the white-light image (Fig. 3B). The left part of the image where surface oxides were removed by acidic etching shows the pristine Cu surface. To the right of the image, outside the area of the acid droplet, CuBTC has visibly grown and is spectroscopically detectable. Raman band integration of the 800 to 850 cm−1 region provides the relative amounts of CuBTC at different sample positions (Fig. 3C; error bars are standard errors of the mean of three measurements taken at roughly the top, middle and bottom of the white-light image). This facile oxide-based pre-patterning approach can serve as starting point to develop improved synthesis protocols for CuBTC patterning. Current patterning approaches require the use of Cu electrodes in the desired shape (i.e. printed circuit boards8 or Cu meshes20). Our approach offers the possibility to subsequently activate parts of a Cu substrate. It is easy to imagine how established ways to deposit Cu2O as nanoparticles or films32,33 or to selectively remove Cu oxide, as shown here, can be converted into a versatile basis for the fabrication of arbitrarily patterned surfaces on nm to mm length scales to create devices covered with MOF of controlled sizes at predetermined spots.
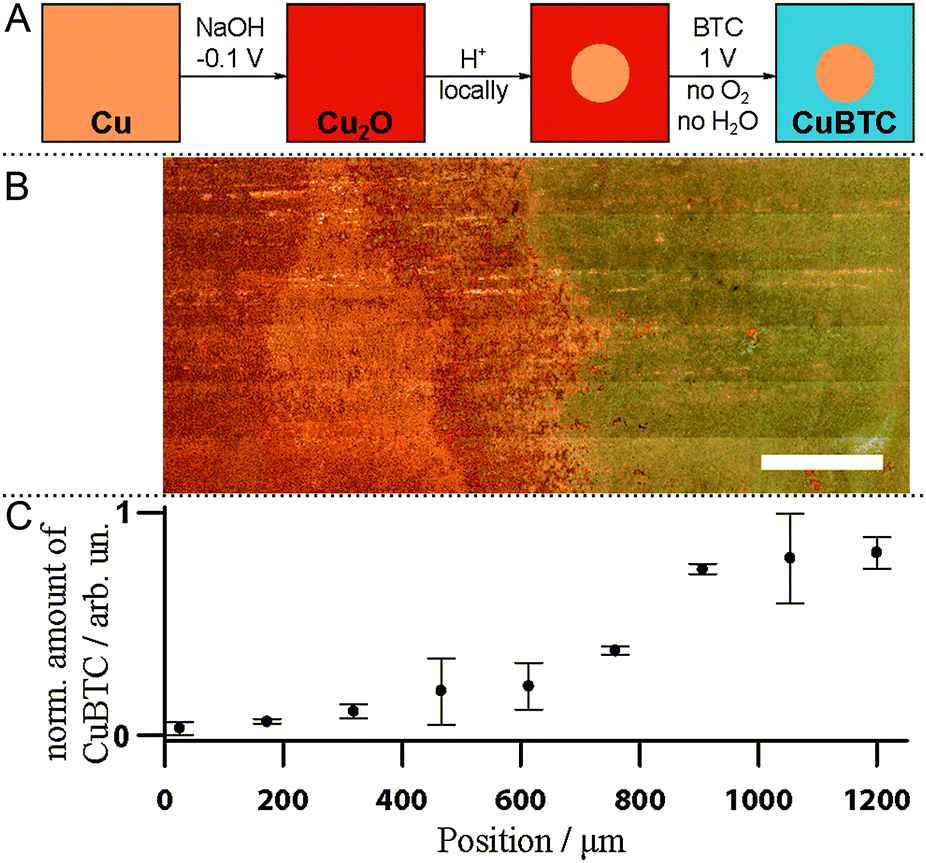 | ||
| Fig. 3 A: Scheme for the fabrication of patterned CuBTC devices by oxide patterning; B: light-microscopy image of a Cu sample (left) partly covered with CuBTC (right). Scale bar: 200 μm; C: normalized amount of CuBTC across sample. | ||
To conclude, we unravelled CuBTC electrosynthesis to proceed in a two-step oxidation mechanism at the electrode surface: Cu is first oxidized to Cu1+2O in the presence of H2O or O2. Cu2O is further oxidized to Cu2+BTC in presence of the linker at the cuprite–electrolyte interface. The MOF octahedrons nucleate directly at the electrode surface and not in solution, rendering the ec mechanism different from the one of solvothermal syntheses with metal salts.34,35 We demonstrate how the gained knowledge can be used for a novel quick and versatile approach to produce patterned CuBTC devices.
We thank G. Glasser for SEM measurements, M. Steiert and B. Norder for XRD measurements and the Max Planck Graduate Center and the Studienstiftung des deutschen Volkes for funding. KFD acknowledges generous support through the Emmy Noether Program of the DFG (#DO1691/1-1).
Notes and references
- J. Teufel, H. Oh, M. Hirscher, M. Wahiduzzaman, L. Zhechkov, A. Kuc, T. Heine, D. Denysenko and D. Volkmer, Adv. Mater., 2013, 25, 635–639 CrossRef CAS PubMed.
- A. U. Czaja, N. Trukhan and U. Müller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- X. Zhang, W. Wang, Z. Hu, G. Wang and K. Uvdal, Coord. Chem. Rev., 2015, 284, 206–235 CrossRef CAS.
- M. A. Nasalevich, M. van der Veen, F. Kapteijn and J. Gascon, CrystEngComm, 2014, 16, 4919 RSC.
- M. G. Goesten, F. Kapteijn and J. Gascon, CrystEngComm, 2013, 15, 9249 RSC.
- Y.-R. Lee, J. Kim and W.-S. Ahn, Korean J. Chem. Eng., 2013, 30, 1667–1680 CrossRef CAS.
- A. Martinez Joaristi, J. Juan-Alcañiz, P. Serra-Crespo, F. Kapteijn and J. Gascon, Cryst. Growth Des., 2012, 12, 3489–3498 CAS.
- R. Ameloot, L. Stappers, J. Fransaer, L. Alaerts, B. F. Sels and D. E. De Vos, Chem. Mater., 2009, 21, 2580–2582 CrossRef CAS.
- I. Hod, W. Bury, D. M. Karlin, P. Deria, C. W. Kung, M. J. Katz, M. So, B. Klahr, D. N. Jin, Y. W. Chung, T. W. Odom, O. K. Farha and J. T. Hupp, Adv. Mater., 2014, 26, 6295–6300 CrossRef CAS PubMed.
- M. Li and M. Dincă, Chem. Sci., 2014, 5, 107 RSC.
- M. Li and M. Dincă, Chem. Mater., 2015, 27, 3203–3206 CrossRef CAS.
- I. Stassen, M. Styles, T. R. C. Van Assche, N. Campagnol, J. Fransaer, J. F. M. Denayer, J. C. Tan, P. Falcaro, D. E. De Vos and R. Ameloot, Chem. Mater., 2015, 27, 1801–1807 CrossRef CAS.
- N. Campagnol, T. R. C. Van Assche, T. Boudewijns, J. F. M. Denayer, K. Binnemans, D. E. De Vos and J. Fransaer, J. Mater. Chem. A, 2013, 1, 5827 CAS.
- M. Li and M. Dincă, J. Am. Chem. Soc., 2011, 133, 12926–12929 CrossRef CAS PubMed.
- H. Al-Kutubi, J. Gascon, E. J. R. Sudhölter and L. Rassaei, ChemElectroChem, 2015, 2, 462–474 CrossRef CAS.
- T. Asadi, M. R. Ehsani, A. M. Ribeiro, J. M. Loureiro and A. E. Rodrigues, Chem. Eng. Technol., 2013, 36, 1231–1239 CrossRef CAS.
- J.-W. Yoon, I.-T. Jang, K.-Y. Lee, Y.-K. Hwang and J.-S. Chang, Bull. Korean Chem. Soc., 2010, 31, 220–223 CrossRef CAS.
- J. Gascon and F. Kapteijn, Angew. Chem., Int. Ed., 2010, 49, 1530–1532 CrossRef CAS PubMed.
- B. Panella, M. Hirscher, H. Pütter and U. Müller, Adv. Funct. Mater., 2006, 16, 520–524 CrossRef CAS.
- T. R. C. Van Assche, G. Desmet, R. Ameloot, D. E. De Vos, H. Terryn and J. F. M. Denayer, Microporous Mesoporous Mater., 2012, 158, 209–213 CrossRef CAS.
- T. R. C. Van Assche and J. F. M. Denayer, Chem. Eng. Sci., 2013, 95, 65–72 CrossRef CAS.
- I. Platzman, R. Brener, H. Haick and R. Tannenbaum, J. Phys. Chem. C, 2008, 112, 1101–1108 CAS.
- L. C. Hall, C. P. Major, J. J. Steppan, J. A. Roth and B. K. Vaughen, J. Electrochem. Soc., 1987, 134, 1902 CrossRef CAS.
- Q. Min Wang, D. Shen, M. Bülow, M. Ling Lau, S. Deng, F. R. Fitch, N. O. Lemcoff and J. Semanscin, Microporous Mesoporous Mater., 2002, 55, 217–230 CrossRef.
- C. Prestipino, L. Regli, J. G. Vitillo, F. Bonino, a. Damin, C. Lamberti, a. Zecchina, P. L. Solari, K. O. Kongshaug and S. Bordiga, Chem. Mater., 2006, 18, 1337–1346 CrossRef CAS.
- B. Stypuła, J. Banaś, M. Starowicz, H. Krawiec, A. Bernasik and A. Janas, J. Appl. Electrochem., 2006, 36, 1407–1414 CrossRef.
- G. Majano and J. Perez-Ramirez, Adv. Mater., 2013, 25, 1052–1057 CrossRef CAS PubMed.
- H. Y. H. Chan, C. G. Takoudis and M. J. Weaver, J. Phys. Chem. B, 1999, 103, 357–365 CrossRef CAS.
- N. Campagnol, T. R. C. Van Assche, L. Stappers, J. F. M. Denayer, K. Binnemans, D. E. De Vos and J. Fransaer, ECS Trans., 2014, 61, 25–40 CrossRef.
- B. Van de Voorde, R. Ameloot, I. Stassen, M. Everaert, D. E. De Vos and J.-C. Tan, J. Mater. Chem. C, 2013, 1, 7716 RSC.
- J. C. Hamilton, J. C. Farmer and R. J. Anderson, J. Electrochem. Soc., 1986, 133, 739 CrossRef CAS.
- P. E. de Jongh, D. Vanmaekelbergh, J. J. Kelly and P. E. D. Jongh, Chem. Mater., 1999, 11, 3512–3517 CrossRef CAS.
- D. P. Singh, N. R. Neti, A. S. K. Sinha and O. N. Srivastava, J. Phys. Chem. C, 2007, 111, 1638–1645 CAS.
- V. Stavila, J. Volponi, A. M. Katzenmeyer, M. C. Dixon and M. D. Allendorf, Chem. Sci., 2012, 3, 1531 RSC.
- O. Shekhah, H. Wang, D. Zacher, R. A. Fischer and C. Wöll, Angew. Chem., Int. Ed., 2009, 48, 5038–5041 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods (Raman, XRD, SEM data); characterization of Samples F. Current transients. See DOI: 10.1039/c6cc00534a |
| This journal is © The Royal Society of Chemistry 2016 |