
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A fluorescent surrogate of thymidine in duplex DNA†
Guillaume
Mata
,
Olivia P.
Schmidt
and
Nathan W.
Luedtke
*
Department of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: nathan.luedtke@chem.uzh.ch
First published on 25th February 2016
Abstract
DMAT is a new fluorescent thymidine mimic composed of 2′-deoxyuridine fused to dimethylaniline. DMAT exhibits the same pKa and base pairing characteristics as native thymidine residues, and its fluorescence properties are highly sensitive to nucleobase ionization, base pairing and metal binding.
Nucleobase analogs constitute an important family of fluorescent probes.1 They can be positioned in nucleic acid structures with high precision, and their photophysical properties are highly sensitive to local polarity,2 viscosity,3 and pH.4 These features facilitate specific monitoring of biochemical transformations,5 conformational changes,6 metal binding,7 and base pairing interactions.8
Nucleobase ionization can mediate proton-coupled folding,9 metal binding,10 and/or the catalytic activities of certain nucleic acids at neutral pH.11 Thymidine (T) and uracil (U) are among the most inherently acidic residues,12 but fluorescent analogs capable of reporting pyrimidine ionization in nucleic acids are scarce. Previously reported examples utilized biaryl or triaryl fluorophores,4c,d having unreported or highly perturbed pKa values (pKa ≈ 6.8) as compared to unmodified T and U residues (pKa ≈ 9.5).12 Here we report “N,N-dimethylaniline-2′-deoxythymidine” or “DMAT” that exhibits the same pKa and base pairing characteristics as thymidine, as well as fluorescence properties that can be used to monitor nucleobase ionization, base pairing and metal binding reactions in DNA.
DMAT was designed to have the same Watson–Crick face and pKa as thymidine. To generate a push–pull fluorophore, an electron donating group was incorporated at the C6 position of a quinazoline core.9,13 This position was selected because it is not in conjugation with N3–H, and therefore expected to have little or no impact on its acidity. Molecular orbital calculations predicted charge transfer from a dimethylaniline-centered HOMO to a pyrimidine-centered LUMO with a HOMO–LUMO energy gap (ΔE) = 2.44 eV (Fig. 1). DMAT was therefore predicted to be a “push–pull” fluorophore in its neutral form. In contrast, the DMAT anion has a pyrimidine-centered HOMO, and a larger ΔE = 2.81 eV. We therefore expected a blue-shift in DMAT fluorescence upon its deprotonation.
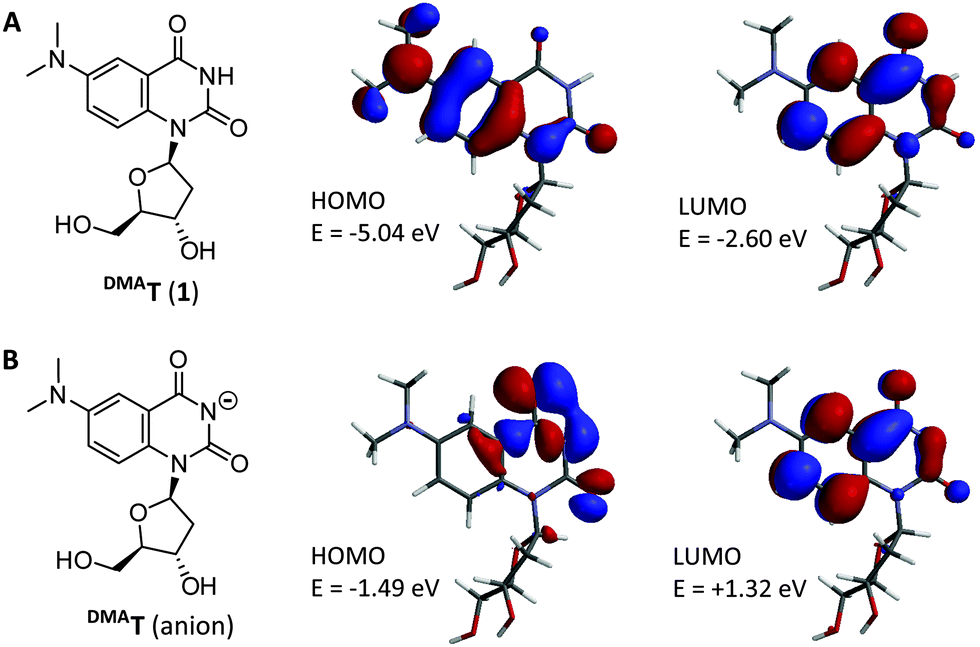 | ||
| Fig. 1 Structure of the DMAT nucleoside (A), and its conjugate base (B). HOMOs, LUMOs, and their relative energies were calculated from DFT-optimized geometries using LSDA/pBP86/DN**. | ||
The synthesis of DMAT (1) commenced from the previously-reported nucleoside 2 (Scheme 1).14 Buchwald–Hartwig coupling with Me2NH gave the known compound 3 in 82% yield.9 Addition of fluoride ions to 3 afforded the new nucleoside DMAT (1) in a 62% yield (see ESI† for synthetic details and characterization). 1H–1H ROESY cross-peaks were observed between H8–H5′, H8–H3′, and H8–H2′β of DMAT (1), indicating an anti-conformation of the nucleobase (Fig. 2). Cross-peaks were also observed between H1′–H4′, which confirmed the β-stereochemistry of the anomeric position.14DMAT (1) therefore possesses the same glycosidic bond conformation and stereochemistry as 2′-deoxythymidine.
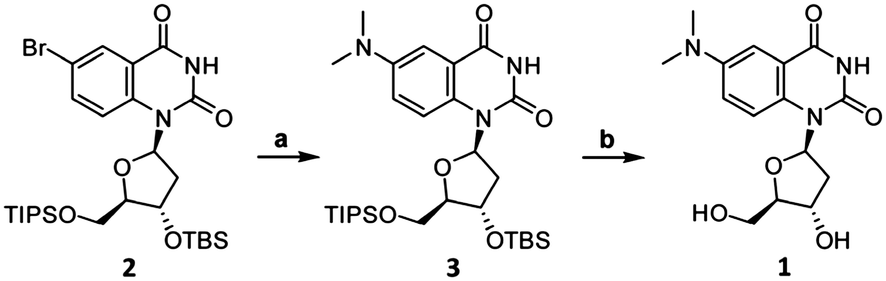 | ||
| Scheme 1 Synthesis of DMAT (1). Reaction conditions: (a) Me2NH, Pd2(dba)3 (5 mol%), JohnPhos (20 mol%), KOtBu, THF, dioxane, 60 °C, 2 h, 82% yield. (b) TBAF, THF, 23 °C, 2 h, 62% yield. | ||
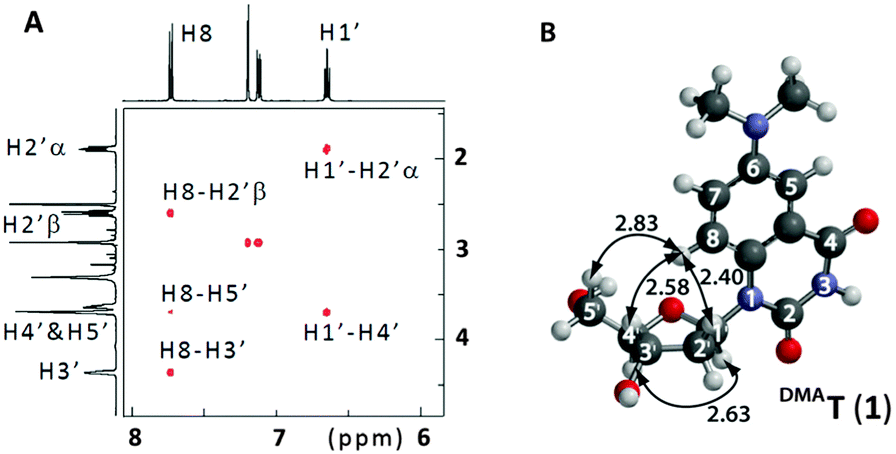 | ||
| Fig. 2 (A) Partial 1H–1H 2D ROESY of DMAT (1) in DMSO-d6. See ESI† for complete spectrum. (B) Energy-minimized conformation of DMAT according to DFT calculations (B3LYP/6-311G*). 1H–1H distances are shown in Å. | ||
Under neutral conditions, DMAT (1) exhibits an exceptionally large Stoke's shift, with an absorbance maximum (λabs) = 357 nm and emission maximum (λem) = 522 nm (Table 1). To characterize its environmental sensitivity, the λabs and λem of DMAT (1) were measured in various water/dioxane mixtures (Table S2 and Fig. S1, ESI†). A linear correlation (R2 = 0.980) with a large slope of 177 cm−1 kcal−1 mol−1 was obtained by plotting the Stoke's shift of DMAT (1) against Reichardt's solvent polarity parameter (E30T).15,16 Together these results confirm that DMAT (1) is a push–pull fluorophore. Interestingly, DMAT (1) exhibits a two-fold higher quantum yield in D2O (ϕ = 0.07) than H2O (ϕ = 0.03, Table 1), suggesting that proton transfer with bulk solvent provides an effective nonradiative decay pathway.7a
| Solvent | λ abs | λ em | ε (260) | ε (λabs) | ϕ |
|---|---|---|---|---|---|
| a Absorbance maxima (λabs) in nm. b Emission maxima (λem) in nm. c Extinction coefficients (ε) in 103 M−1 cm−1 were measured at 260 nm and at λabs. d Quantum yields (ϕ) were calculated using quinine hemisulfate in 0.5 M H2SO4 as a fluorescent standard.17 Reproducibility is within ±10% of each reported ϕ value. | |||||
| H2O | 357 | 522 | 15.0 | 2.9 | 0.03 |
| pH = 11.0 | 345 | 480 | 17.1 | 3.9 | 0.09 |
| D2O | 355 | 520 | 13.0 | 2.7 | 0.07 |
| pD = 11.0 | 345 | 480 | 14.3 | 3.4 | 0.19 |
To evaluate the fluorescence sensitivity of DMAT (1) towards nucleobase ionization, its absorbance and emission spectra were recorded at different pH values. Consistent with DFT calculations, the emission maximum of DMAT (1) shifted towards the blue with increasing pH (Fig. 3A). This was accompanied by a dramatic increase in fluorescence intensity. The absorbance and fluorescence changes were plotted against pH to determine a pKa = 9.5 ± 0.1, Fig. 3B. This value corresponds to the pKa of thymidine and uracil.12
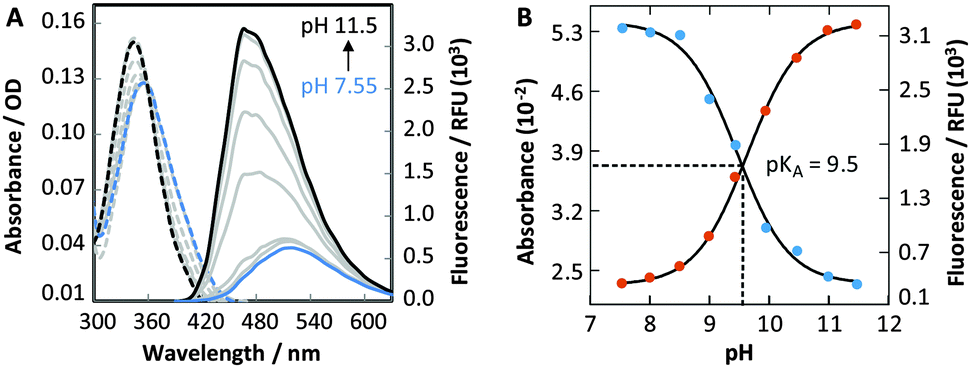 | ||
Fig. 3 (A) Absorbance (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) and emission (—) spectra (λex = 360 nm) of 40 μM DMAT (1) in phosphate citric acid buffer (200 mM of Na2HPO4, 100 mM of citric acid and 100 mM NaCl). (B) Single-wavelength analyses of the pH-dependent absorbance (400 nm, blue dots) and emission (470 nm, red dots) of DMAT (1). ) and emission (—) spectra (λex = 360 nm) of 40 μM DMAT (1) in phosphate citric acid buffer (200 mM of Na2HPO4, 100 mM of citric acid and 100 mM NaCl). (B) Single-wavelength analyses of the pH-dependent absorbance (400 nm, blue dots) and emission (470 nm, red dots) of DMAT (1). | ||
To facilitate the site-specific incorporation of DMAT into DNA, phosphoramidite 5 was prepared in two steps by standard DMT-protection and phosphitylation reactions (Scheme 2). Using automated DNA synthesis, DMAT was incorporated at one of four positions within the same, 21-residue DNA sequence (Table S3, ESI†). Three positions near the middle of the sequence (X13, X14 and X15) and a single position near the 5′ terminus (X2) were selected in order to evaluate the impacts of variable flanking sequences and DNA end “breathing” motions, respectively.‡ The identity and purity of the purified oligonucleotides were confirmed using analytical HPLC and HR-MS (Table S4 and Fig. S2, ESI†).
 | ||
| Scheme 2 Synthesis of DMAT phosphoramidite (5). Reaction conditions: (a) DMT-Cl, pyridine, 23 °C, 45 min, 79% yield. (b) 2-Cyanoethyl-N,N-diisopropyl-chlorophosphoramidite, DIPEA, CH2Cl2, 0 °C to 23 °C, 45 min, 86% yield. See ESI† for synthetic details and characterizations. | ||
Circular dichroism (CD) and thermal denaturation experiments were used to assess the impact of DMAT on the global structure and stability of duplex DNA. DMAT-containing duplexes were prepared by heating and slow cooling with 1.1 equiv. of the complementary strand to give CD spectra consistent with the formation of B-form helices (Fig. S3, ESI†).18 CD spectra were monitored as a function of temperature (Fig. S4, ESI†) to determine the melting temperature of each duplex (Tm, Table 2). DMAT–A-containing duplexes exhibited nearly identical Tm values as the corresponding wild-type duplexes containing T–A base pairs (ΔTm = −0.2 to −1.7 °C). In contrast, duplexes containing a single C–A, A–A, DMAT–T or T–T mismatch at positions X13–X15 caused a large loss in thermal stability (ΔTm = −4.8 to −7.9 °C). End breathing motions of duplex DNA explain the relatively small changes when the mismatches were positioned at X2 (Table 2).
| Position | T–A | DMAT–A | C–A | A–A | DMAT–T | T–T |
|---|---|---|---|---|---|---|
| a All samples contained 5 μM of DNA in phosphate citric acid buffer (200 mM of Na2HPO4, 100 mM of citric acid, and 100 mM NaCl or NaNO3) at pH = 7.35. Reproducibility is within ±0.3 °C of each reported value. See Table S3, ESI for duplex sequences. | ||||||
| X2 | 69.0 | 68.8 | 67.3 | 67.5 | 68.0 | 67.0 |
| X13 | 68.8 | 67.1 | 60.9 | 62.4 | 62.3 | 61.5 |
| X14 | 68.7 | 67.3 | 61.3 | 63.9 | 62.5 | 61.5 |
| X15 | 68.9 | 68.0 | 61.2 | 62.7 | 64.0 | 62.8 |
The fluorescence properties of DMAT were highly sensitive to the global structure of the DNA containing it (Fig. S5, ESI,† and Table 3). At all three internal positions X13–X15, DMAT exhibited a two-fold higher quantum yield and blue-shifted λem in duplex versus single-stranded DNA. Decreased probe hydration upon duplex formation is probably responsible for these differences, because the DMAT nucleoside (1) exhibited higher quantum yields and blue-shifted λem in organic versus aqueous solvents (Table S2 and Fig. S1, ESI†). At all four positions of incorporation, higher fluorescence anisotropy was observed in duplex DNA (r = 0.09–0.18) as compared to unfolded structures (r = 0.03–0.06), consistent with large losses in dynamic motions of the probe upon duplex formation (Table 3).
| Structure | Position | λ abs | λ em | r | ϕ |
|---|---|---|---|---|---|
| a Absorbance maxima (λabs) in nm. b Emission maxima (λem) in nm. c Fluorescence anisotropy (r) with λex = 375 nm and λem = 500 nm. d Quantum yields (ϕ) calculated using the DMAT nucleoside (ϕ = 0.03) as a fluorescent standard. Reproducibility is within ±30% of each reported ϕ value. All samples contained 4 μM of DNA in a buffer containing 20 mM of Na2HPO4, 10 mM of citric acid and 10 mM NaCl (pH = 7.35). See Table S3, ESI for duplex sequences. | |||||
| Duplex | X2 | 355 | 505 | 0.18 | 0.03 |
| X13 | 355 | 504 | 0.09 | 0.20 | |
| X14 | 355 | 492 | 0.14 | 0.13 | |
| X15 | 355 | 486 | 0.15 | 0.11 | |
| Unfolded | X2 | 365 | 515 | 0.03 | 0.05 |
| X13 | 365 | 515 | 0.05 | 0.09 | |
| X14 | 365 | 516 | 0.06 | 0.06 | |
| X15 | 365 | 515 | 0.05 | 0.05 | |
The photophysical properties of DMAT were sensitive to matched versus mismatched base pairing in duplex DNA. DMAT exhibited a higher quantum yield and blue-shifted λabs and λem in DMAT–A base pairs as compared to DMAT–T, DMAT–G and DMAT–C mismatches (Table 4). Changes in probe hydration and base stacking are likely responsible for these differences, because similar trends were also observed when comparing duplex versus single-stranded DNA containing DMAT (Table 3). Taken together with the thermal denaturation results (Table 2), these data provide additional evidence that DMAT exhibits the same base pairing specificity as T.
| Position | Pairing | λ abs | λ em | r | ϕ |
|---|---|---|---|---|---|
| a See Table 3 footnotes for experimental details and symbol definitions. | |||||
| X13 | DMAT–A | 355 | 504 | 0.09 | 0.20 |
| DMAT–T | 365 | 505 | 0.09 | 0.13 | |
| DMAT–G | 365 | 508 | 0.10 | 0.09 | |
| DMAT–C | 365 | 498 | 0.10 | 0.12 | |
| X15 | DMAT–A | 355 | 486 | 0.15 | 0.11 |
| DMAT–T | 365 | 500 | 0.12 | 0.08 | |
| DMAT–G | 370 | 505 | 0.12 | 0.06 | |
| DMAT–C | 370 | 502 | 0.17 | 0.05 | |
Metal-mediated base pairing interactions serve as important recognition motifs in biological and material sciences.10 For example, HgII ions specifically bind to opposing thymine residues to form T–Hg–T base pairs19 that can cause miscoding of DNA synthesis in vitro and possibly in vivo.20 To evaluate the ability of DMAT–T to mimic T–T in duplex DNA, Tm values were measured in the presence or absence of 1.0 equiv. of HgII (Table 5 and Fig. S6–S8, ESI†). Only small increases in thermal stabilities (ΔTm = +0.8 to +1.5 °C) were observed when HgII was added to duplexes containing a DMAT–T or T–T at position X2, whereas much larger increases were observed at positions X13–X15 (ΔTm = +2.9 to +6.0 °C). At all four positions, the same Tm values were obtained for duplexes containing DMAT–Hg–T as T–Hg–T. In contrast, the addition of 1.0 equiv. of HgII to duplexes containing a C–T mismatch resulted in no increase in thermal stability as compared to the mismatch alone (Table 5). Taken together, these results demonstrate the excellent mimicry of DMAT for thymidine residues in the demanding context of T–HgII–T base pairs.
The fluorescence properties of DMAT can be utilized to monitor site-specific binding of HgII ions to DMAT–T sites. Bi-phasic fluorescence quenching was observed upon addition of HgII to duplex DNA containing a DMAT–T mismatch, giving a 95% decrease in fluorescence intensity upon addition of 3 equiv. of HgII (Fig. 4). Similar results were obtained when DMAT was located at all four positions X2, X13, X14 and X15 (Table S5 and Fig. S9, ESI†). The biphasic quenching (Fig. 4B) mirrors the increases in Tm values obtained when HgII is added to duplexes containing a T–T mismatch (Fig. S6, ESI†). A comparison of these results reveals that the steep slopes observed between 0.0 to 1.0 equiv. of added HgII are a result of T–T-specific binding, and the shallow slopes between 2.0 to 3.0 equiv. are due to non-specific interactions. In contrast to DMAT–T, very little fluorescence quenching (−20%) was observed upon the addition of HgII ions to duplex DNA containing a DMAT–A base pair, or a DMAT–G mismatch (Table S6 and Fig. S10, ESI†). Conversely, the addition of ZnII, CuII, MgII, FeII, CaII, AgI, CdII, PdII and NiII ions to duplexes containing a DMAT–T mismatch resulted in little or no change in DMAT fluorescence (Fig. S11, ESI†). These results are consistent with previous studies demonstrating a high degree of specificity between T–T mismatches and HgII ions using thermal denaturation.19 To the best of our knowledge, our results provide the first example of using a fluorescent nucleobase analog to monitor a specific binding reaction between DNA and HgII ions. DMAT will therefore enable detailed kinetics analyses and large-scale screening efforts that are not feasible using other analytical techniques.10d
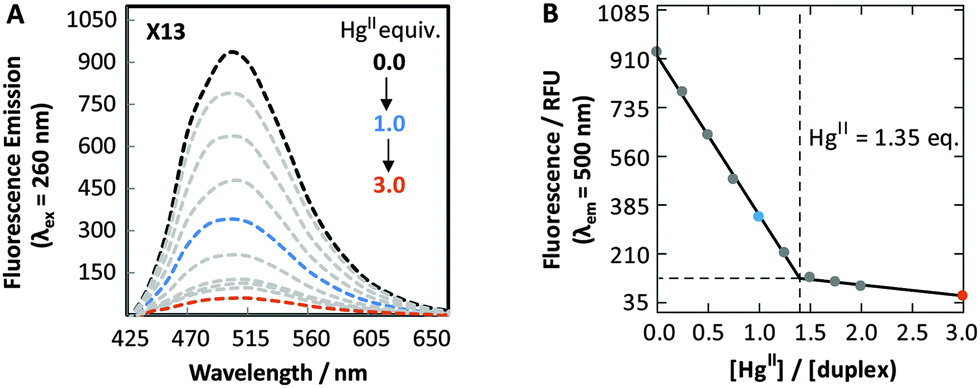 | ||
| Fig. 4 (A) Fluorescence spectra (λex = 260 nm) of duplex X13 in the absence (black) and in the presence of variable Hg(ClO4)2. (B) Plot of fluorescence intensity (λem = 500 nm) versus equiv. of HgII ions. All samples contained 5 μM of DNA in phosphate citric acid buffer (200 mM of Na2HPO4, 100 mM of citric acid and 100 mM NaNO3) at pH = 7.35. See Table S3, ESI† for duplex sequences. | ||
A variety of fluorescent nucleoside analogs are available for solid-phase synthesis of DNA and RNA.1–8 However, the vast majority of these probes are quenched by their incorporation into duplex nucleic acids, where they can disrupt duplex stability by as much as a base pair mismatch. Here we report a new fluorescent thymidine mimic composed of 2′-deoxyuridine fused to dimethylaniline. According to thermal denaturation, fluorescence, and metal binding studies, DMAT exhibits the same base pairing characteristics as native thymidine residues. The quantum yield of DMAT (ϕ = 0.03 in water) increases upon its incorporation into duplex DNA (ϕ = 0.11–0.20), where its fluorescence properties are highly sensitive to nucleobase hydration, ionization, base pairing, and metal binding. Taken together, these results demonstrate that DMAT will enable a wide variety of studies aimed at characterizing biochemical transformations, conformational changes, site-specific metal binding, and base pairing interactions with single-nucleotide resolution.
Notes and references
- (a) M. E. Hawkins, Cell Biochem. Biophys., 2001, 34, 257 CrossRef CAS PubMed; (b) A. T. Krueger, H. Lu, A. H. Lee and E. T. Kool, Acc. Chem. Res., 2007, 40, 141 CrossRef CAS PubMed; (c) R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579 CrossRef CAS PubMed; (d) L. M. Wilhelmsson, Q. Rev. Biophys., 2010, 43, 159 CrossRef CAS PubMed; (e) A. A. Tanpure, M. G. Pawar and S. G. Srivatsan, Isr. J. Chem., 2013, 53, 366 CrossRef CAS; (f) M. Weinberger, F. Berndt, R. Mahrwald, N. P. Ernsting and H. A. Wagenknecht, J. Org. Chem., 2013, 78, 2589 CrossRef CAS PubMed; (g) A. Matarazzo and R. H. E. Hudson, Tetrahedron, 2015, 71, 1627 CrossRef CAS.
- R. W. Sinkeldam, N. J. Greco and Y. Tor, ChemBioChem, 2008, 9, 706 CrossRef CAS PubMed.
- R. W. Sinkeldam, A. J. Wheat, H. Boyaci and Y. Tor, ChemPhysChem, 2011, 12, 567 CrossRef CAS PubMed.
- (a) K. M. Sun, C. K. McLaughlin, D. R. Lantero and R. A. Manderville, J. Am. Chem. Soc., 2007, 129, 1894 CrossRef CAS PubMed; (b) D. Jiang and F. Seela, J. Am. Chem. Soc., 2010, 132, 4016 CrossRef CAS PubMed; (c) J. Riedl, R. Pohl, L. Rulisek and M. Hocek, J. Org. Chem., 2012, 77, 1026 CrossRef CAS PubMed; (d) R. W. Sinkeldam, P. A. Hopkins and Y. Tor, ChemPhysChem, 2012, 13, 3350 CrossRef CAS PubMed.
- (a) R. A. Mizrahi, D. Shin, R. W. Sinkeldam, K. J. Phelp, A. Fin, D. J. Tantillo, Y. Tor and P. A. Beal, Angew. Chem., Int. Ed., 2015, 54, 8713 CrossRef CAS PubMed; (b) A. C. Jones and R. K. Neely, Q. Rev. Biophys., 2015, 48, 244 CrossRef CAS PubMed.
- (a) F. Godde, J. J. Toulmé and S. Moreau, Biochemistry, 1998, 37, 13765 CrossRef CAS PubMed; (b) Y. Hikida, M. Kimoto, S. Yokoyama and I. Hirao, Nat. Protoc., 2010, 5, 1312 CrossRef CAS PubMed; (c) A. Dumas and N. W. Luedtke, Nucleic Acids Res., 2011, 39, 6825 CrossRef CAS PubMed; (d) A. Dumas and N. W. Luedtke, ChemBioChem, 2011, 12, 2044 CrossRef CAS PubMed; (e) M. Sholokh, R. Sharma, D. Shin, R. Das, O. A. Zaporozhets, Y. Tor and Y. Mély, J. Am. Chem. Soc., 2015, 137, 3185 CrossRef CAS PubMed; (f) A. A. Tanpure and S. G. Srivatsan, Nucleic Acids Res., 2015, 43, e149 CrossRef PubMed.
- (a) A. Dumas and N. W. Luedtke, Chem. – Eur. J., 2012, 18, 245 CrossRef CAS PubMed; (b) A. Omumi, C. K. McLaughlin, D. Ben-Israel and R. A. Manderville, J. Phys. Chem. B, 2012, 116, 6158 CrossRef CAS PubMed; (c) S. K. Jana, X. Guo, H. Mei and F. Seela, Chem. Commun., 2015, 51, 17301 RSC.
- (a) J. Michel, G. Gueguen, J. Vercauteren and S. Moreau, Tetrahedron, 1997, 53, 8457 CrossRef CAS; (b) A. Okamoto, K. Tainaka and I. Saito, J. Am. Chem. Soc., 2003, 125, 4972 CrossRef CAS PubMed; (c) H.-A. Wagenknecht, Ann. N. Y. Acad. Sci., 2008, 1130, 122 CrossRef CAS PubMed; (d) Y. Xie, A. V. Dix and Y. Tor, J. Am. Chem. Soc., 2009, 131, 17605 CrossRef CAS PubMed; (e) N. J. Greco, R. W. Sinkeldam and Y. Tor, Org. Lett., 2009, 11, 1115 CrossRef CAS PubMed; (f) X. Ming and F. Seela, Chem. – Eur. J., 2012, 18, 9590 CrossRef CAS PubMed; (g) Y. Saito, A. Suzuki, Y. Okada, Y. Yamasaka, N. Nemoto and I. Saito, Chem. Commun., 2013, 49, 5684 RSC.
- G. Mata and N. W. Luedtke, J. Am. Chem. Soc., 2015, 137, 699 CrossRef CAS PubMed.
- (a) G. H. Clever, C. Kaul and T. Carell, Angew. Chem., Int. Ed., 2007, 46, 6226 CrossRef CAS PubMed; (b) J. Müller, Metallomics, 2010, 2, 318–327 RSC; (c) G. H. Clever and M. Shionoya, Met. Ions Life Sci., 2012, 10, 269 CAS; (d) Y. Tanaka, J. Kondo, V. Sychrovský, J. Šebera, T. Dairaku, H. Saneyoshi, H. Urata, H. Torigoe and A. Ono, Chem. Commun., 2015, 51, 17343 RSC.
- (a) P. L. Nixon and D. P. Giedroc, J. Mol. Biol., 2000, 296, 659 CrossRef CAS PubMed; (b) S. R. Das and J. A. Piccirilli, Nat. Chem. Biol., 2005, 1, 45 CrossRef CAS PubMed.
- R. M. Izatt, J. J. Christensen and H. Rytting, Chem. Rev., 1971, 71, 439 CrossRef CAS PubMed.
- (a) Y. Xie, T. Maxson and Y. Tor, J. Am. Chem. Soc., 2010, 132, 11896 CrossRef CAS PubMed; (b) G. Mata and N. W. Luedtke, Org. Lett., 2013, 15, 2462 CrossRef CAS PubMed; (c) D. Liu, Z. Y. Zhang, H. Z. Zhang and Y. Wang, Chem. Commun., 2013, 49, 10001 RSC; (d) S. Achelle, J. Rodriguez-Lopez and F. Robin-le Guen, J. Org. Chem., 2014, 79, 7564 CrossRef CAS PubMed.
- G. Mata and N. W. Luedtke, J. Org. Chem., 2012, 77, 9006 CrossRef CAS PubMed.
- (a) C. Reichardt, Chem. Rev., 1994, 94, 2319 CrossRef CAS; (b) R. W. Sinkeldam and Y. Tor, Org. Biomol. Chem., 2007, 5, 2523 RSC.
- (a) T. L. Netzel, M. Zhao, K. Nafisi, J. Headrick, M. S. Sigman and B. E. Eaton, J. Am. Chem. Soc., 1995, 117, 9119 CrossRef CAS; (b) R. S. Butler, P. Cohn, P. Tenzel, K. A. Abboud and R. K. Castellano, J. Am. Chem. Soc., 2009, 131, 623 CrossRef CAS PubMed.
- W. H. Melhuish, J. Phys. Chem., 1961, 65, 229 CrossRef CAS.
- (a) D. M. Gray, R. L. Ratliff and M. R. Vaughan, Methods Enzymol., 1992, 211, 389 CAS; (b) J. Kypr, I. Kejnovska, D. Renciuk and M. Vorlickova, Nucleic Acids Res., 2009, 37, 1713 CrossRef CAS PubMed.
- Y. Miyake, H. Togashi, M. Tashiro, H. Yamaguchi, S. Oda, M. Kudo, Y. Tanaka, Y. Kondo, R. Sawa, T. Fujimoto, T. Machinami and A. Ono, J. Am. Chem. Soc., 2006, 128, 2172 CrossRef CAS PubMed.
- H. Urata, E. Yamaguchi, T. Funai, Y. Matsumura and S. Wada, Angew. Chem., Int. Ed., 2010, 49, 6516 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc09552b |
| ‡ DNA sequences: X2: CXC-TAA-CCC-TAA-CCC-TAA-CCC; X13: CCC-TAA-CCC-TAA-XCC-TAA-CCC; X14: CCC-TAA-CCC-TAA-CXC-TAA-CCC; X15: CCC-TAA-CCC-TAA-CCX-TAA-CCC; (X = DMAT, T (wt), C or A). |
| This journal is © The Royal Society of Chemistry 2016 |