
Unambiguous characterization of anisotropic foldamer packing in a foldecture with an elongated hexagonal plate shape†
Eunyoung
Yoon
,
Jintaek
Gong
,
Yoonchul
Jung
,
Wonchul
Lee
,
Russell W.
Driver
and
Hee-Seung
Lee
*
Department of Chemistry, KAIST, Daejeon, 305-701, Korea. E-mail: hee-seung_lee@kaist.ac.kr
First published on 15th March 2016
Abstract
Herein we correlate secondary structure perturbation with changes in the solid-state molecular architectures of an elongated hexagonal plate-shaped foldecture derived from the self-assembly of rigid 12-helical β-peptide foldamers to which a flexible C-terminus α-leucine moiety has been appended. This study provides the first complete characterization of the directional molecular packing patterns of individual foldamer components within a foldecture, from which a 3D molecular-level picture of the entire foldecture was unambiguously constructed.
Peptide self-assembly is an efficient way to make topologically complex molecular systems that mimic protein-like structures and functions.1 Precise control over the shape and size of self-assembled peptide morphologies is crucial because most applications require a uniform sample of nanostructures.2 However, the self-assembly of short peptides routinely gives rise to inhomogeneous and low dimensional shapes with poor size distribution. To tackle this nontrivial problem, it is necessary to design structurally well-defined peptides that interact with adjacent partners in the solid state in predictable ways through non-covalent interactions such as hydrogen bonding and hydrophobic packing.3
Our group has demonstrated that a diverse set of three dimensional (3D) foldamer-derived molecular architectures (i.e. foldectures) can be synthesized by the controlled self-assembly of β-peptide foldamers.4 Foldectures are expected to be useful as anisotropic core materials, drug delivery vehicles and protein mimetics, inter alia. By taking advantage of the predictable solid and solution state conformational preferences of β-peptide foldamers, systematic investigations of the rules governing self-assembly behavior in foldectures are possible. For example, previous studies from our group have found that subtle changes in the length of β-peptides with identical 12-helical secondary structures results in different molecular packing motifs, which in turn control the symmetry of the self-assembled shapes.
In our initial studies of foldectures, we used rigid β-peptide foldamers composed exclusively of a well-studied β-amino acid, trans-ACPC (trans-2-aminocyclopentanecarboxylic acid),5 as a tractable model system. Subsequently, we have begun to investigate another fundamental question – how structural perturbation of the β-peptide foldamer affects the pattern of solid-state molecular associations and thereby the self-assembled shape. Herein we address this question in the context of a 12-helical β-peptide foldamer to which a short but flexible helical α-peptide segment has been appended, a “graft foldamer”, and report the self-assembly of a new foldecture with an elongated hexagonal plate shape. The knowledge gained from this study may provide new routes to foldectures possessing useful functions through the use of graft foldamers to position functional groups onto the surfaces or inside foldectures during self-assembly.
Many different graft foldamers composed of ACPC and α-amino acids were initially investigated (Fig. S3, ESI†); graft foldamers with several α-leucine residues appended to the C-terminus of a solution-stable ACPC hexamer core (harboring four intramolecular hydrogen bonds) were found to possess the best physical and self-assembly properties. In preparation for a comprehensive study correlating graft foldamer flexibility with foldecture morphology, herein we present a rigorous structural analysis of a hexagonal plate-shaped graft foldamer-derived foldecture through powder X-ray diffraction (PXRD), selected area electron diffraction (SAED), high resolution transmission electron microscopy (HRTEM), and grazing-incidence wide angle X-ray scattering (GIWAXS) techniques. This study provides the first complete characterization of the directional molecular packing pattern of individual foldamer components within a foldecture, from which a 3D molecular-level picture of the entire foldecture was unambiguously constructed.
Foldamer 1, an octameric graft foldamer, was synthesized by coupling a β-peptide hexamer composed of (S,S)-ACPC with a α-(L)-leucine dimer (Fig. 1a and ESI† for Experimental details). The circular dichroism (CD) spectrum of 1 (0.1 mM in methanol) displayed a minimum at λ = 204 nm and a maximum at λ = 222 nm, typical of 12-helical β-peptides (Fig. S4, ESI†).6 A single crystal X-ray structure was obtained from methanol and revealed that 1 adopts a 12-helix through the ACPC region, but forms a 11-membered hydrogen bonded ring at the C-terminus (Fig. S5 and Table S2 see ESI†).
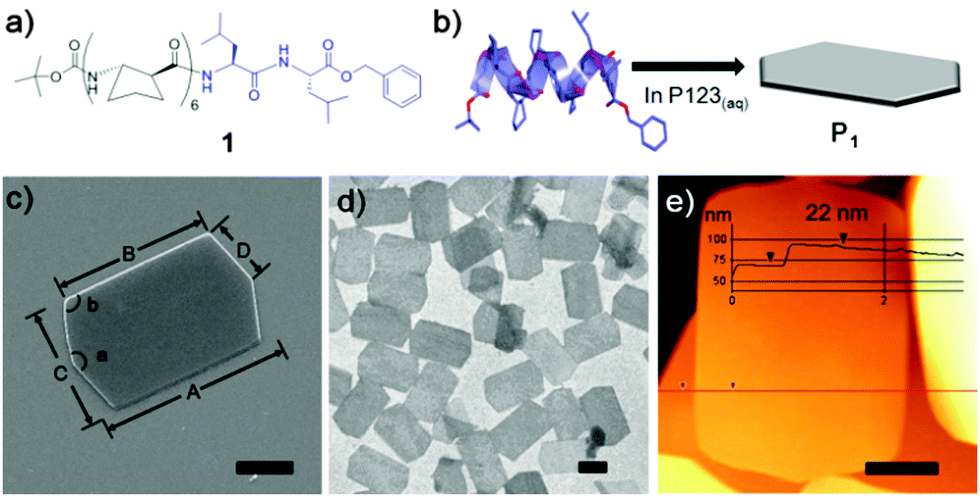 | ||
| Fig. 1 (a) Molecular structure of an octameric graft foldamer, Boc-(S,S)-ACPC6Leu2-OBn (1). Molecular structure showing ACPC (black, β-amino acid) and Leu (blue, α-amino acid) residues, respectively. (b) Schematic representation of the formation of foldecture P1 by the self-assembly of 1 in aqueous P123 solution. (c) SEM image of an elongated hexagonal plate, P1, with distance and angle profiles. A = 8 μm, B = 6 μm, C = 5 μm, D = 2.5 μm, a = 145°, b = 108°. (d) TEM image of P1. (e) AFM image of P1 with thickness information. Scale bars = 2 μm. | ||
The surfactant-assisted self-assembly of 1 was performed by our standard protocol in aqueous solution in the presence of P123 (Pluronic P123, (ethylene glycol)20-(propylene glycol)70-(ethylene glycol)20) surfactant to give a hexagonal plate (P1) with very high homogeneity of size and shape. SEM analysis of the self-assembled structure of 1 at a P123 concentration of 1.0 gL−1 revealed that the elongated hexagonal microplate P1 is symmetrical along the midline (corresponding to measurement ‘A’ in Fig. 1c) and has micro-scale side lengths. Analysis by TEM (Fig. 1d) and atomic force microscopy (AFM) (Fig. 1e) showed that P1 is a flat and uniformly thin plate with a thickness of 22 nm.
To investigate the relationship between the molecular packing motif and morphology of the self-assembled plate, a high-resolution PXRD pattern was collected at the SPring-8 synchrotron (Hyogo, Japan). The foldecture was found to have an orthorhombic unit cell (a = 22.5473(5) Å, b = 30.2016(5) Å and c = 9.6366(1) Å, α = β = γ = 90.0°, V = 6562.21(22) Å3) and the space group was determined to be P21212. The secondary structure of 1 was adopted from a single crystal structure, and bond distances and angles were restrained during the structure calculation process (see ESI† for details); low-cost solutions were used as starting geometries for Rietveld refinement, which was carried out in a way identical to previous studies. A spherical harmonic preferential orientation approximation (ODF)7 was used during refinement.8 The final R-factors (Rwp = 0.0436, Rp = 0.0313) showed a good quality of fitting. The refined structure is shown in Fig. 2a along with the indexed faces of the unit cell (for the detailed experimental parameters, see ESI†). The secondary structure of foldamers in the foldecture and single-crystal are almost identical (Fig. S6, ESI†).
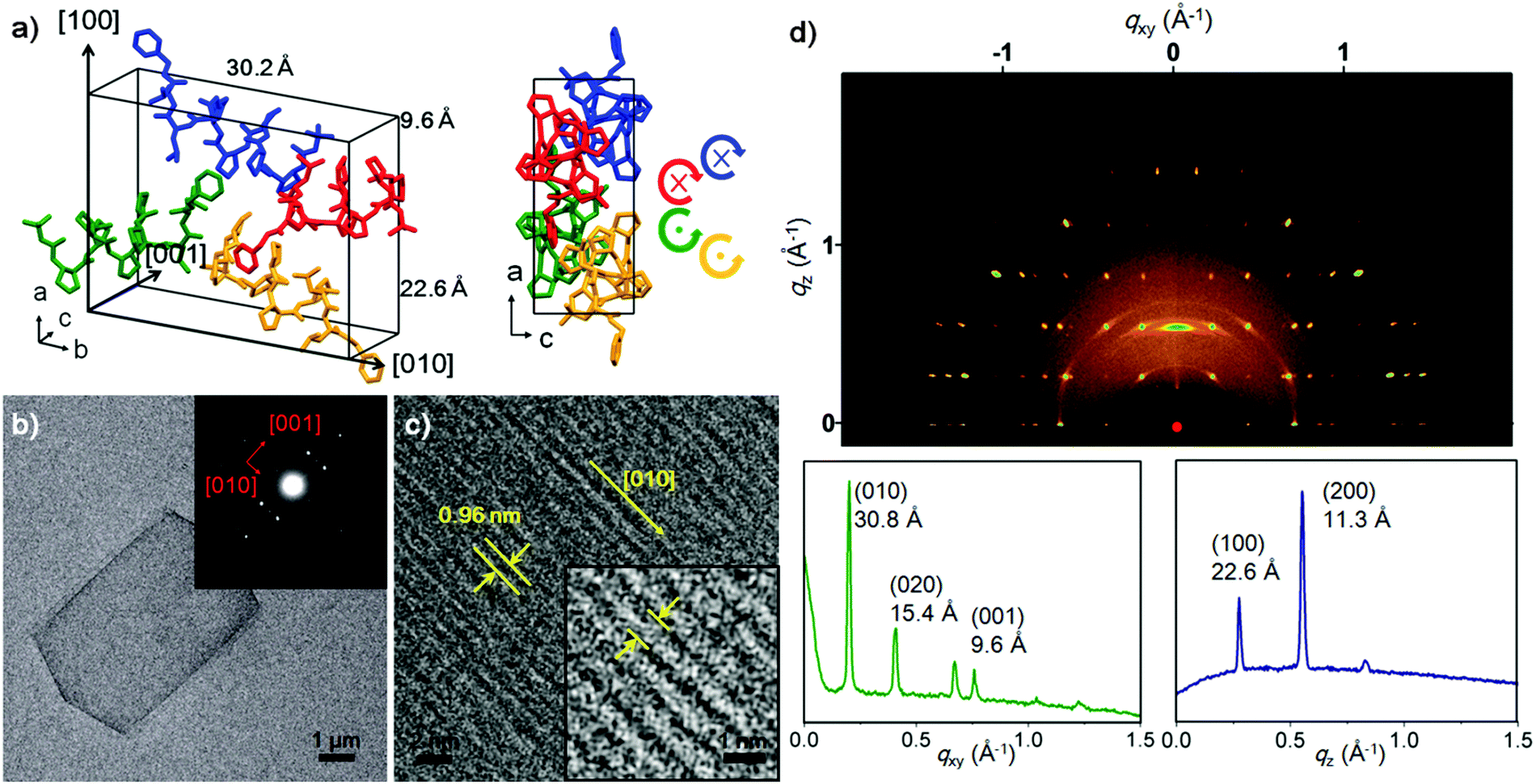 | ||
| Fig. 2 Structural characterization of foldecture P1. (a) Molecular packing structure of P1 in the unit cell. A stick model (left) is shown with unit cell dimensions. Individual foldamers are shown in blue, red, green and yellow. A stick model from a view along the b-axis (right) shows the helical handedness and direction of individual foldamers (N to C). (b) TEM image of P1 with SAED pattern (inset). Red arrows indicate plane direction. (c) HRTEM image of P1 taken at the [001] zone axis and a magnified view (inset) of the plate at an intermediate range. The lattice fringe and direction correspond to 0.96 nm and [010], respectively. (d) GIWAXS plot of P1 (top), and in-plane (bottom left) at qz = 0.27 Å−1 and out-of-plane (bottom right) lineshapes extracted from the GIWAXS patterns. | ||
To identify which unit cell axis corresponds to the out-of-plane direction of the plate shape, we prepared a stacked sample of P1 on glass (Fig. S8b, ESI†) for out-of-plane XRD analysis. Data showed enhancement of a peak at 2θ = 7.8° (i.e. d = 11.3 Å), one half of the unit cell length of a-axis, suggesting that the out-of-plane axis of P1 corresponds to the (100) direction of the unit cell (Fig. S8a, ESI†). Accordingly, this plane was assigned to be the bc plane of the unit cell, which was then confirmed by in-plane XRD analysis. The d-spacing peak values (d = 15.1 Å and 4.9 Å) of the in-plane XRD spectrum correspond to one half of the unit cell lengths along the b- and c-axes, respectively (Fig. S8b, ESI†). From this analysis we can conclude that foldamers are laid parallel with the in-plane axis and the thickness of P1 (22 nm) is comparable to a theoretical stacking of 20 molecules (22.6 nm).
To discover the absolute orientation of the foldamer helical axes, we examined the surface of a single plate by using SAED9 and HRTEM.10Fig. 2b shows the TEM image along with the SAED pattern (Fig. 2b inset). The bright SAED spot pattern indicated that P1 was highly crystalline. Analysis of the diffraction pattern revealed two major reflections orthogonal to (001) and (010), which is supported by the in-plane XRD analysis. The HRTEM image of a P1 shows a highly ordered array of molecules on the surface, which provided the direction of the helical axes of foldamers through a measurement of the lattice fringe (0.96 nm), which is identical to the c-axis length and corresponds to the (001) plane (Fig. 2c). Most peptide materials are unstable under the electron beam irradiation applied during SAED and HRTEM, and rapidly decompose. However, the excellent stability of P1 allowed visualization the lattice fringes.
The scanning TEM analysis of three different points of a P1 sample provided an identical SAED pattern, suggesting a single molecular packing mode in P1 (Fig. S9, ESI†). To examine the entire area of the sample, we further carried out the surface characterization of P1 by using 2D GIWAXS technique. GIWAXS provides clear evidence of molecular packing and orientation for the x-, y- and z-directions at a glance.11 As shown in Fig. 2d, we obtained a bright and complex pattern of discreet reflections, indicating that the sample is flat and periodic in all three dimensions.11b The GIWAXS pattern exhibits four intense peaks along the qxy axis at q = 0.201 Å−1, 0.407 Å−1, 0.654 Å−1 and 0.780 Å−1, at qz = 0.27 Å−1. The calculated lattice spacing was 30.8 Å, 15.4 Å, 9.6 Å and 8.0 Å, matching the (010), (020), (001) and (022) planes, respectively. The qz region, with sharp peaks at q = 0.275 Å−1 and q = 0.551 Å−1, correlated with d = 22.6 Å, (100) and d = 11.3 Å (200).
The solution-state secondary structure of 1 was subsequently examined through variable temperature, hydrogen/deuterium isotope exchange and 2D 1H ROESY experiments (pyridine-d5, 298 K, 10 mM) on a Bruker 900 MHz NMR spectrometer at Korea Basic Sciences Institute. We calculated the solution-state conformational ensemble with Xplor-NIH12 from distance restraints derived from interatomic ROE cross-peak intensities, dihedral angle restraints derived from 3JNH–H coupling constants, and hydrogen bonding restraints (see ESI† for details). Following initial ROE analysis and preliminary calculations without hydrogen bonding restraints, ACPC core residues were assigned an (i, i + 3) hydrogen bonding motif, but to avoid prejudice, leucine amide hydrogen bonding partners were not assigned. Simulated annealing refinement gave a convergent family of structures (no dihedral angle violations >4.0° nor NOE violations >0.3 Å; backbone RMSD 0.472 Å over the 30/44 lowest-energy structures) featuring two distinct conformations through the leucine residues. The major conformer (22/30 structures) is positioned to form consecutive 12- and 11-membered hydrogen bonds, and is similar to the single crystal X-ray structure of 1 (Fig. 3a, representative av. pairwise RMSD 1.163 Å over 30 backbone atoms). The minor conformer (5/30 structures), which is not observed in P1, is positioned to form a bifurcated 12- and 15-membered hydrogen bond (Fig. 3b and Fig. S10, ESI†). NMR analysis suggested that despite the potential for a minor conformational isomer in solution, structural perturbation at the C-terminus of 12-helical β-peptide foldamer by appending a flexible α-leucine dimer is an effective design strategy for new foldectures.
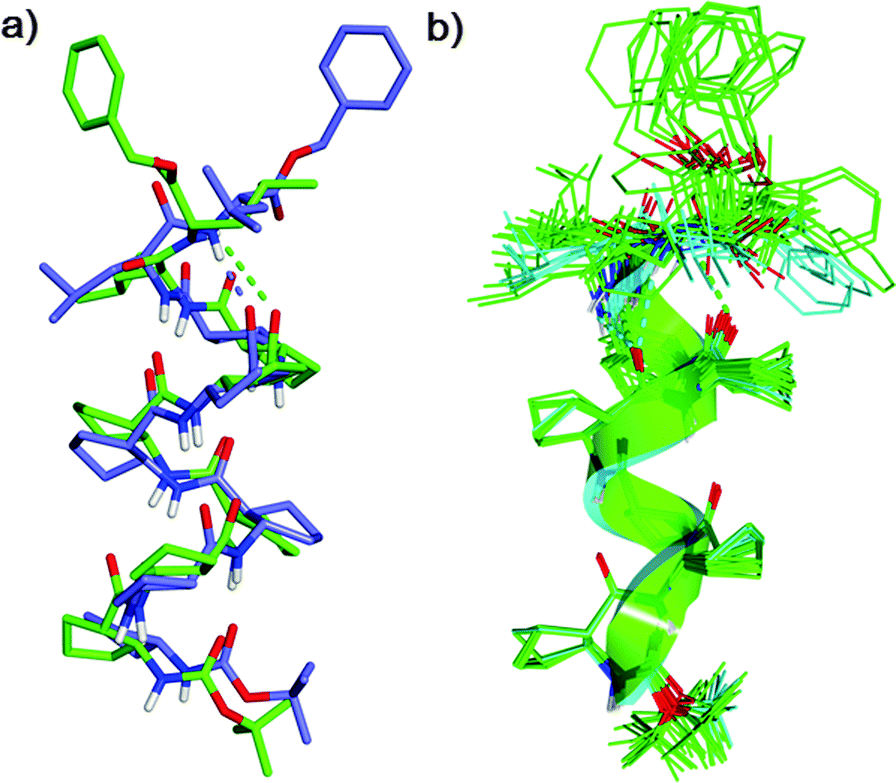 | ||
| Fig. 3 (a) Overlay of the singe crystal X-ray (purple) and NMR major conformer (green) of foldamer 1. (b) Overlay of the solution state ensemble showing both major (green) and minor (blue) conformers. | ||
Through comprehensive structural characterization we were able to elucidate the molecular architecture of elongated hexagonal plate shaped foldecture P1 (Fig. 4). The different molecular association patterns along each direction, which are responsible for the anisotropic shape of P1, can be clearly observed. Both intermolecular hydrogen bonding and hydrophobic interactions exert important influences on plate morphology. Analysis of the molecular network shows that foldamers are associated by intermolecular hydrogen bonding along the b-axis, and well-packed through hydrophobic interactions along the a- and c-axis in an antiparallel and a parallel arrangement, respectively. This is the first complete characterization of the directional molecular packing patterns of individual foldamer components in a foldecture.
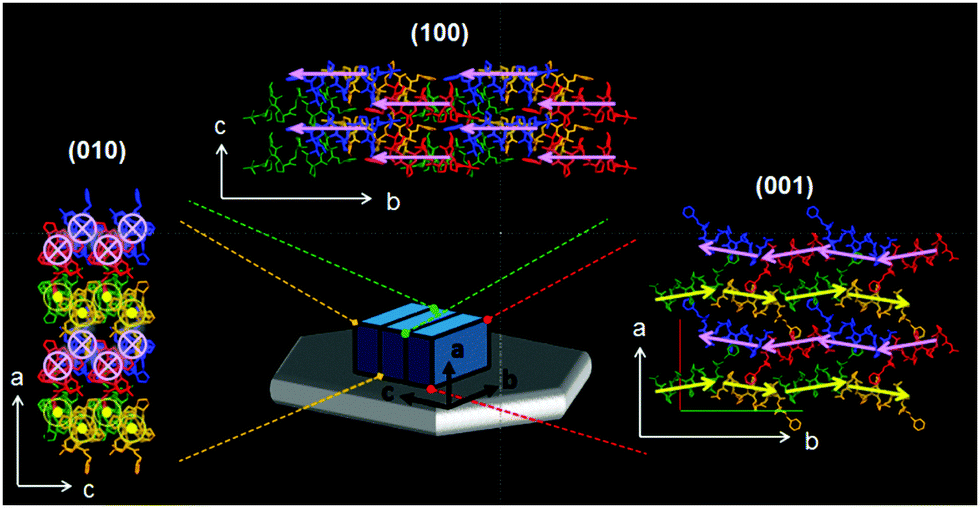 | ||
Fig. 4 The complete molecular arrangement of foldecture P1. A blue box representing a unit cell is laid on the hexagonal plate (gray) alongside the absolute axes. Extended molecular networks (2 × 2 × 2) as viewed along the a- (top), b- (left), and c-axes (right) show the molecular-level packing. The pink and yellow arrows and symbols ( and and  ) superimposed on each foldamer indicate the helical direction (N to C). ) superimposed on each foldamer indicate the helical direction (N to C). | ||
This unambiguous structural information helped us to understand further the self-assembly behavior of 1. SEM analysis of the self-assembled structures of 1 at different P123 concentrations revealed that the hexagonal plate evolved from a thin rod with identical PXRD and SAED patterns (implying structural homology) (Fig. S12 and S13, ESI†). As the concentration of P123 was increased, the molecular packing along the c-axis was strongly retarded, while the relative assembly rates along a-axis and b-axis were constant, resulting in the formation of a thicker hexagonal plate (Fig. S14, ESI†). This uneven directional effect by P123 surfactant is attributed to the anisotropic intermolecular packing mode.
In summary, a new foldecture with an elongated hexagonal plate shape was synthesized from the self-assembly of graft foldamer (Boc-(S,S)-ACPC6Leu2-OBn, 1), which adopts a 12-helical secondary structure with 11-membered hydrogen bonding at the C-terminus in both the solid and solution states. Micro-sized plates P1 are not like a mono/bilayer thin film, but instead have high crystallinity, and thus it was possible to employ multiple analytical methods to obtain comprehensive structural characterization. From these results, the orientation and anisotropic packing mode of foldamers in P1 was unambiguously revealed. This study will pave the way for the evaluation of new functional foldectures4d,13 through the design of graft foldamer with many other combinations of α-amino acid residues.
This research was supported by the National Research Foundation (NRF) of Korea grant funded by Ministry of Science, ICT, and Future Planning (2013R1A2A1A01008358, 2009-0083525). We would like to thank Dr H. Baik in Korea Basic Science Institute (KBSI) for help with TEM experiments.
Notes and references
- (a) E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC; (b) M. Zelzer and R. V. Ulijn, Chem. Soc. Rev., 2010, 39, 3351–3357 RSC; (c) Z. N. Mahmoud, S. B. Gunnoo, A. R. Thomson, J. M. Fletcher and D. N. Woolfson, Biomaterials, 2011, 32, 3712–3720 CrossRef CAS PubMed.
- (a) H. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS PubMed; (b) A. Makino, Polym. J., 2014, 46, 783–791 CrossRef CAS; (c) T. P. J. Knowles, T. W. Oppenheim, A. K. Buell, D. Y. Chirgadze and M. E. Welland, Nat. Nanotechnol., 2010, 5, 204–207 CrossRef CAS PubMed; (d) M. P. Del Borgo, A. I. Mechler, D. Traore, C. Forsyth, J. A. Wilce, M. C. J. Wilce, M. I. Aguilar and P. Perlmutter, Angew. Chem., Int. Ed., 2013, 52, 8266–8270 CrossRef CAS PubMed.
- (a) J. M. Lehn, Angew. Chem., Int. Ed., 1988, 27, 89–112 CrossRef; (b) J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed; (c) S. E. Paramonov, H. W. Jun and J. D. Hartgerink, J. Am. Chem. Soc., 2006, 128, 7291–7298 CrossRef CAS PubMed; (d) J. Fremaux, L. Mauran, K. P. Ziach, B. Kauffmann, B. Odaert and G. Guichard, Angew. Chem., Int. Ed., 2015, 54, 9816–9820 CrossRef CAS PubMed.
- (a) S. Kwon, A. Jeon, S. H. Yoo, I. S. Chung and H.-S. Lee, Angew. Chem., Int. Ed., 2010, 49, 8232–8236 CrossRef CAS PubMed; (b) S. Kwon, H. S. Shin, J. Gong, J. H. Eom, A. Jeon, S. H. Yoo, I. S. Chung, S. J. Cho and H.-S. Lee, J. Am. Chem. Soc., 2011, 133, 17618–17621 CrossRef CAS PubMed; (c) J. Kim, S. Kwon, S. H. Kim, C.-K. Lee, J.-H. Lee, S. J. Cho, H.-S. Lee and H. Ihee, J. Am. Chem. Soc., 2012, 134, 20573–20576 CrossRef CAS PubMed; (d) S. H. Yoo, T. Eom, S. Kwon, J. Gong, J. Kim, S. J. Cho, R. W. Driver, Y. Lee, H. Kim and H.-S. Lee, J. Am. Chem. Soc., 2015, 137, 2159–2162 CrossRef CAS PubMed.
- (a) H.-S. Lee, P. R. LePlae, E. A. Porter and S. H. Gellman, J. Org. Chem., 2001, 66, 3597–3599 CrossRef CAS PubMed; (b) D. Appella, L. A. Christianson, I. L. Karle, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1999, 121, 6206–6212 CrossRef CAS.
- (a) D. Appella, L. A. Christianson, I. L. Karle, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1999, 121, 6206–6212 CrossRef CAS; (b) J. J. Barch, X. Husang, D. H. Appella, L. A. Chistianson, S. R. Durell and S. H. Gellman, J. Am. Chem. Soc., 2000, 122, 2711–2718 CrossRef.
- (a) R. B. Von Dreele, J. Appl. Crystallogr., 1997, 30, 517–525 CrossRef CAS; (b) B. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- (a) M. D. Capracotta, R. M. Sullivan and J. D. Martin, J. Am. Chem. Soc., 2006, 128, 13463–13473 CrossRef CAS PubMed; (b) A. D. Fortes, E. Suard, M.-H. Lenée-Cailleau, C. J. Pickard and R. J. Neods, J. Am. Chem. Soc., 2009, 131, 13508–13515 CrossRef CAS PubMed.
- (a) P. Sikorski, E. D. T. Atkins and L. C. Serpell, Structure, 2003, 11, 915–926 CrossRef CAS PubMed; (b) T. Jiang, C. Xu, X. Zuo and V. P. Conticello, Angew. Chem., Int. Ed., 2014, 53, 8367–8371 CrossRef CAS PubMed.
- K. T. Nam, S. A. Shelby, P. H. Choi, A. B. Marciel, R. Chen, L. Tan, T. K. Chu, R. A. Mesch, B. C. Lee, M. D. Connolly, C. Kisielowski and R. N. Zuckermann, Nat. Mater., 2010, 9, 454–460 CrossRef CAS PubMed.
- (a) S. S. Magidovich, M. Lee, V. Vaiser, B. Struth, S. H. Gellman and H. Rapaport, Chem. – Eur. J., 2011, 17, 14857–14866 CrossRef PubMed; (b) M. Ree, Macromol. Rapid Commun., 2014, 35, 930–959 CrossRef CAS PubMed.
- C. D. Schwieters, J. J. Kuszewski and G. M. Clore, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 48, 47–62 CrossRef CAS.
- S. Kwon, B. J. Kim, H.-K. Lim, K. Kang, S. H. Yoo, J. Gong, E. Yoon, J. Lee, I. S. Choi, H. Kim and H.-S. Lee, Nat. Commun., 2015, 6, 8747 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, experimental procedures and characterization results. CCDC 1446030. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc00502k |
| This journal is © The Royal Society of Chemistry 2016 |