
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic oxidation of iron(II) complexes by dioxygen using 9-mesityl-10-methylacridinium ions†
Takeshi
Tsudaka
a,
Kei
Ohkubo
*ab and
Shunichi
Fukuzumi
*abc
aDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, ALCA and SENTAN, Japan Science and Technology Agency (JST), Suita, Osaka 565-0871, Japan. E-mail: ookubo@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7370
bDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul 120-750, Korea. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp
cFaculty of Science and Technology, Meijo University, ALCA and SENTAN, Japan Science and Technology Agency (JST), Nagoya, Aichi 468-8502, Japan
First published on 30th March 2016
Abstract
Photocatalytic oxidation of iron(II) complexes by dioxygen occurred using the organic photocatalysts, 9-mesityl-10-methylacridinium ions (Acr+-Mes) and 2-phenyl-4-(1-naphthyl) quinolinium ions (QuPh+-NA), in the presence of triflic acid in acetonitrile under visible light irradiation. The electron-transfer state of Acr+-Mes produced upon photoexcitation oxidized the iron(II) complexes, whereas it reduced dioxygen with protons to produce iron(III) complexes and H2O2.
Metal complexes are usually oxidized by inorganic oxidants such as cerium ammonium nitrate and lead dioxide.1–5 In such cases, stoichiometric amounts of inorganic oxidants are required to obtain oxidized metal complexes, producing inorganic wastes which cause environmental problems. The ideal oxidant, which is environmentally benign, is dioxygen (O2), producing only hydrogen peroxide or water as the reduced product. However, the oxidation of metal complexes by O2 is often endergonic even in the presence of an acid. Thus, an appropriate photocatalyst is required for the oxidation of metal complexes by O2 in the presence of an acid. Ruthenium(II) complexes, such as [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) are known to be oxidized by O2 in the presence of an acid to yield the corresponding Ru(III) complexes.6,7 On the other hand, organic photocatalysts have merited increasing attention for a variety of oxidation reactions.8–14 However, there has been no report on photocatalytic oxidation of metal complexes by O2 using organic photocatalysts.
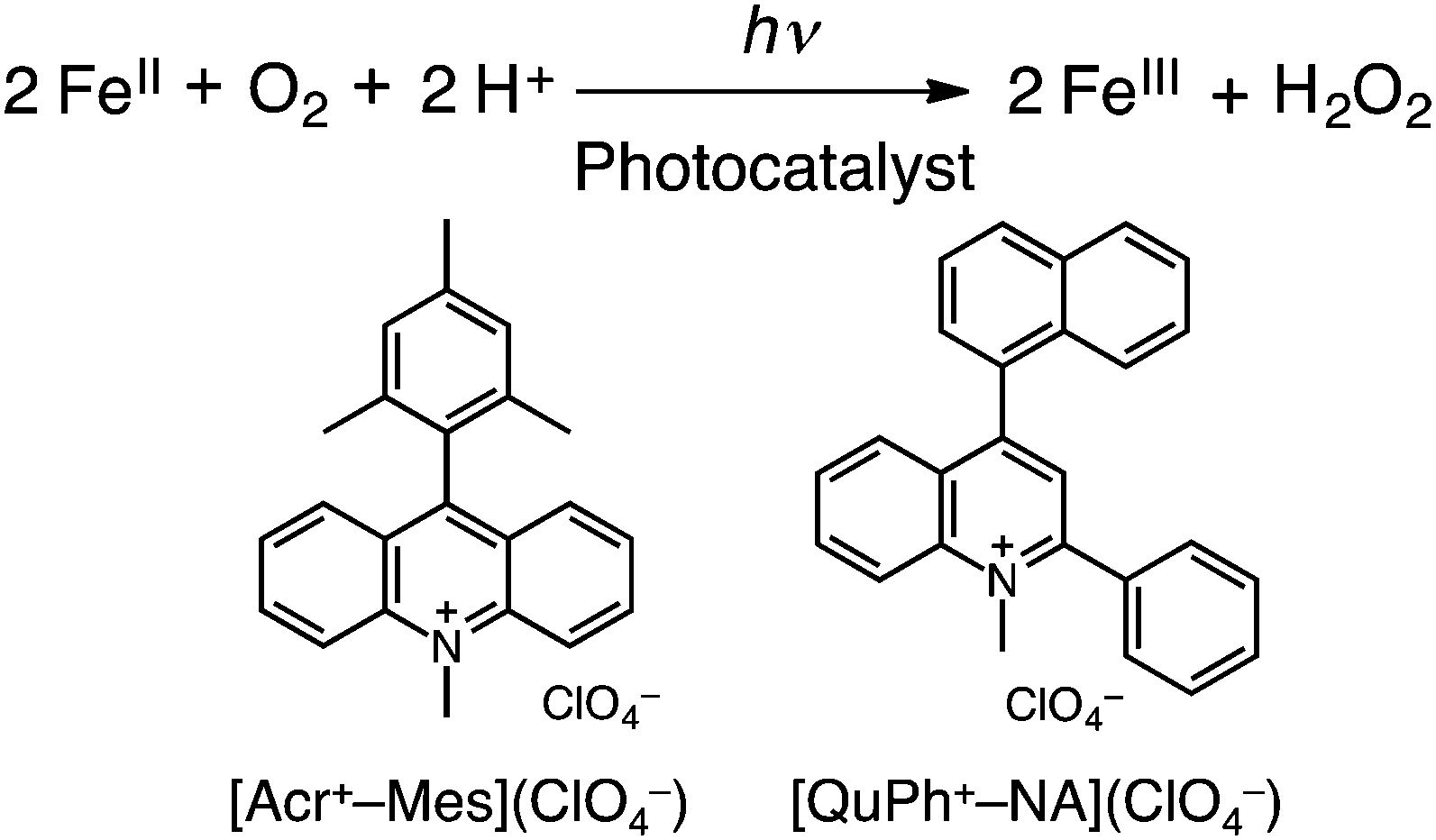
We report herein the photocatalytic oxidation of iron(II) complexes by O2 using 9-mesityl-10-methylacridinium ions (Acr+-Mes)15 and 2-phenyl-4-(1-naphthyl)quinolinium ions (QuPh+-NA)16 as organic photocatalysts in the presence of triflic acid (HOTf) in acetonitrile (MeCN) under visible light irradiation [eqn (1)]. Visible light irradiation of O2-saturated acetonitrile is shown in Fig. 1, where the absorption band at 520 nm due to [FeII(bpy)3]2+ decreased, accompanied by the increase in absorption at 650 nm due to [FeIII(bpy)3]3+. [FeII(bpy)3]2+ was not oxidized without Acr+-Mes under irradiation (Fig. S1 and S2 in the ESI†). The reduced product of O2 was H2O2, which was detected by spectral titration with the use of the oxo[5,10,15,20-tetra(4-pyridyl)porphyrinato]titanium(IV) complex (see the Experimental section in the ESI†).17
 | (1) |
 | ||
| Fig. 1 Visible absorption change in photocatalytic oxidation of [FeII(bpy)3]2+(PF6−)2 (2.0 mM) by O2 in O2-saturated MeCN in the presence of [Acr+-Mes](ClO4−) (0.20 mM) and HOTf (0.10 M) at 298 K under visible light irradiation using a xenon lamp with a cut filter (λ < 390 nm). | ||
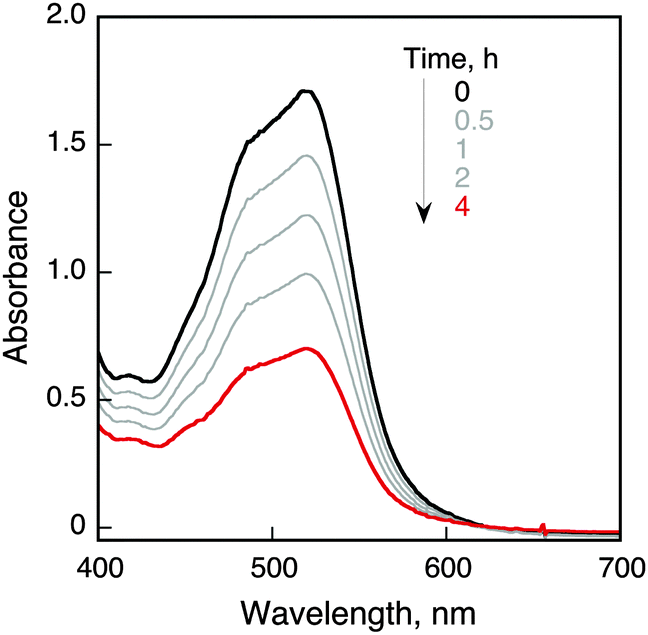
The photocatalytic oxidation of other iron(II) complexes by O2 was examined in the presence of HOTf in O2-saturated MeCN (Fig. S3–S6 in the ESI†). The quantum yields of formation of iron(III) complexes were determined using a ferrioxalate actinometer (see Fig. S7–S9 and the Experimental section in the ESI†).18 The quantum yields (Φ) of photocatalytic oxidation of iron(II) complexes by O2 in the presence of HOTf in MeCN are listed in Table 1 together with the concentrations of iron(II) complexes, HOTf and O2, the one-electron oxidation potentials of iron(II) complexes (Eox)19 and the free energy change of the oxidation (ΔGox). The ΔGox values were evaluated from the Eox values and the Ered value of O2 in the presence of an acid in MeCN (0.75 V vs. SCE) [eqn (2)].20
| ΔGox = −2e(Ered − Eox) | (2) |
| Entry | FeII complex | E ox vs. SCEa, V | ΔGox, eV | Acr+-Mes yieldd, % | TON | Φ , % | QuPh+-NA yielde, % | TON | Φ , % |
|---|---|---|---|---|---|---|---|---|---|
| Clphen = 5-chloro-1,10-phenanthroline, bpy = 2,2′-bipyridine, Me2bpy = 4,4′-dimethyl-2,2′-bipyridine, BrC5H4 = bromocyclopentadienyl, C5H5 = cyclopentadienyl, reaction conditions: [photocatalyst] = 0.20 mM; [HOTf] = 0.10 M; [[FeII(Clphen)3]2+(PF6−)2] = 1.0 mM, [[FeII(bpy)3]2+(PF6−)2] = 2.0 mM, [[FeII(Me2bpy)3]2+(PF6−)2] = 2.0 mM, [FeII(BrC5H4)2] = 1.0 mM, [FeII(BrC5H4)(C5H5)] = 2.0 mM.a Taken from ref. 19.b MeCN (0.40 mL), cell path length 0.1 cm.c MeCN (3.0 mL), cell path length 1 cm.d Photoirradiation (λ > 390 nm).e (λ > 300 nm).f See the Experimental section in the ESI. | |||||||||
| 1b | [FeII(Clphen)3]2+ | 1.20 | 0.90 | 5 | 0.25 | 0.11 | 30 | 1.5 | 0.34 |
| 2b | [FeII(bpy)3]2+ | 1.06 | 0.62 | 42 | 4.2 | 0.32 | 32 | 3.5 | 0.73 |
| 3b | [FeII(Me2bpy)3]2+ | 0.88 | 0.26 | 22 | 2.1 | 1.6 | 30 | 3.0 | 5.2 |
| 4c | FeII(BrC5H4)2 | 0.72 | −0.06 | 81 | 4.1 | 7.2 | 87 | 4.2 | 13 |
| 5c | FeII(BrC5H4)(C5H5) | 0.53 | −0.44 | 81 | 4.1 | 19 | 60 | 3.0 | 26 |
Judging from the ΔGox values, the photocatalytic oxidation of [FeII(Clphen)3]2+, [FeII(bpy)3]2+ and [FeII(Me2bpy)3]2+ is endergonic, whereas that of FeII(BrC5H4)2 and FeII(BrC5H4)(C5H5) is exergonic. The Φ values increased with the decreasing the Eox values of iron(II) complexes as the free energy change of the reaction in eqn (1) decreased to be thermodynamically more favourable. Dependence of Φ on concentrations of [FeII(bpy)3]2+(PF6−)2, HOTf and O2 is shown in Fig. 2 (parts a, b and c, respectively). The Φ value became constant with the increasing concentrations of [FeII(bpy)3]2+(PF6−)2, HOTf and O2, respectively. The photocatalytic oxidation of iron(II) complexes is enhanced by using QuPh+-NA instead of Acr+-Mes as shown in Table 1. (Fig. S10–S14 in the ESI†).
 | ||
| Fig. 2 Dependence of Φ on concentrations of (a) [FeII(bpy)3]2+, (b) HOTf and (c) O2. Standard conditions: [[Acr+-Mes](ClO4−)] = 1.0 mM; [[FeII(bpy)3]2+(PF6−)2] = 2.0 mM; [HOTf] = 0.15 M; [O2] = 2.6 mM; dehydrated MeCN (0.40 mL); cell path length 0.1 cm; excitation wavelength: 420 nm. | ||
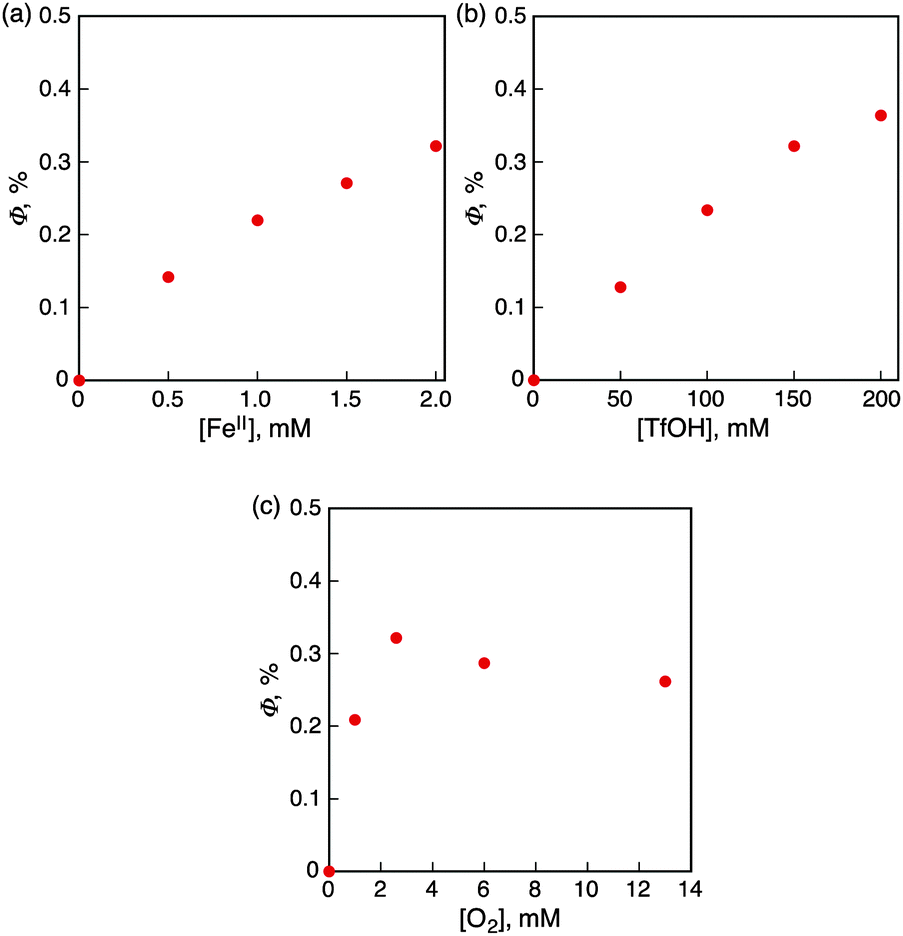
Nanosecond laser flash photolysis measurements were performed in order to clarify the catalytic mechanism for photocatalytic oxidation of iron(II) complexes by O2 using Acr+-Mes in the presence of HOTf in MeCN. Transient absorption spectra were taken after the nanosecond laser excitation at 355 nm of a deaerated MeCN solution of Acr+-Mes in the absence and presence of [FeII(bpy)3]2+(PF6−)2 as shown in Fig. 3. The transient absorption band at 490 nm is due to the electron-transfer state of Acr+-Mes. In the presence of [FeII(bpy)3]2+ the absorption at 490 nm decayed more rapidly and the decay rate increased with the increasing concentration of [FeII(bpy)3]2+. The decay rate obeyed pseudo-first-order kinetics and the pseudo-first order rate constant increased linearly with the increasing concentration of [FeII(bpy)3]2+(PF6−)2. From the slope the rate constant (kox) of electron transfer from [FeII(bpy)3]2+ to the electron-transfer state of Acr+-Mes was determined to be 3.7 × 108 M−1 s−1 as shown in the inset of Fig. 3. Similarly the kox values of other iron(II) complexes were determined (see Fig. S15–S23 in the ESI†) as listed in Table 2 together with the Eox values. The kox value increases with the decreasing Eox values. In the presence of O2, electron transfer from the electron-transfer state of Acr+-Mes to O2 is known to occur with a rate constant of 6.8 × 108 M−1 s−1.21 The rate constant of electron transfer from the electron-transfer state of QuPh+-NA to O2 was determined to be 6.3 × 108 M−1 s−1 (see Fig. S24 in the ESI†). Thus, the photocatalytic oxidation of iron(II) complexes (FeII) by O2 in the presence of HOTf proceeds as shown in Scheme 1. Photoexcitation of Acr+-Mes results in the formation of the electron-transfer state of Acr+-Mes, which oxidizes iron(II) complexes to iron(III) complexes and reduces O2 with protons to produce HO2˙, which disproportionates to yield H2O2.
 | ||
| Fig. 3 Transient absorption decay at 490 nm due to the electron-transfer state of [Acr+-Mes](ClO4−) with various concentrations of [FeII(bpy)3]2+(PF6−)2. Inset: Decay rate constant versus concentrations of [FeII(bpy)3]2+(PF6−)2. | ||
| Entry | FeII complex | E ox vs. SCE, V | Acr+-Mes ket, M−1 s−1 | QuPh+-NA ket, M−1 s−1 |
|---|---|---|---|---|
| 1 | [FeII(Clphen)3]2+ | 1.20 | 1.4 × 108 | 4.9 × 108 |
| 2 | [FeII(bpy)3]2+ | 1.06 | 3.7 × 108 | 4.5 × 108 |
| 3 | [FeII(Me2bpy)3]2+ | 0.88 | 4.5 × 108 | 5.8 × 108 |
| 4 | FeII(BrC5H4)2 | 0.72 | 7.6 × 109 | 7.2 × 109 |
| 5 | FeII(BrC5H4)(C5H5) | 0.53 | 8.5 × 109 | 7.9 × 109 |
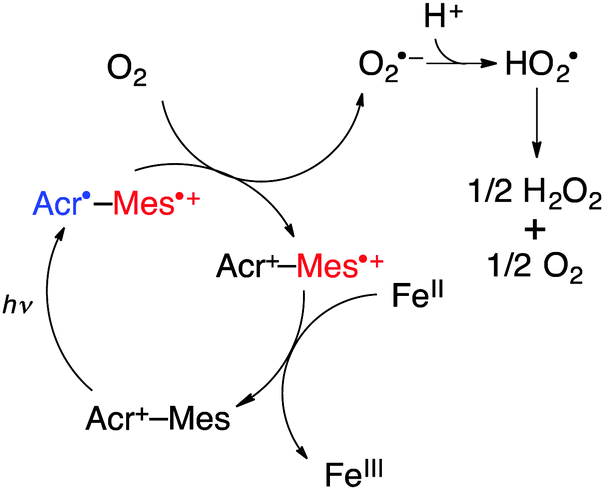 | ||
| Scheme 1 Photocatalytic cycle for oxidation of iron(II) complexes to iron(III) by O2 with Acr+-Mes. | ||
In conclusion, iron(II) complexes are oxidized to iron(III) complexes by O2 using Acr+-Mes as an organic photocatalyst in the presence of HOTf in MeCN under visible light irradiation via electron-transfer oxidation of iron(II) complexes and reduction of O2 by the electron-transfer state of Acr+-Mes produced upon photoexcitation of Acr+-Mes, respectively. The present study provides an environmentally benign approach for oxidation of metal complexes by O2 to obtain the oxidised metal complexes and hydrogen peroxide (H2O2). Because there are many synthetically useful oxidation reactions using H2O2,22 this study has paved a new way for photocatalytic oxidation of substrates by O2 with organic photocatalysts and iron(II) complexes.
This work was supported by Grants-in-Aid (no. 26620154 and 26288037 to K. O.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT); ALCA and SENTAN projects from JST, Japan (to S. F.).
Notes and references
- (a) L. Duan, A. Fischer, Y. Xu and L. Sun, J. Am. Chem. Soc., 2009, 131, 10397 CrossRef CAS PubMed; (b) T. Ishizuka, S. Ohzu and T. Kojima, Synlett, 2014, 1667 Search PubMed.
- (a) M. Murakami, D. Hong, T. Suenobu and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 11605 CrossRef CAS PubMed; (b) T. Kojima, K. Nakayama, K. Ikemura, T. Ogura and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 11692 CrossRef CAS PubMed.
- D. Hong, S. Mandal, Y. Yamada, Y.-M. Lee, W. Nam, A. Llobet and S. Fukuzumi, Inorg. Chem., 2013, 52, 9522–9531 CrossRef CAS PubMed.
- (a) G. P. McDermott, P. Jones, N. W. Barnett, D. N. Donaldson and P. S. Francis, Anal. Chem., 2011, 83, 5453 CrossRef CAS PubMed; (b) M. P. Patel, S. A. Varnum, D. Gandla, M. J. Zdilla and C. J. Martoff, Anal. Methods, 2014, 6, 5818 RSC.
- (a) S. Fukuzumi, C. L. Wong and J. K. Kochi, J. Am. Chem. Soc., 1980, 102, 2928 CrossRef CAS; (b) S. Fukuzumi, K. Miyamoto, T. Suenobu, E. Van Caemelbecke and K. M. Kadish, J. Am. Chem. Soc., 1998, 120, 2880–2889 CrossRef CAS; (c) S. Fukuzumi, H. Miyao and T. Suenobu, J. Phys. Chem. A, 2005, 109, 3285–3294 CrossRef CAS PubMed.
- A. Das, V. Joshi, D. Kotkar, V. S. Pathak, V. Swayambunathan, P. V. Kamat and P. K. Ghosh, J. Phys. Chem. A, 2001, 105, 6945–6954 CrossRef CAS.
- S. Kato, J. Jung, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2013, 6, 3756–3764 CAS.
- (a) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725 CrossRef CAS PubMed; (b) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97 RSC.
- (a) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355–360 CrossRef CAS; (b) D. A. Nicewicz and D. S. Hamilton, Synlett, 2014, 1191–1196 CrossRef.
- D. Ravelli and M. Fagnoni, ChemCatChem, 2012, 4, 169 CrossRef CAS.
- (a) K. Ohkubo and S. Fukuzumi, Bull. Chem. Soc. Jpn., 2009, 82, 303 CrossRef CAS; (b) S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561 RSC.
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999 RSC.
- (a) M. L. Marin, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710 CrossRef CAS PubMed; (b) M. L. Marin, A. Miguel, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Photochem. Photobiol. Sci., 2007, 6, 848 RSC.
- Y. Pan, S. Wang, C. W. Kee, E. Dubuisson, Y. Yang, K. P. Loh and C.-H. Tan, Green Chem., 2011, 13, 3341 RSC.
- (a) S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600 CrossRef CAS PubMed; (b) S. Fukuzumi, H. Kotani and K. Ohkubo, Phys. Chem. Chem. Phys., 2008, 10, 5159 RSC.
- (a) H. Kotani, K. Ohkubo and S. Fukuzumi, Faraday Discuss., 2012, 155, 89 RSC; (b) Y. Yamada, T. Miyahigashi, H. Kotani, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 16136 CrossRef CAS PubMed.
- C. Matsubara, N. Kawamoto and K. Takamura, Analyst, 1992, 117, 1781 RSC.
- C. G. Hatchard and C. A. Parker, Proc. R. Soc. London, Ser. A, 1956, 235, 518–536 CrossRef CAS.
- (a) J. Park, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 5052 CrossRef CAS PubMed; (b) H. Yoon, Y.-M. Lee, X. Wu, K.-B. Cho, R. Sarangi, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 9186 CrossRef CAS PubMed.
- R. Cofé and D. T. Sawyer, Inorg. Chem., 1986, 25, 2089 CrossRef.
- H. Kotani, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2004, 126, 15999 CrossRef CAS PubMed.
- (a) A. Fingerhut, O. V. Serdyuk and S. B. Tsogoeva, Green Chem., 2015, 17, 2042 RSC; (b) Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 114, 8199 CrossRef CAS PubMed; (c) S. Fukuzumi and K. Ohkubo, Asian J. Org. Chem., 2015, 4, 836 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and kinetic details. See DOI: 10.1039/c6cc00359a |
| This journal is © The Royal Society of Chemistry 2016 |