
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterization, and properties of a benzofuran-based cage-shaped borate: photo activation of Lewis acid catalysts†
Akihito
Konishi
ab,
Ryosuke
Yasunaga
a,
Kouji
Chiba
c and
Makoto
Yasuda
*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: yasuda@chem.eng.osaka-u.ac.jp
bCenter for Atomic and Molecular Technologies, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
cScience and Technology System Division, Ryoka Systems Inc., Tokyo Skytree East Tower, 1-1-2 Oshiage, Sumida-ku, Tokyo 131-0045, Japan
First published on 21st January 2016
Abstract
A cage-shaped borate with benzofuran moieties was synthesized. This borate showed a higher degree of catalytic activity for Mukaiyama-aldol type reactions than a simple benzene-based cage-shaped borate induced by self-aggregation. Moreover, the exposure of the complex to black-light irradiation enhanced the catalytic activity.
The precise control of Lewis acidity is very important for organic synthesis because activation of molecules mainly depends on the character of the Lewis acid.1 The Lewis acidity is often controlled by the steric or electronic factors of the species. Recently, we developed cage-shaped borate esters that have a triphenolic ligand with a tripod-chelated structure such as 2, which shows significantly different characteristics than those of the planar structure of a common borate 1.2 As shown in Scheme 1, a cage-shaped borate 3 can be tuned by the introduction of substituents, R, such as halogens3 or aryl groups.4 The structure of the cage shape in 3 allows the precise control of the dihedral angles of C–O–B–O in the cage and LUMO levels by changing a tether atom, X, from carbon to silicon.5 These factors lead to the formation of Lewis acids with the same type of template but a variety of characteristics. A complete replacement of the benzene rings with heteroaromatic rings or with a partial embedding of heteroatoms in the π-conjugated system has been an attractive approach to control either the physical or the chemical properties.6 Thus, the ring-fusion of heteroaromatic moieties to the aromatic portions in the cage-shaped borates would cause electronic perturbations in the Lewis acid center without a steric effect, which would allow the precise control of the Lewis acidity. In the present study, heteroaromatic moieties in the form of benzofuran-frameworks were introduced into the cage structure, which conferred characteristics that differed from those of the previously synthesized borates 2 and 3. A benzofuran moiety has an intrinsic dipole7 due to the ring oxygen with a polarity that affects the Lewis acidity of the complex.
 | ||
| Scheme 1 Chemical structures of planar and cage-shaped triphenolic borates. | ||
We planned to prepare a benzofuran-based triphenolic ligand from a commercially available 5-methoxybenzofuran 5 (Scheme 2). As our previously reported synthetic route for the simple cage-shaped borate 2,2a 5-methoxybenzofuran 5 was deprotonated by nBuLi and treated with ethyl chloroformate, giving an undesired regioisomeric carbinol 6 bound at the 2-position of the methoxybenzofuran. Then, as a regioselective bromination at the ortho-position of phenols has been reported,8 5-hydroxybenzofuran 7 was prepared via the deprotection of a methoxy group of 5 by BBr3. The bromination of 78 by NBS/iPr2NH successfully afforded 4-bromo-5-hydroxybenzofuran 8. After the protection of OH in OMe, the addition of nBuLi for a halogen–lithium exchange followed by treatment with ethyl chloroformate gave the desired triarylmethanol 10 selectively bound at the 4-position of methoxy-4-benzofuran. A reduction of 10 under acidic conditions in THF/MeCN9 and the deprotection of the methoxy groups by BBr3 afforded the tris(5-hydroxy-4-benzofuranyl)methane 12. A mixture of 12 with BH3·THF in THF readily generated the cage-shaped borate 4·THF (Scheme 2).
 | ||
| Scheme 2 Synthesis of benzofuran-based triphenolic ligand 12 and cage-shaped borate 4. | ||
The NMR spectroscopy of 4·THF showed typical signals for a cage-shaped borate; the upfield/downfield shift of the δ(1H)/δ(13C) of C–H at the tether position was compared with those of the ligand 12: (δ(1H) 6.69 → 6.32 ppm, δ(13C) 38.9 → 43.2 ppm).3 The coordinating THF showed broad signals at the lower chemical shifts compared with those of free THF. The recrystallization of 4·THF from hexane/CH2Cl2 gave a suitable crystal for X-ray analysis. The ORTEP drawings are shown in Fig. 1.10 The bond length of B–O(THF) was 1.575(3) Å, which was shorter than that (1.595(8) Å) of the simple cage-shaped borate 2·THF. The sum of the bond angles of O–B–O (O: ligand) was 343.96° for 4·THF, while the sum of the bond angles (344.81°) for 2·THF was slightly larger. These structural features indicated that 4·THF had a slightly higher Lewis acidity than 2·THF.
 | ||
| Fig. 1 ORTEP drawing of 4·THF at the 50% probability level (aryl hydrogens are omitted for clarity) (a) side view and (b) top view (THF is omitted for clarity). Selected bond lengths (Å): B–O(1) 1.441(3), B–O(2) 1.435(3), B–O(3) 1.435(3), B–O(4) 1.575(3). Selected bond angles (deg.): O(1)–B–O(2) 114.3(2), O(2)–B–O(3) 114.5(2), O(3)–B–O(1) 115.2(2), O(1)–B–O(4) 104.2(2), O(2)–B–O(4) 103.6(2), O(3)–B–O(4) 103.1(2). | ||
Lewis acidity was also estimated via the NMR chemical shift δ(13C) and infrared stretching frequency of 2,6-dimethyl-γ-pyrone 13 ligated to borates 4, as shown in Table 1.3 The Δδ(13C) shift of C3 and the stretching frequency of the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups in 13 clearly show the degree of Lewis acidity. The borate 4 showed a large Δδ (13C) and Δν(CO), which indicated a Lewis acidity that was slightly higher than that of 2.
O) groups in 13 clearly show the degree of Lewis acidity. The borate 4 showed a large Δδ (13C) and Δν(CO), which indicated a Lewis acidity that was slightly higher than that of 2.
|
|
|||
|---|---|---|---|
| Entry | B compound | Δδ(13C) of C3/ppm | Δν(CO)/cm−1 |
| 1 | 2·THF | 6.78 | 13.69 |
| 2 | 4·THF | 6.82 | 13.90 |
| 3 | BBr3 | — | 25.07 |
| 4 | BF3·OEt2 | 8.71 | — |
| 5 | B(OPh)31 | 0.77 | 2.89 |

The DFT calculations of 4 at the B3PW91/6-31+G(d,p) level showed it to be a ligand-free, three-coordinated structure, which would contribute to an active Lewis acid species during the reaction course (Fig. 2 and Table S1 in the ESI†). The dihedral angles C–O–B–O, which reflect the shape of the cage, were almost the same as those of the species 2.2a However, an effective conjugation between the B atom and the benzofuran skeleton in 4 was estimated, which would lead to a decrease in the energy level of the unoccupied orbital of a Lewis acid (in this case, the next-LUMO level), while the orbital of 2 is located closer to its B atom. The energy level of the next-LUMO was −21.6 kcal mol−1 (−0.94 eV) for 4, which was lower than those of the benzene-based borates 2 (−18.3 kcal mol−1) and 3c (−16.8 kcal mol−1) (Table S1, ESI†).3 Although a lower next-LUMO level of 4 was expected to contribute to a large reaction rate, the pyridine-complexation energy, ΔE, reflected a catalytic turnover of 4 (−18.8 kcal mol−1) that was similar to that of 2 (−19.2 kcal mol−1). Generally, when a next-LUMO energy level becomes lower, the stabilization energy shows larger (larger negative for ΔE), which means that the Lewis acidity and catalytic turnover have a trade-off relationship. It should be noted that the borate 4 showed an unexpected relationship that resulted in a high affinity for the substrate and a high catalytic turnover. Significant differences between 4 and 2 were found in the magnitude of the dipole moment and in the charge distribution (Fig. S1, ESI†). Both compounds had a dipole moment that was directed from the cage center to the tether position, but the magnitude of the dipole of 4 is 1.5 times larger than that of 2, presumably due to the effective fixation of the dipole vector of each furan ring that resulted from the formation of its rigid cage-structure. The Mulliken charge distribution also reflected the difference in dipole; the Mulliken charge of the B atom for 4 (+1.265 a.u.) was more positive than that of 2 (+1.161 a.u.) (Fig. S2, ESI†).
 | ||
| Fig. 2 Optimum structures and MO diagrams of 4 and 2. | ||
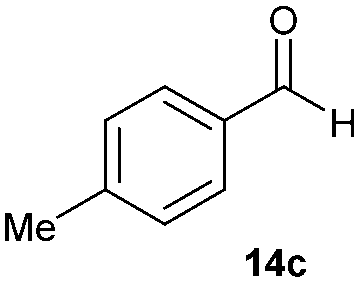
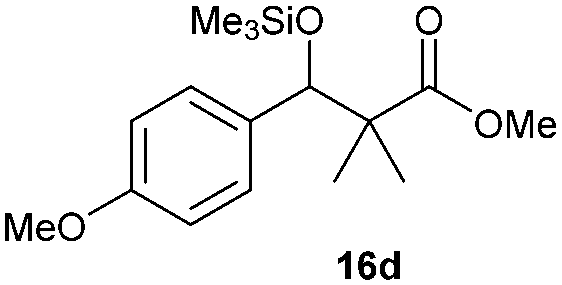
Precise tuning of the electronic features of 4 clearly characterized its catalytic activities. To estimate the Lewis acid catalytic ability, the Mukaiyama-type reaction11 of a variety of aldehydes 14a–e with silylketene acetal 15 was examined in the presence of borate catalysts 4·THF and 2·THF.12 The borate 4·THF gave a much higher yield of the product 16a–d compared with that given by 2·THF for aromatic aldehydes 14a–d in a short time, although prolonging the reaction time led to high yields even in the catalytic reaction with 2·THF (Table 2). Moreover, the borate 4·THF showed high tolerance to a variety of functional groups. While 4 would have limited applicability to aliphatic aldehydes (entry 5), it should be noted that 4 catalyzed a wide range of aromatic aldehydes to give the desired products in moderate yields in a short time. As expected from the theoretical analyses, the borate 4 showed high affinity for the substrate and high catalytic turnover, which was accomplished via an effective π-conjugation between the Lewis acid center and the aromatic core with a large dipole moment in the cage structure.









To further explore the catalytic turnover, the ligand-exchange rate was investigated. The mixture of the complex 4·DMAP in a pyridine-d5 solution was observed via1H-NMR (Table S4, ESI†). The kinetic equation based on the first order of the borate in our previous report3 unreasonably showed a large negative activation entropy, ΔS‡ (−33.4 cal K−1 mol−1),13 and, thus, the first-order hypothesis was found to be incorrect because a step for releasing the DMAP from the ligated complex led to an increase in entropy. Based on these results, ligand dissociation does not proceed via a simple dissociation of the ligand but includes the assistance of another boron complex to release the DMAP. Based on the bimolecular contribution of the borate-to-ligand dissociation, kinetic analysis gave the activation parameters ΔG‡ 25.4 kcal mol−1, ΔH‡ 20.3 kcal mol−1, and ΔS‡ −17.2 cal K−1 mol−1. This unusual mechanism came from the intermolecular interaction of 4 (see also the ESI†).
A bimolecular process requires the presence of an intermolecular interaction in the course of a ligand-exchange pathway. We performed UV measurements of 4·THF under variable concentrations, since the electronic interactions between the chromophores cause spectral changes, which in most cases results in a hypochromic shift.14 The absorption spectra of 4·THF were recorded in CH2Cl2 in a concentration range from ca. 2.0 × 10−5 to 2.0 × 10−4 M. Under these conditions, the longest absorption bands of 4·THF at 330–280 nm, which were mainly assigned to the HOMO → LUMO transition (π → π*) via time-dependent DFT calculation (Fig. S3, ESI†), exhibited remarkable spectral changes that resulted in hypochromic effects with slight red shifts at 330–300 nm and hyperchromic effects with slight blue shifts at 300–280 nm (Fig. S4(A), ESI†). On the other hand, the spectra of 2·THF in CH2Cl2 exhibited no remarkable concentration dependence (Fig. S4(B), ESI†).15 These results suggest that the benzofuran-based 4·THF would benefit from self-intermolecular stabilization, presumably due to its dipole–dipole interaction.16,17
Finally, we describe the effect of photo-irradiation on the control of Lewis acidity. A Mukaiyama-type aldol reaction of 14c with 15 upon exposure to black-light (centred at 365 nm) irradiation at room temperature under the slightly modified conditions was performed. Although no significant change in reactivity was observed in the presence of borate 2·THF, borate 4·THF afforded 16c in a 1.7-fold higher yield under the black-light irradiated conditions (Table 3). While the mechanistic details of the effect of the photo-irradiation have not been clarified, the excitation energy of 4 was lower than that of 2 as a result of the expanded π-conjugation, as well as the molecular geometry changes and charge reorientations in the first excited state, and this may have played a key role in controlling the Lewis acidity.

We synthesized a cage-shaped borate 4 composed of a benzofuran framework, characterized its molecular geometry via X-ray crystallography, and estimated its electronic features via DFT calculations. The synthesized complex exhibited a high Lewis acidity with a high catalytic turnover, which was confirmed via Mukaiyama-aldol type reaction, ligand-exchange rate determination, and the concentration dependence of the electronic absorptions. For the benzofuran-based caged borate 4, an effective lowering of the next-LUMO level and an intermolecular stabilization of the ligand-free complex were achieved via photo-activation, which allowed us to successfully balance two conflicting aims for Lewis acid catalysts: acidity and catalytic turnover. Although there is still room for improvement in the versatilities of the reactions catalyzed by 4, the precise tuning of Lewis acid catalytic activity in association with the introduction of heteroaromatic moieties into the cage-shaped borate core holds the potential to construct photo- or electro-responsive Lewis acid catalysts. Theoretical studies as well as further experimental investigations of the obtained Lewis acid catalyst by our group are ongoing.
This work was supported by the Ministry of Education, Culture, Sports, Science and Technology (Japan). This work was also supported by the Nagase Science and Technology Foundation. A. K. wishes to thank the Research Capability Development Project (Professional development program for assistant professors) of Frontier Research Center of Graduate School of Engineering, Osaka University. We thank Dr Nobuko Kanehisa for the valuable advice regarding X-ray crystallography. Thanks are due to Mr H. Moriguchi, Faculty of Engineering, Osaka University, for assistance in obtaining the MS spectra.
Notes and references
- (a) M. T. Reetz, Acc. Chem. Res., 1993, 26, 462 CrossRef CAS; (b) B. B. Snider, Acc. Chem. Res., 1980, 13, 426 CrossRef CAS; (c) H. Yamamoto, Lewis Acids in Organic Synthesis, Wiley-VCH, 2000 Search PubMed.
- (a) M. Yasuda, S. Yoshioka, S. Yamasaki, T. Somyo, K. Chiba and A. Baba, Org. Lett., 2006, 8, 761 CrossRef CAS PubMed. Triphenolic methane-based metal complexes see also (b) T. Matsuo and H. Kawaguchi, Chem. Lett., 2004, 33, 640 CrossRef CAS; (c) M. B. Dinger and M. J. Scott, Inorg. Chem., 2001, 40, 856 CrossRef CAS; (d) J. Kobayashi, Y. Domoto and T. Kawashima, Chem. Lett., 2010, 39, 134 CrossRef CAS; (e) U. Verkerk, M. Fujita, T. L. Dzwiniel, R. McDonald and J. M. Stryker, J. Am. Chem. Soc., 2002, 124, 9988 CrossRef CAS PubMed; (f) M. Fujita, O. C. Lightbody, M. J. Ferguson, R. McDonald and J. M. Stryker, J. Am. Chem. Soc., 2009, 131, 4568 CrossRef CAS PubMed; (g) S. M. Raders and J. G. Verkade, J. Org. Chem., 2009, 74, 5417 CrossRef CAS PubMed.
- M. Yasuda, H. Nakajima, R. Takeda, S. Yoshioka, S. Yamasaki, K. Chiba and A. Baba, Chem. – Eur. J., 2011, 17, 3856 CrossRef CAS PubMed.
- H. Nakajima, M. Yasuda, R. Takeda and A. Baba, Angew. Chem., Int. Ed., 2012, 51, 3867 CrossRef CAS PubMed.
- M. Yasuda, S. Yoshioka, H. Nakajima, K. Chiba and A. Baba, Org. Lett., 2008, 10, 929 CrossRef CAS PubMed.
- For some recent examples of heteroaromatic conjugated systems, see: (a) K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347 CrossRef CAS PubMed; (b) J.-S. Wu, S.-W. Cheng, Y.-J. Cheng and C.-S. Hsu, Chem. Soc. Rev., 2015, 44, 1113 RSC; (c) A. Fukazawa and S. Yamaguchi, Chem. – Asian J., 2009, 4, 1386 CrossRef CAS PubMed; (d) A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616 RSC; (e) X.-Y. Wang, A. Narita, X. Feng and K. Müllen, J. Am. Chem. Soc., 2015, 137, 7668 CrossRef CAS PubMed; (f) V. M. Hertz, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2015, 54, 8800 CrossRef CAS PubMed; (g) Q. Ye and C. Chi, Chem. Mater., 2014, 26, 4046 CrossRef CAS; (h) U. H. F. Bunz, Acc. Chem. Res., 2015, 48, 1676 CrossRef CAS PubMed; (i) M. Watanabe, W.-T. Su, Y. J. Chang, T.-H. Chao, Y.-S. Wen and T. J. Chow, Chem. – Asian J., 2013, 8, 60 CrossRef CAS PubMed.
- (a) R. D. Brown and B. A. W. Coller, Theor. Chim. Acta, 1967, 7, 259 CrossRef CAS; (b) A. Maris, B. M. Giuliano, S. Melandri, P. Ottaviani, W. Caminati, L. B. Faverob and B. Velinoc, Phys. Chem. Chem. Phys., 2005, 7, 3317 RSC; (c) G. Buemi, F. Zuccarello and G. Romeo, J. Mol. Struct., 1983, 94, 115 CrossRef.
- J. Velder, T. Robert, I. Weidner, J.-M. Neudörfl, J. Lex and H.-G. Schmalz, Adv. Synth. Catal., 2008, 350, 1309 CrossRef CAS.
- M. Wada, K. Kirishima, Y. Oki, M. Miyamoto, M. Asahara and T. Erabi, Bull. Chem. Soc. Jpn., 1999, 72, 779 CrossRef CAS.
- The analytical data of the borate complex 4·THF is included in the ESI.† CCDC 1440145.
- (a) T. Mukaiyama, K. Narasaka and K. Banno, Chem. Lett., 1973, 1011 CrossRef CAS; (b) T. Mukaiyama, Angew. Chem., Int. Ed., 2004, 43, 5590 CrossRef CAS PubMed.
- (a) O. Lefebvre, T. Brigaud and C. Portella, J. Org. Chem., 2001, 66, 1941 CrossRef CAS PubMed; (b) C. Le Roux, H. Gaspard-Iloughmane, J. Dubac, J. Jaud and P. Vignaux, J. Org. Chem., 1993, 58, 1835 CrossRef CAS; (c) N. Takenaka, J. P. Abell and H. Yamamoto, J. Am. Chem. Soc., 2007, 129, 742 CrossRef CAS PubMed.
- ΔG‡ 24.6 kcal mol−1, ΔH‡ 14.9 kcal mol−1, ΔS‡ −33.4 cal K−1 mol−1.
- For example: (a) F. Diederich, in Cyclophanes, ed. J. F. Stoddart, The Royal Society of Chemistry, Cambridge, 1991, pp. 19–21 Search PubMed; (b) Y. Tobe, N. Utsumi, K. Kawabata, A. Nagano, K. Adachi, S. Araki, M. Sonoda, K. Hirose and K. Naemura, J. Am. Chem. Soc., 2002, 124, 5350 CrossRef CAS PubMed; (c) C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Chem. Soc., Perkin Trans. 2, 2001, 651 RSC; (d) C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525 CrossRef CAS.
- 1H NMR measurments with variable concentrations were also performed, and, no remarkable peak shift was observed even at high concentration; see Fig. S5 (ESI†) while the UV spectra showed the shift.
- The basicity of the oxygen atoms in benzofuran, as well as the dipolar interaction between benzofuran cores, could contribute to the intermolecular stabilization. see: (a) J. R. Andreatta, G. B. Cieslinski, M. Batool, X. Z. Sun, M. W. George, E. N. Brothers, D. J. Darensbourg and A. A. Bengali, Inorg. Chem., 2009, 48, 7787 CrossRef CAS PubMed; (b) We also estimated the stabilization energy of the aggregates assisted by the oxygen atoms to be −1.2 kcal mol−1, details are shown in the ESI†.
- Since the dissociation of THF from the boron center should promote an intermolecular interaction, we attempted to observe the existence of the ligand-free complex under suitable conditions; see the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1440145. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc00291a |
| This journal is © The Royal Society of Chemistry 2016 |