
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced product selectivity promoted by remote metal coordination in acceptor-free alcohol dehydrogenation catalysis†
Marta
Valencia
ab,
Helge
Müller-Bunz
b,
Robert A.
Gossage
ac and
Martin
Albrecht
*ab
aDepartement für Chemie und Biochemie, Universität Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: martin.albrecht@dcb.unibe.ch
bSchool of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland
cDepartment of Chemistry & Biology, Ryerson University, 350 Victoria St., Toronto, ON M5B 2K3, Canada
First published on 20th January 2016
Abstract
A bimetallic [Ir3+]2 complex was synthesized based on a bridging 1,2,3-triazole ligand that coordinates to one Cp*Ir unit as N,N-bidentate chelate, and to the other as a C,C-bidentate ligand. When compared to monometallic homologues, the bimetallic complex shows greatly enhanced product selectivity for the acceptorless dehydrogenation of alcohols; spectroscopic and electrochemical analysis suggest significant alteration of the metal properties in the bimetallic system compared to the monometallic species, which offers a rationale for the observed high selectivity.
Nature achieves high selectivity and reactivity in substrate activation by the application of highly complex molecular enzymes. Many of these systems rely on cooperation between reactive metal centres.1 These metal sites are often arranged in close mutual proximity, in particular in cases where redox chemistry is involved. This necessity is governed by the typical requirement for two-electron transformations that involve bio-available metals with one-electron redox processes as the only energetically feasible option (e.g. Cu+/2+, Fe2+/3+).2 In synthetic systems, judicious ligand design allows the two redox active metal centres to be placed in an enforced proximity. In many cases, this leads to metal–metal bond formation during the redox processes. Hence the influence of one or both metals on one another is intimately connected.3 Less well-known are catalytic systems in which redox active metals are held in a conformation that inhibits metal–metal bond formation.4 Elegant work by Peris using 1,2,4-triazolylidene scaffolds revealed a direct positive effect on catalytic performance.5 Here, we present a bimetallic system that induces greatly enhanced product selectivity when compared to the monometallic homologues, in parts through effective kinetic discrimination. Efficient alteration of the metal properties are demonstrated inter alia by a significant lowering of the potential for iridium(III) oxidation.
We previously reported on the excellent catalytic activity of iridium complexes in oxidation reactions, which is imparted by the unique features of mesoionic triazolylidene ligands6 containing pyridyl-derived substituents.7 Inspired by these results, we have now investigated Cp*Ir complexes of related ligands containing secondary bonding capabilities (Cp* = pentamethylcyclopenta-dienyl, C5Me5−). To this end, 1,2,3-triazole containing two pyridyl substituents at N1 and C4 positions was selectively mono-methylated at the C-bound pyridyl site to afford the pyridinium triazole ligand precursor L1 (ESI†). Metalation of this pyridinium salt with [IrCp*Cl2]2 in the presence of NaOAc at room temperature induced Ctrz–H bond activation and subsequent cyclometalation to afford the bimetallic complex 1 (Scheme 1).8 This complex was air-stable and purified by column chromatography, and was isolated as a red solid in 48% yield. Note that complex 1 contains two non-identical iridium centers in formal +3 oxidation state, one κ2-N,N′ bound and the second displaying a κ2-C,C′ bonding motif; one of these bonding pockets is formally anionic.9
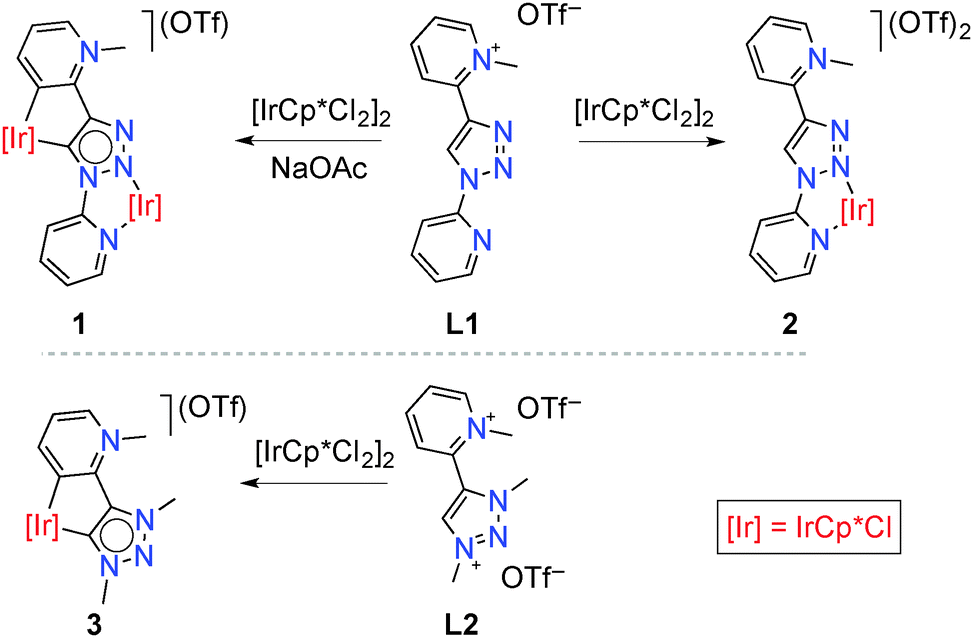 | ||
| Scheme 1 Synthesis of the bis-iridium complex 1, and its monometallic analogues 2 and 3. | ||
Treatment of the pyridinium salt L1 with [IrCp*Cl2]2 under identical conditions but in the absence of NaOAc yielded the monometallic complex 2 (Scheme 1), which was isolated as a light yellow solid (61%). While the N3 position of the triazole heterocycle is generally more basic and hence should coordinate preferably to a Lewis acid,10 chelation via bonding to the pyridyl unit directs the metal to the triazole N2-position. Related metal coordination to N2 in triazolium salts has been established in specific cases, in particular when chelating groups were available as substituents.11 Notably, complex 2 contains an iridium center in essentially the same coordination environment as the N,N-bound iridium center in complex 1. The C,C-bound portion of complex 1 is mimicked by the previously reported7b complex 3 featuring a mesoionic pyridylidene and a mesoionic triazolylidene ligand bonding site (Scheme 1). Even though the ligand in complex 1 is formally an anionic L,X-type system, previous work provides evidence that N-coordination of metal centers to N-heterocycles such as triazoles or imidazoles has very similar effects to N-alkylation.12 In both complexes, the triazole-derived heterocycle is mesoionic in nature, and hence complex 3 is a better mimic of the C,C-chelated iridium center than a rigidly anionic triazolyl ligand site. In support of this notion, the triazol proton resonance shifts by about the same shift difference when the triazole scaffold is alkylated or coordinating to iridium.13
Complexes 1–3 were identified by elemental analysis, 1H and 13C NMR spectroscopy, and single crystal X-ray diffraction (ESI†). The integral ratio of the Cp-bound CH3 groups relative to the singlet of the pyridinium–CH3 resonance gave unambiguous evidence for the presence of two and one [Ir(Cp*)] units per ligand site in complexes 1 and 2, respectively. While pyridylidene and triazolylidene bonding was readily deduced from the multiplicity and chemical shift of the pyridylidene 1H resonances and 13C NMR data (e.g. δC = 160.4 for Ctrz–Ir, δC = 160.7 for Cpy–Ir), C,C-chelation was established unambiguously by X-ray diffraction (Fig. 1). Structural comparison reveals that the Ir–C bonds are consistently shorter than the Ir–N bonds, irrespective of the type of heterocycle (trz or pyr; Table 1). The Ir–N and the Ir–C bond lengths of 1 are essentially identical to the corresponding bond lengths in the monometallic analogues 2 and 3, suggesting that complexes 2 and 3 are appropriate structural mimics for the two different iridium centers in complex 1.
 | ||
| Fig. 1 ORTEP representation of complexes 1–3 (50% probability, hydrogens and non-coordinated OTf− anions omitted for clarity). | ||
| 2 | 1 | 3 | |||
|---|---|---|---|---|---|
| a X = Cl in complex 1, X = NCMe in complex 3. b From ref. 7b. | |||||
| 2.080(5) | Ir–Ntrz | 2.072(2) | 1.993(2) | Ir–Ctrz | 2.017(1) |
| 2.116(6) | Ir–Npy | 2.116(6) | 2.049(2) | Ir–Cpy | 2.049(1) |
| 2.396(2) | Ir–Cl | 2.4039(6) | 2.4088(4) | Ir–Xa | 2.033(2) |
| 75.5(2) | Ntrz–Ir–Npy | 75.53(7) | 77.32(2) | Ctrz–Ir–Cpy | 76.02(2) |
| 84.1(2) | Cl–Ir–Ntrz | 84.69(5) | 91.57(6) | X–Ir–Ctrz | 90.09(6) |
| 87.8(2) | Cl–Ir–Npy | 84.60(5) | 89.39(6) | X–Ir–Cpy | 87.61(6) |
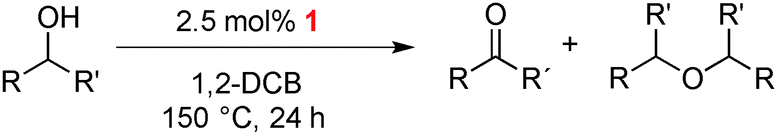
Complexes 1–3 were used as catalyst precursors for the acceptorless oxidation of alcohols14 using benzyl alcohol as a model substrate. Reactions were typically carried out at 150 °C and using 5 mol% [Ir] in 1,2-dichlorobenzene as solvent (Table 2 and Fig. S1, ESI†). Evaluation of these catalytic runs indicates two important and unusual features. Firstly, the initial catalytic activity as well as the alcohol conversion after 24 h are higher for both monometallic complexes 2 and 3 than that of the bimetallic Ir2 complex 1. For example, conversions after 24 h reach 89% and 98% with the N,N-bidentate and C,C-bidentate coordinated iridium centers, respectively, in a monometallic framework, while the bimetallic system only reaches 72% within the same period and at the same concentration of iridium (entries 1–4). Full conversion requires longer reaction times (entry 2). Secondly and more significantly, complete product selectivity towards benzyl aldehyde is obtained using the bimetallic catalyst precursor 1 and conversions and yields are identical even after complete substrate conversion (up to 4 d). In sharp contrast, the monometallic complexes 2 and 3 both afforded a mixture of two products, viz. benzaldehyde from dehydrogenation and, in about equal portions, dibenzyl ether as a result of alcohol dehydration. Similar etherification has been noted with related complexes.15 Of note, runs involving a combination of complex 2 and 3 (2.5 mol% of each complex to obtain the same 5 mol% loading) under the same catalytic conditions yielded essentially identical results as if only one of the mono-Ir complexes is employed, that is, higher activity yet vastly inferior selectivity than 1 (entry 5).16 We thus attribute the remarkably high selectivity of the catalyst derived from complex 1 to the unique electronic configuration imparted by the presence of two metal centers. This bimetallic configuration effectively suppresses dehydration and decelerates also dehydrogenation, thus producing benzaldehyde at lower rate, but with exquisite selectivity.
|
|
||||
|---|---|---|---|---|
| Entry | [Ir] | Time (h) | Conv'nb (%) | BnCHO/Bn2Oc (%) |
| a Conditions: alcohol (0.2 mmol), [Ir] (0.01 mmol, 5 mol% based on Ir), 1,2-dichlorobezene (2 mL), 150 °C. b Determined by 1H NMR spectroscopic analysis in CDCl3 with hexamethylbenzene as internal standard. c Ratio of products given in percent. d 5 μmol of 2 plus 5 μmol of 3. | ||||
| 1 | 1 | 24 | 72 | 100/0 |
| 2 | 1 | 96 | 95 | 100/0 |
| 3 | 2 | 24 | 89 | 42/58 |
| 4 | 3 | 24 | 98 | 57/43 |
| 5 | 2 + 3d | 24 | 92 | 46/54 |

To shed some light on the unique selectivity of the bimetallic complex 1, the spectroscopic and physical properties of 1–3 were examined in more detail. Structural (ground-state) comparison of bimetallic 1 to both monometallic complexes 2 and 3 reveals little significant differences (see X-ray data above). The 1H NMR spectrum (CD2Cl2) of complex 1 shows two singlet resonances for the two magnetically inequivalent Cp* ligands (δH = 1.83 and 1.60). The lower field singlet resonates at a similar frequency to that observed for both complexes 2 and 3 (δH = 1.82 and 1.84, respectively), whereas the higher field singlet indicates a significantly more shielded environment of one IrCp* unit. Nuclear Overhauser experiments unambiguously demonstrated that the shielded Cp* unit belongs to the C,C-bidentate coordinated ligand,17 thus offering a rationale for the altered catalytic activity and selectivity, and suggesting that the triazolylidene-bound iridium is the catalytically active site.
The UV-vis spectra of complexes 1–3 (CH2Cl2: Fig. 2) revealed notable differences between the nature of complex 1 with respect to that of 2 or 3. The monometallic complexes 2 or 3 feature only a single absorption band at 288 or 326 nm, respectively, while complex 1 displays three absorption bands, which are clearly not simple superimpositions of the bands of complexes 2 and 3. Most relevant is the new charge transfer band at 460 nm, which has no counterpart in the monometallic complexes. Presumably, the planar organization of the three heterocycles in complex 1, entailed by the coordination to two iridium centers, increases the donor properties and thus enhance LMCT interactions. The intra-ligand charge transfer bands (π–π* transitions) have energies that are similar to those observed in the monometallic complexes and are located at 324 and 260 nm (cf. 326 and 288 nm for 3 and 2, respectively).
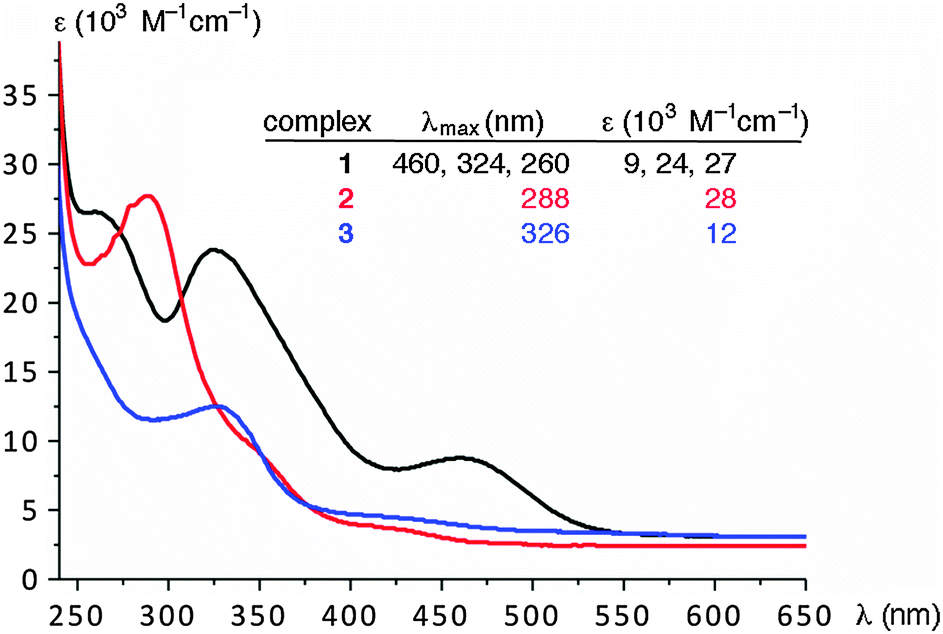 | ||
| Fig. 2 UV/vis spectra of complexes 1–3 (in CH2Cl2, ca. 10−5 M). | ||
Probably the most remarkable feature of the bimetallic complex 1 is the fully reversible oxidation process as established by electrochemical studies using cyclic voltammetry (CV; Fig. 3). While no oxidation wave is observed in either complex 2 nor 3, a reversible single-electron oxidation process is present in the bimetallic complex 1 at E1/2 = + 1.03 V vs. SCE. Cathodic and anodic peak currents are essentially equal at various scan rates (see Fig. S3 and Table S2, ESI†). The complete lack of redox behavior of both monometallic complexes 2 and 3 is unsurprising18 and effectively negates ligand-centered redox processes,19 thus demonstrating the high redox stability of the ligand framework and of the formal +3 oxidation state of the iridium center in both complexes. In contrast, the fully reversible nature of the one-electron redox cycle of complex 1 likely involves the C,C-chelated iridium center and thus corroborates the UV-vis behaviour and the higher electron density as surmised from NMR spectroscopy. Considering the otherwise high similarity of the iridium centers in complex 1 with those of the monometallic complexes 2 and 3, we suggest that the electronic configuration imparted by the N,N-bound iridium center significantly affects the electronic properties of the C,C-bound iridium site, and thus likely constitutes a primary reason for the observed catalytic selectivity.
 | ||
| Fig. 3 Cyclic voltammogram of complexes 1–3 in CH2Cl2 (ca. 10−3 M) with Ag/AgCl (3 M) as reference and ferrocene as internal standard (E1/2 (Fc/Fc+) = 0.46 V vs. SCE). Complex 1: E1/2 = 1.03 V, Ipa ≈ Ipc. | ||
The high selectivity towards alcohol dehydrogenation imparted by complex 1 was further examined with a small selection of representative primary and secondary alcohols (Table 3). In all cases, the ketone/aldehyde is the exclusive product and no traces of the corresponding ether was detected that would point to dehydration activity (Fig. S2, ESI†). Hence high product selectivity is an intrinsic feature of the bimetallic triazolylidene complex 1. Substrate variation suggests that aromatic substituents enhance the catalytic activity (entries 1–3 vs. entries 4 and 5), and that secondary alcohols are faster converted than primary alcohols (e.g. entry 1 vs. 3, or 4 vs. 5). Specifically, phenylethanol and diphenylmethanol are easier dehydrogenated (89% and 80% yield, entries 1 and 2) than benzylalcohol (72%, entry 3). Aliphatic alcohols such as 2-butanol produced the corresponding ketone in 70% yield (entry 4). Using primary and aliphatic alcohols such as 1-octanol afford the lowest conversion (37%, entry 5). These results indicate that the selectivity can be tailored even further to differentiate effectively between aliphatic primary and aromatic secondary alcohols.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | R′ | Time (h) | Conv'nb (%) | Ketone/etherc (%) |
| a Conditions: alcohol (0.2 mmol), complex 1 (0.01 mmol, 2.5 mol%, 5 mol% based on Ir), 1,2-dichlorobezene (2 mL), 150 °C. b Determined by 1H NMR spectroscopic analysis with hexamethylbenzene as internal standard. c % of product ratio. | |||||
| 1 | Ph | CH3 | 24 | 89 | 100/0 |
| 2 | Ph | Ph | 24 | 80 | 100/0 |
| 3 | Ph | H | 24 | 72 | 100/0 |
| 4 | Et | CH3 | 24 | 70 | 100/0 |
| 5 | nOct | H | 24 | 37 | 100/0 |
In conclusion, a bimetallic [Ir3+]2 complex containing a bridging triazolylidene ligand has been developed. This bimetallic complex displays superior catalytic selectivity for the acceptorless dehydrogenation of alcohols when compared to closely related mono-metallic analogues. Spectroscopic and electrochemical analyses reveal a unique electronic setting of the C,C-bound iridium center that is imparted by the N,N-coordinated metal unit, and these features presumably entail the high selectivity. When considering the slightly lower reaction rates, it is plausible that the high selectivity originates from an effective suppression of the dehydration pathway, which results in lower activity, yet higher selectivity. Cooperative substrate binding is less probable when considering the activity of the monometallic complexes. While synergistic interactions have been known to provide access to enhanced catalytic activity, the effects on product selectivity are much less developed and the results presented here may stimulate further work along these lines.
The authors gratefully acknowledge financial support from the European Commission (ERC CoG 615653, Marie Sklodowska-Curie Action 660929 to M. V.).
Notes and references
- (a) S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, Mill Valley, CA 1994 Search PubMed; (b) A. Sigel, H. Sigel and R. K. O. Sigel, Metal Ions in Life Science, Royal Society of Chemistry, London, UK, 2009 Search PubMed; (c) E. I. Solomon, R. K. Szilagyi, S. DeBeer George and L. Basumallick, Chem. Rev., 2004, 104, 419 CrossRef CAS PubMed; (d) E. I. Solomon, M. J. Baldwin and M. D. Lowery, Chem. Rev., 1992, 92, 521 CrossRef CAS.
- (a) N. Sträter, W. N. Lipscomb, T. Klabunde and B. Krebs, Angew. Chem., Int. Ed. Engl., 1996, 35, 2024 CrossRef; (b) H. Steinhagen and G. Helmchen, Angew. Chem., Int. Ed. Engl., 1996, 35, 2339 CrossRef CAS; (c) R. G. Wilkins, Chem. Soc. Rev., 1992, 21, 171 RSC; (d) J. B. Vincent, G. L. Olivier-Lilley and B. A. Averill, Chem. Rev., 1990, 90, 1447 CrossRef CAS.
- For selected examples, see: (a) P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28 CrossRef CAS PubMed; (b) S. M. Inamdar, V. S. Shinde and N. T. Patil, Org. Biomol. Chem., 2015, 13, 8116 RSC; (c) J. A. Mata, F. E. Hahn and E. Peris, Chem. Sci., 2014, 5, 1723 RSC; (d) N. P. Mankad, Synlett, 2014, 1197 CrossRef; (e) L. Stegbauer, F. Sladojevich and D. J. Dixon, Chem. Sci., 2012, 3, 942 RSC; (f) J. I. van der Vlugt, Eur. J. Inorg. Chem., 2012, 363 CrossRef CAS; (g) M. Delferro and T. J. Marks, Chem. Rev., 2011, 111, 2450 CrossRef CAS PubMed; (h) H. Li and T. J. Marks, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15295 CrossRef CAS PubMed; (i) M. Shibasaki, M. Kanai, S. Matsunaga and N. Kumagai, Top. Organomet. Chem., 2011, 37, 1 CrossRef CAS; (j) N. Kielland, E. C. Escudero-Adán, M. Martínez Belmonte and A. W. Kleij, Dalton Trans., 2013, 42, 1427 RSC; (k) U. J. Schele, S. Dechert and F. Meyer, Chem. – Eur. J., 2008, 14, 5112 CrossRef PubMed; (l) S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029 CrossRef CAS; (m) C. Sens, I. Romero, M. Rodriguez, A. Llobet, T. Parella and J. Benet-Buchholz, J. Am. Chem. Soc., 2004, 126, 7798 CrossRef CAS PubMed; (n) P. Haack and C. Limberg, Angew. Chem., Int. Ed., 2014, 53, 4282 CrossRef CAS PubMed.
- (a) S. Sabater, J. A. Mata and E. Peris, Organometallics, 2012, 31, 6450 CrossRef CAS; (b) M. Nussbaum, O. Schuster and M. Albrecht, Chem. – Eur. J., 2013, 19, 17517 CrossRef CAS PubMed.
- (a) A. Zanardi, J. A. Mata and E. Peris, J. Am. Chem. Soc., 2009, 131, 14531 CrossRef CAS PubMed; (b) S. Sabater, J. A. Mata and E. Peris, Nat. Commun., 2013, 4, 3553 Search PubMed.
- (a) P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534 CrossRef CAS PubMed; (b) G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759 CrossRef CAS PubMed; (c) J. D. Crowley, A.-L. Lee and K. J. Kilpin, Aust. J. Chem., 2011, 64, 1118 CrossRef CAS; (d) K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145 RSC.
- (a) R. Lalrempuia, N. D. McDaniel, H. Müller-Bunz, S. Bernhard and M. Albrecht, Angew. Chem., Int. Ed., 2010, 49, 9765 CrossRef CAS PubMed; (b) J. A. Woods, R. Lalrempuia, A. Petronilho, N. D. McDaniel, H. Müller-Bunz, M. Albrecht and S. Bernhard, Energy Environ. Sci., 2014, 7, 2316 RSC; (c) I. Corbucci, A. Petronilho, H. Müller-Bunz, L. Rocchigiani, M. Albrecht and A. Macchioni, ACS Catal., 2015, 5, 2714 CrossRef CAS.
- A. Petronilho, J. A. Woods, H. Mueller-Bunz, S. Bernhard and M. Albrecht, Chem. – Eur. J., 2014, 20, 15775 CrossRef CAS PubMed.
- Note that the resonance structure in Scheme 1 suggests a carbanionic triazolyl binding site, though an alternative resonance structure featuring a Ir
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C carbene unit leads to a nitrogen anion and to an L,X-type N,N-bidentate coordination site.
C carbene unit leads to a nitrogen anion and to an L,X-type N,N-bidentate coordination site. - J. R. Wright, P. C. Young, N. T. Lucas, A.-L. Lee and J. L. Crowley, Organometallics, 2013, 32, 7065 CrossRef CAS PubMed.
- (a) Y. Tulchinsky, M. A. Iron, M. Botoshansky and M. Gandelman, Nat. Chem., 2011, 3, 525 CrossRef CAS PubMed; (b) W. K. C. Lo, G. S. Huff, J. R. Cubanski, A. D. W. Kennedy, C. J. McAdam, D. A. McMorran, K. C. Gordon and J. D. Crowley, Inorg. Chem., 2015, 54, 1572 CrossRef CAS PubMed; (c) P. Thongkam, S. Jindabot, S. Prabpai, P. Kongsaeree, T. Wititsuwannakul, P. Surawatanawong and P. Sangtrirutnugul, RSC Adv., 2015, 5, 55847 RSC.
- This notion is supported also by the isolobal relationship of the [CH3]+ unit in 3 with the [IrCp*Cl]+ fragment in 1. For previous evidence, see: (a) O. Karagiaridi, M. B. Lalonde, W. Bury, A. A. Sarjean, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 18790 CrossRef CAS PubMed; (b) M. B. Lalonde, O. K. Farha, K. A. Scheidt and J. T. Hupp, ACS Catal., 2012, 2, 1550 CrossRef CAS; (c) H. M. Bass, S. A. Cramer, A. S. McCullough, K. J. Bernstein, C. R. Murdock and D. M. Jenkins, Organometallics, 2013, 32, 2160 CrossRef CAS.
- The triazole proton shifts typically about 0.50 ppm downfield upon N-methylation (see ref. 6–8). Iridium coordination to N2 shifts the Htrz resonance from δH = 9.39 in L1 to δH = 10.22 in complex 2 (Δδ = 0.83), suggesting a similar electronic perturbation of the heterocycle upon methylation and iridium coordination.
- For pioneering work on acceptorless alcohol oxidation, see: (a) J. Zhang, M. Gandelman, L. J. W. Shimon, H. Rozenberg and D. Milstein, Organometallics, 2004, 23, 4026 CrossRef CAS; (b) H. Junge and M. Beller, Tetrahedron Lett., 2005, 46, 1031 CrossRef CAS; (c) G. R. A. Adair and J. M. J. Williams, Tetrahedron Lett., 2005, 46, 8233 CrossRef CAS for recent examples, see: ; (d) B. Saha, S. M. Wahidur Rahaman, P. Daw, G. Sengupta and J. K. Bera, Chem. – Eur. J., 2014, 20, 6542 CrossRef CAS PubMed; (e) S. Dinda, A. Genest and N. Rösch, ACS Catal., 2015, 5, 4869 CrossRef CAS; (f) H. Zhao, Q. Chen, L. Wei, Y. Jiang and M. Cai, Tetrahedron, 2015, 71, 8725 CrossRef CAS ; for examples based on triazolylidene ligands, see: ; (g) A. Prades, E. Peris and M. Albrecht, Organometallics, 2011, 30, 1162 CrossRef CAS; (h) M. Delgado-Rebollo, D. Canseco-Gonzalez, M. Hollering, H. Müller-Bunz and M. Albrecht, Dalton Trans., 2014, 43, 4462 RSC.
- (a) A. Prades, R. Corberan, M. Poyatos and E. Peris, Chem. – Eur. J., 2008, 14, 11474 CrossRef CAS PubMed; (b) A. Petronilho and M. Albrecht, in preparation.
- This result suggests that the enhanced selectivity is not due to intermolecular synergistic effects of the two iridium centers. Moreover, catalytic runs with complex 3 in the presence of 5 mol% KOH did not change the selectivity, which excludes a Brønsted acid catalyzed etherification (Table S1, ESI†). Complex 2 is unstable in the presence of base and undergoes a C–H activation process.
- The resonance at 1.83 ppm shows a nOe with the N–CH3 group (δH = 4.71), while the signal at 1.60 ppm displays a nOe with the pyridylidene C3–H proton (δH = 8.53). Both Cp–CH3 singlets show an nOe with pyridyl protons, the resonance at 1.83 ppm with C3–H (δH = 8.78), the signal at 1.60 ppm with C6–H (δH = 9.04).
- A. Petronilho, A. Llobet and M. Albrecht, Inorg. Chem., 2014, 53, 12896 CrossRef CAS PubMed.
- For a recent example of oxidation involving the ligand, see: K. Jeya Prathap and G. Maayan, Chem. Commun., 2015, 51, 11096 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, representative catalytic runs and time conversion profiles, crystallographic details. CCDC 1434920 (1) and 1434921 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc00267f |
| This journal is © The Royal Society of Chemistry 2016 |