
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluoride-assisted synthesis of bimodal microporous SSZ-13 zeolite†
Xiaochun
Zhu
a,
Nikolay
Kosinov
a,
Jan P.
Hofmann
 a,
Brahim
Mezari
a,
Qingyun
Qian
b,
Roderigh
Rohling
a,
Bert M.
Weckhuysen
b,
Javier
Ruiz-Martínez
b and
Emiel J. M.
Hensen
*a
a,
Brahim
Mezari
a,
Qingyun
Qian
b,
Roderigh
Rohling
a,
Bert M.
Weckhuysen
b,
Javier
Ruiz-Martínez
b and
Emiel J. M.
Hensen
*a
aInorganic Materials Chemistry Group, Schuit Institute of Catalysis, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology 5600 MB, Eindhoven, The Netherlands. E-mail: e.j.m.hensen@tue.nl
bInorganic Chemistry and Catalysis Group, Debye Institute for Nanomaterials Science, Department of Chemistry, Faculty of Science, Utrecht University 3584 CG, Utrecht, The Netherlands
First published on 20th January 2016
Abstract
The presence of small amount of fluoride in alkaline hydrothermal synthesis of SSZ-13 zeolite yields bimodal microporous particles with substantially improved performance in the methanol-to-olefins (MTO) reaction. Hydrocarbon uptake measurements and fluorescence microspectroscopy of spent catalysts demonstrate enhanced diffusion through micropores at the grain boundaries of nanocrystals running through the zeolite particles. Fluoride-assisted SSZ-13 synthesis is a cheap and scalable approach to optimize the performance of MTO zeolite catalysts.
Zeolites are crystalline microporous aluminosilicates owing their importance in industrial catalysis to molecule-sized pores, high surface area, outstanding chemical and thermal stability, and adsorptive and catalytic properties.1 They are widely used for shape-selective conversion of hydrocarbons in well-established and large scale petroleum refinery processes such as fluid catalytic cracking, hydroisomerization, hydrocracking and alkylation as well as novel chemical processes such as methanol conversion to hydrocarbons.2,3 The catalytic potential of zeolites is often seriously hampered by diffusion limitations, because the crystal dimensions are by far larger than the micropores. Besides limiting the conversion of bulky reactants,4 slow diffusion can cause formation of undesired by-products such as deposits that deactivate the catalyst. A common method to overcome these limitations is to introduce larger pores in the zeolite crystals. A wide range of methods to introduce additional pores in zeolites have been explored in the last two decades.5–7 Top-down and bottom-up approaches are distinguished to obtain hierarchical zeolites. Though cheap and scalable, demetallation8–10 often negatively affects catalytic performance, as the mesopores are often not uniformly distributed over the zeolite crystals.8 Bottom-up approaches, on the other hand, depend on mesopore-filling agents and allow better control over the location and size of mesopores.11–14 The mesoporogens are often expensive organic compounds, making these approaches less scalable.
In this work, we show that fluoride anions in alkaline hydorthermal synthesis of chabazite (CHA) zeolite results in highly crystalline SSZ-13 particles with additional micropores that are larger than the inherent CHA micropores. This bimodal microporous SSZ-13 zeolite shows unexpectedly high resistance to coking and outperforms bulk SSZ-13 zeolite in the industrially important MTO reaction. The MTO reaction is a key step in the conversion of fossil feedstock into building blocks for the chemical industry. In practice, the zeotype SAPO-3415,16 is preferred over its aluminosilicate SSZ-13 analogue, because strong zeolite acidity results in rapid coking deactivation. If deactivation can be slowed down, SSZ-13 would allow operating the MTO process at lower temperature.17 This can be achieved by pore hierarchization of SSZ-13 employing expensive organic templates.18 Our novel approach, on the other hand, is simple and inexpensive and introduces an additional micropore system in the SSZ-13 zeolite particles. The larger pores decrease diffusional resistances, resulting in complete utilization of the micropore space of this bimodal microporous zeolite during methanol conversion reactions. Zeolite synthesis in the presence of fluoride is usually carried out at neutral pH,19 with little efforts to establish its effect in alkaline conditions.20–22 Here, we synthesized SSZ-13 zeolites from gels containing N,N,N-trimethyl-1-adamantammonium hydroxide as the structure-directing agent and varying amounts of NaF (details in ESI†). The resulting zeolites are denoted by SSZ-13-xF where x is the molar F/SiO2 ratio (x = 0, 5, 10, 20). XRD (Fig. S1, ESI†) and SEM and TEM images (Fig. S2, ESI†) show that all samples are highly crystalline CHA zeolites of similar large crystal size except for SSZ-13-20F. At too high F/SiO2 ratio quartz is obtained (Fig. S1d and S2d, ESI†), which is a known impurity in SSZ-13 synthesis at high Si/Al ratio.2319F NMR spectroscopy shows that calcination removes all fluoride species from as-synthesized SSZ-13-10F (Fig. S3, ESI†).
Fig. 1a compares the MTO performance of the zeolite catalysts as a function of the F content. All catalysts initially convert methanol completely and then deactivate to a low conversion level. The catalyst lifetime of SSZ-13-10F is highest (Table S1, ESI†); this catalyst converts nearly twice as much methanol as SSZ-13-0F. The lifetimes to deactivation to 50% conversion level were 5.1 h, 7.3 h and 10.1 h for SSZ-13-0F, SSZ-13-5F and SSZ-13-10F, respectively. The main reaction products were ethylene and propylene (Table S1, ESI†). As the coke content of spent catalysts increased with increasing gel F content of the gel (Fig. 1b), we speculate that the micropores of fluoride-prepared zeolites were better utilized.
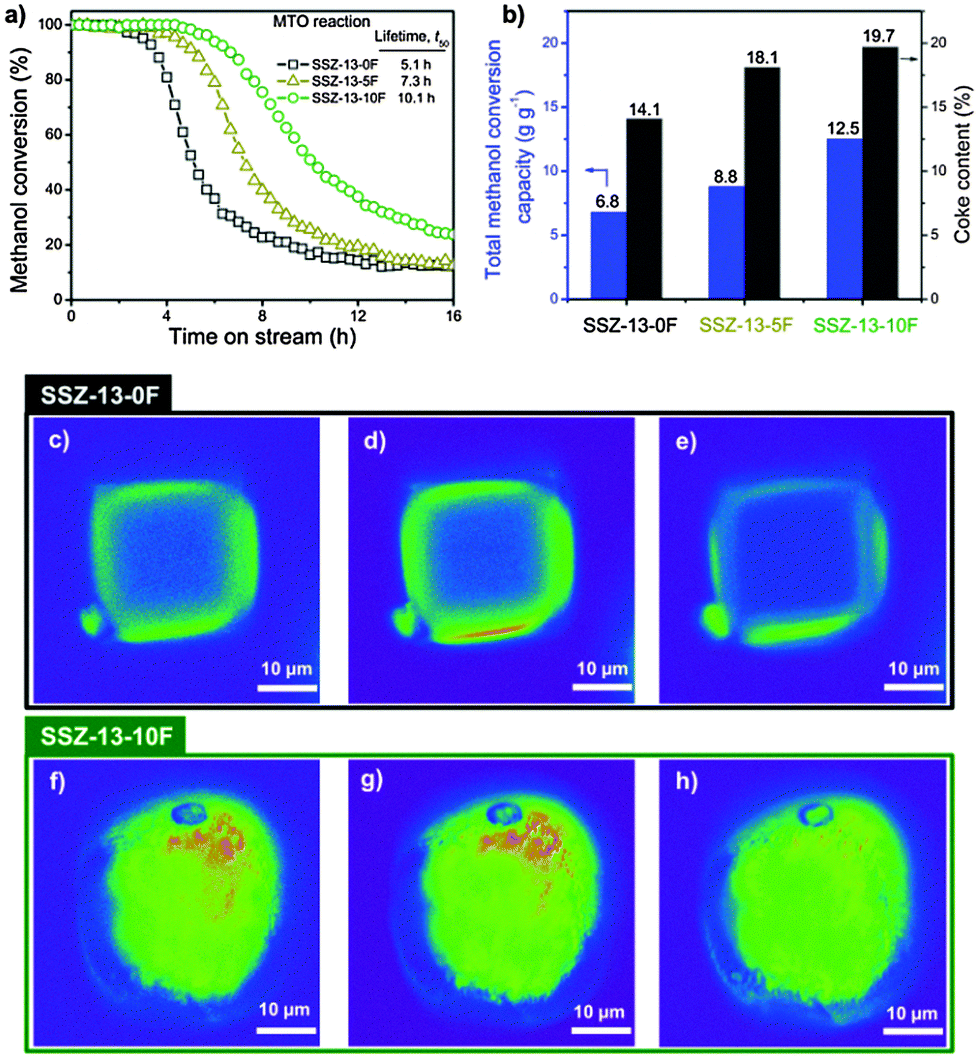 | ||
Fig. 1 MTO reaction and spatial distribution of coke species in single crystals of spent crystals. (a) Methanol conversion as a function of time on stream for SSZ-13-0F (■), SSZ-13-5F ( ), and SSZ-13-10F ( ), and SSZ-13-10F ( ) (MTO reaction: WHSV = 0.8 h−1; T = 350 °C). (b) Coke content of SSZ-13 zeolite catalysts after 24 h time on stream and the total methanol conversion capacity of zeolites (Fig. S4, ESI†). Confocal fluorescence microphotographs of (c–e) SSZ-13-0F and (f–h) SSZ-13-10F crystals collected after the MTO reaction (reaction conditions are shown in ESI†). The images, in false colors, were taken in the middle plane. For the visualization of coke species, 488 nm (c and f), 561 nm (d and g) and 635 nm (e and h) lasers were used with a detection range of 510–550 nm, 570–620 nm and 662–737 nm, respectively. ) (MTO reaction: WHSV = 0.8 h−1; T = 350 °C). (b) Coke content of SSZ-13 zeolite catalysts after 24 h time on stream and the total methanol conversion capacity of zeolites (Fig. S4, ESI†). Confocal fluorescence microphotographs of (c–e) SSZ-13-0F and (f–h) SSZ-13-10F crystals collected after the MTO reaction (reaction conditions are shown in ESI†). The images, in false colors, were taken in the middle plane. For the visualization of coke species, 488 nm (c and f), 561 nm (d and g) and 635 nm (e and h) lasers were used with a detection range of 510–550 nm, 570–620 nm and 662–737 nm, respectively. | ||
Differences in the type and location of carbonaceous deposits in the zeolite crystals formed in the MTO reaction are highlighted by in situ visible light microspectroscopy (Fig. S5, ESI†) and ex situ confocal fluorescence microscopy (Fig. 1c–h). In the spectra of SSZ-13-0F, a broad band around 400 nm due to polymethylated benzene species dominates the spectra.24,25 For SSZ-13-10F, additional strong absorption bands at higher wavelengths are seen, which are ascribed to bulkier polycyclic aromatic hydrocarbon (PAH) species. The spectra were deconvoluted into two main component spectra by non-negative matrix factorization analysis (Fig. S5b and e, ESI†).26 The first component spectrum is mainly made up by the 400 nm feature and the second one by higher wavelength features, representing polymethylated benzenes and other methylated PAHs, respectively.24 The contribution of these two components with time on stream are shown in Fig. S5c and f (ESI†). For SSZ-13-0F, the intensity of polymethylated benzenes goes through a maximum with time on stream and PAHs form later. The amount of these PAHs remains relatively low, although it is difficult to put relative numbers on these observations, as extinction coefficients of these two groups of species are unknown. The spectra for SSZ-13-10F are qualitatively similar, but the amount of PAHs increases for a longer time than for SSZ-13-0F. These observations are consistent with the larger amount of methanol converted by SSZ-13-10F in catalytic activity measurements and also with the higher amount of coke accumulated in this sample.
The spatial coke distribution in spent catalysts used in these microspectroscopy experiments was also investigated by confocal fluorescence microscopy (Fig. 1c–h). Different laser lines allow imaging the distribution of distinct aromatics in the middle plane of spent crystals. Longer excitation wavelengths visualise larger aromatic species.24,27 For spent SSZ-13-0F crystal (Fig. 1c–e), the aromatic species are mainly located in the outer regions of the zeolite crystal in a zone with a thickness of 2–4 μm. The absence of fluorescence originating from inner regions and, thus, the absence of aromatics indicates that only the crystal periphery was used for converting methanol. In stark contrast, the fluorescent images of a spent SSZ-13-10F crystal (Fig. 1f–h) show that aromatic species are homogeneously distributed throughout the crystal. These images demonstrate the better utilization of the SSZ-13-10F particles in the MTO reaction.
In attempting to understand the catalytic differences, we characterized bulk SSZ-13-0F zeolite and the optimal SSZ-13-10F zeolite in more detail. Their Si/Al ratios determined by elemental analysis and 29Si MAS NMR (Fig. S6, ESI†) are very similar (Table S2, ESI†). 27Al 3QMAS NMR spectra (Fig. 2c and d) did not reveal differences in the Al speciation and confirms that nearly all Al atoms are built into the zeolite framework in line with 27Al 1D MAS NMR spectra (Fig. S6, ESI†). 1H MAS NMR spectra (Fig. 2a and Fig. S7, ESI†) proved that SSZ-13-0F and SSZ-13-10F contain comparable amounts of acid sites (Table S3, ESI†). Their similar acid strength is confirmed by the same shift of the bridging OH stretching frequency upon CO adsorption (Fig. S8, ESI†). EDX line scans of FIB cuts of zeolite crystals (Fig. 2b and Fig. S9, ESI†) evidence that the Al gradient across the zeolite particles is similar. All these results prove that there are no chemical or, more specifically, acidity differences between SSZ-13-10F and SSZ-13-0F to which the performance difference can be linked.
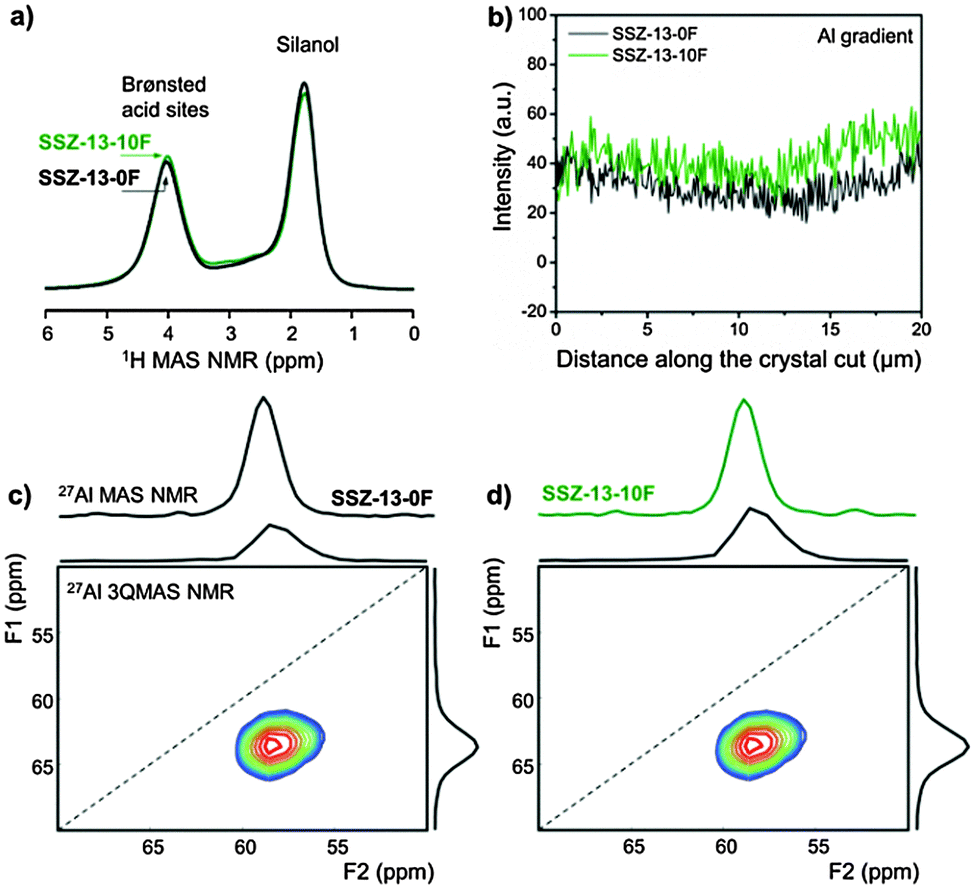 | ||
| Fig. 2 (a) 1H MAS NMR spectra, (b) Al gradient as determined by SEM-EDX line scans of FIB-cut crystals, and (c and d) 27Al MAS and 27Al 3QMAS NMR spectra. The NMR spectra were normalized to the sample weight. | ||
Textural properties obtained by high resolution Ar physisorption (Fig. S10, ESI†) show that the zeolites have a pre-dominantly microporous texture with only few mesopores. The isotherms (Fig. 3a) reveal that, compared with SSZ-13-0F, SSZ-13-10F takes up Ar at lower partial pressures and the adsorption capacity of the micropores continues to be higher at increased partial pressure.
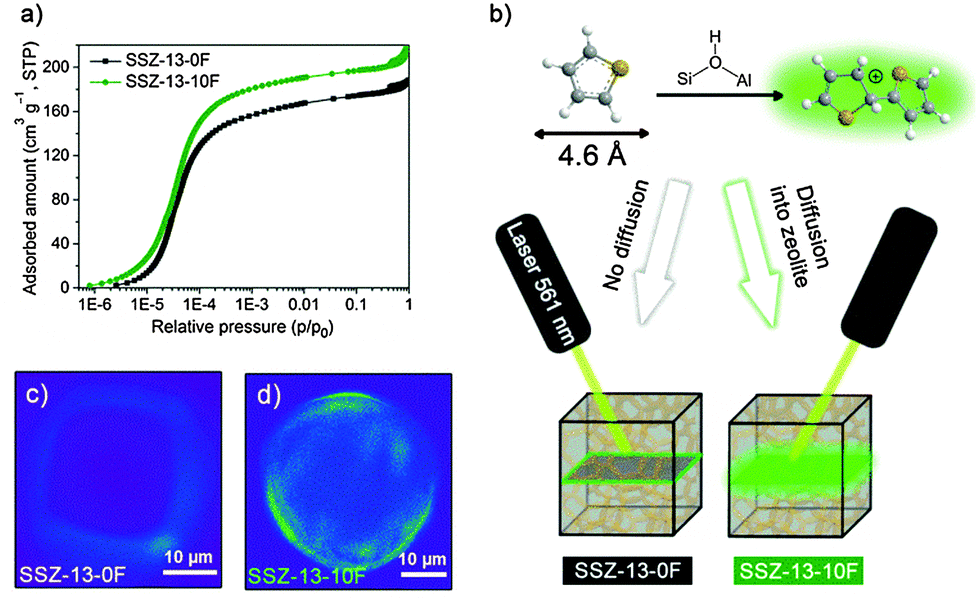 | ||
| Fig. 3 (a) High-resolution Ar physisorption isotherms, (b) acid-catalyzed thiophene oligomerization results in fluorescent species and confocal fluorescence microphotographs, in false colours, taken in the middle plane of single crystals of (c) SSZ-13-0F and (d) SSZ-13-10F after thiophene oligomerization at 120 °C. The excitation wavelength of the laser is 561 nm and detection was in the 570–620 nm range. | ||
Consistent with this, the micropore volume of SSZ-13-10F is slightly higher than that of SSZ-13-0F (Table S4, ESI†). The main contribution to the micropore volume in all samples stems from micropores with an average size of ∼0.54 nm (the effective CHA pore diameter, see Fig. S11, ESI†).28,29 The NLDFT micropore size distribution show that SSZ-13-10F also contains a small amount of micropores with a size of ∼0.5 nm. These micropores are also seen for SSZ-13-5F, but not for SSZ-13-0F. The micropore volume and MTO performance of SSZ-13-5F lie between values for SSZ-13-0F and SSZ-13-10F.
Convincing evidence for these additional micropores is provided by carrying out thiophene oligomerization in the zeolite crystals. Thiophene will react with Brønsted acid sites and form fluorescent oligomers, whose location can be resolved by confocal fluorescence microscopy.30,31 The pore CHA pore apertures are too small (0.38 nm) to allow diffusion of thiophene (kinetic diameter = 0.46 nm) inside SSZ-13. This explains why in Fig. 3c the fluorescent species are only formed on the surface of a SSZ-13-0F particle. On contrary, the fluorescent species are seen throughout the zeolite particle of SSZ-13-10F (Fig. 3d). These results support the conclusion that SSZ-13-10F contains pores that are larger than the size of the CHA pore apertures. These micropores contain Brønsted acid sites that catalyse thiophene oligomerization.
To establish whether these pores improve mass transport, we compared the room-temperature uptake of several organic molecules of varying kinetic diameters in SSZ-13-0F and SSZ-13-10F (Fig. S12, ESI†). Fig. 4 shows the initial uptake of methanol, butanols and o-xylene. The uptake of methanol, the reactant in the MTO reaction, was nearly unaffected by the presence of additional micropores in SSZ-13-10F. This result and the similar total methanol uptake demonstrate that pore obstruction by deposits present in fresh SSZ-13-0F can be excluded as a cause of the lower catalytic performance.
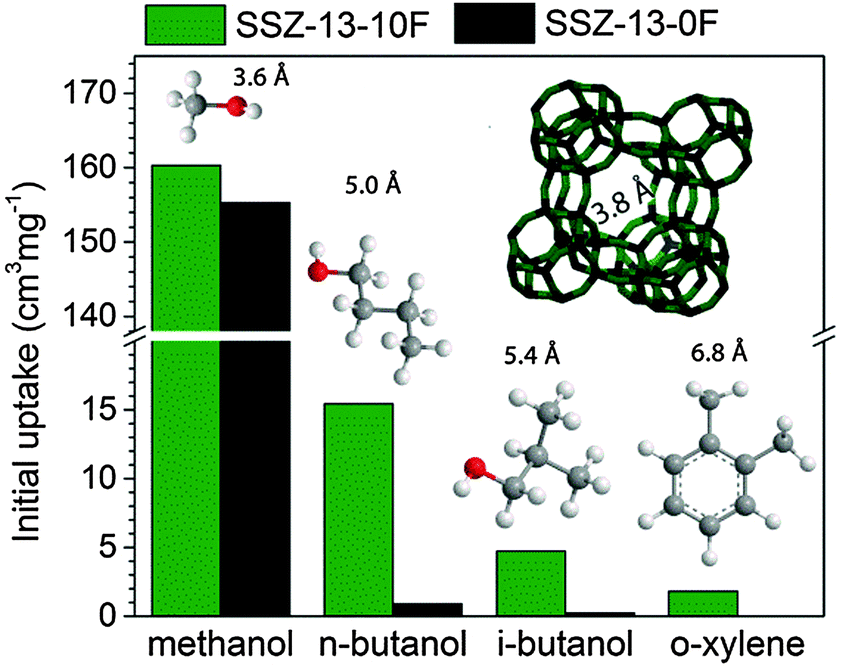 | ||
| Fig. 4 Initial uptakes of methanol, n-butanol, i-butanol, and o-xylene in dehydrated SSZ-13-0F and SSZ-13-10F. Uptake measurements were performed at 30 °C in a TGA apparatus. Extrapolation of the linear part of the uptake curve (Fig. S12, ESI†) to zero time defines the initial uptake. | ||
The uptake of n-butanol, on the other hand, is very different for the two zeolites (Fig. S12b, ESI†). Whereas SSZ-13-0F only slowly adsorbs this larger alcohol in the cage-window pore system of the CHA zeolite in agreement with data for linear alcohols adsorption in SAPO-34,32 the uptake of n-butanol is much faster in SSZ-13-10F, especially directly after exposure to n-butanol and it continues to be faster later on. The amount of n-butanol adsorbed during the initial steep uptake (0.028 cm3 g−1) for SSZ-13-10F is comparable to the difference in micropore volume between SSZ-13-0F and SSZ-13-10F (0.03 cm3 g−1). Similar differences were seen for uptake measurements of bulkier i-butanol (Fig. S12c, ESI†) and o-xylene (Fig. S12d, ESI†). Initial uptake values (Fig. 4) underpin the more accessible pore structure of SSZ-13-10F. By taking into account the sizes of the adsorbates, we estimate the size of the additional pores between 4.6 and 7 Å.
We also investigated the uptake of propylene, one of the main MTO reaction products. At room temperature, propylene is adsorbed much faster in SSZ-13-10F than in SSZ-13-0F and the total uptake is also higher (Fig. S13a, ESI†). The total uptake in SSZ-13-10F amounts to 3–4 propylene molecules per Brønsted acid site, which is due to oligomerization of propylene on acid sites33 as confirmed by in situ IR spectroscopy (Fig. S13b, ESI†).34 SSZ-13-10F contains more of these oligomers than SSZ-13-0F. Confocal fluorescence microspectroscopy data of these two samples after room-temperature exposure to propylene (Fig. S13c and d, ESI†) shows that propylene oligomerization took place throughout the entire SSZ-13-10F crystal, whereas this reaction was limited to the outer regions of the SSZ-13-0F crystals. From these observations, we infer that the oligomers formed in the outer regions of the SSZ-13-0F crystal block the further diffusion of propylene into the inner parts of the crystal. The additional micropores in SSZ-13-10F provide a less hindered diffusion pathway for propylene and uniform oligomerization throughout the zeolite crystals, even if part of the CHA pores become obstructed.
In summary, we describe a novel approach to synthesize optimum SSZ-13 zeolite for the MTO reaction. Addition of a small amount of fluoride to an alkaline synthesis gel results in bimodal microporous SSZ-13. The presence of additional micropores larger than the pore apertures of the CHA pore network follows from high-resolution Ar physisorption, alcohol uptake measurements and thiophene oligomerization-fluorescence microscopy experiments. TEM (Fig. S2, ESI†) excludes formation of hollow structures due to etching as reported in the F-mediated synthesis of SAPO-34.35 SEM (Fig. S2k, ESI†) emphasize the very rough surface topography of the fluoride-prepared zeolite compared with bulk SSZ-13. The rough surface points to adhesive growth of the zeolite driven by high supersaturation resulting from fluorination of inorganic precursor units.36 This growth mode resembles the non-classical growth mechanism for SSZ-13 recently reported by Kumar et al.37 We speculate that the secondary micropore system in fluoride-prepared SSZ-13 is a direct consequence of this growth mechanism; these pores are likely situated at the boundaries of the particles attached to the growing zeolite. These additional micropores lower diffusion resistance through the zeolite framework, even if parts of the CHA micropores blocked by coke deposits. When used in the MTO reaction, this leads to complete utilization of the micropore space of the SSZ-13 zeolite particles. Compared with other methods, the current approach is a cost-effective and scalable approach and we expect it to be also useful for other SSZ-13 catalyzed processes.38
Notes and references
- K. Na, C. Jo, J. Kim, K. Cho, J. Jung, Y. Seo, R. J. Messinger, B. F. Chmelka and R. Ryoo, Science, 2011, 333, 328 CrossRef CAS PubMed.
- B. Smit and T. L. M. Maesen, Nature, 2008, 451, 671 CrossRef CAS PubMed.
- W. J. Roth, P. Nachtigall, R. E. Morris and J. Čejka, Chem. Rev., 2014, 114, 4807 CrossRef CAS PubMed.
- J. Kärger, T. Binder, C. Chmelik, F. Hibbe, H. Krautscheid, R. Krishna and J. Weitkamp, Nat. Mater., 2014, 13, 333 CrossRef PubMed.
- J. Pérez-Ramírez, Nat. Chem., 2012, 4, 250 CrossRef PubMed.
- X. Meng, F. Nawaz and F. S. Xiao, Nano Today, 2009, 4, 292 CrossRef CAS.
- V. Valtchev and L. Tosheva, Chem. Rev., 2013, 113, 6734 CrossRef CAS PubMed.
- S. van Donk, A. H. Janssen, J. H. Bitter and K. P. de Jong, Catal. Rev., 2003, 45, 297 CAS.
- A. H. Janssen, A. J. Koster and K. P. de Jong, Angew. Chem., Int. Ed., 2001, 40, 1102 CrossRef CAS.
- D. Verboekend, T. C. Keller, S. Mitchell and J. Pérez-Ramírez, Adv. Funct. Mater., 2013, 23, 1923 CrossRef CAS.
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Nature, 2009, 461, 246 CrossRef CAS PubMed.
- Y. Seo, S. Lee, C. Jo and R. Ryoo, J. Am. Chem. Soc., 2013, 135, 8806 CrossRef CAS PubMed.
- X. Zhang, D. Liu, D. Xu, S. Asahina, K. A. Cychosz, K. V. Agrawal, Y. Al Wahedi, A. Bhan, S. Al Hashimi, O. Terasaki, M. Thommes and M. Tsapatsis, Science, 2012, 336, 1684 CrossRef CAS PubMed.
- X. Zhu, R. Rohling, G. Filonenko, B. Mezari, J. P. Hofmann, S. Asahina and E. J. M. Hensen, Chem. Commun., 2014, 50, 14658 RSC.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922 CrossRef CAS.
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810 CrossRef CAS PubMed.
- F. Bleken, M. Bjørgen, L. Palumbo, S. Bordiga, S. Svelle, K. P. Lillerud and U. Olsbye, Top. Catal., 2009, 52, 218 CrossRef CAS.
- L. Wu, V. Degirmenci, P. C. M. M. Magusin, B. M. Szyja and E. J. M. Hensen, Chem. Commun., 2012, 48, 9492 RSC.
- A. Corma, M. J. Díaz-Cabañas, J. Martínez-Triguero, F. Rey and J. Rius, Nature, 2002, 418, 514 CrossRef CAS PubMed.
- Y. Kalvachev, M. Jaber, V. Mavrodinova, L. Dimitrov, D. Nihtianova and V. Valtchev, Microporous Mesoporous Mater., 2013, 177, 127 CrossRef CAS.
- Z. Qin, L. Lakiss, L. Tosheva, J.-P. Gilson, A. Vicente, C. Fernandez and V. Valtchev, Adv. Funct. Mater., 2013, 24, 257 CrossRef.
- O. Larlus and V. P. Valtchev, Chem. Mater., 2005, 17, 881 CrossRef CAS.
- E. A. Eilertsen, M. H. Nilsen, R. Wendelbo, U. Olsbye and K. P. Lillerud, Stud. Surf. Sci. Catal., 2008, 174, 265 CrossRef.
- V. Van Speybroeck, K. Hemelsoet, K. De Wispelaere, Q. Qian, J. Van der Mynsbrugge, B. De Sterck, B. M. Weckhuysen and M. Waroquier, ChemCatChem, 2013, 5, 173 CrossRef CAS.
- L. Palumbo, F. Bonino, P. Beato, M. Bjørgen, A. Zecchina and S. Bordiga, J. Phys. Chem. C, 2008, 112, 9710 CAS.
- E. Borodina, F. Meirer, I. Lezcano-González, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, J. Ruiz-Martinez and B. M. Weckhuysen, ACS Catal., 2015, 5, 992 CrossRef CAS.
- Q. Qian, J. Ruiz-Martínez, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, H. E. van der Bij, J. Kornatowski and B. M. Weckhuysen, Chem. – Eur. J., 2013, 19, 11204 CrossRef CAS PubMed.
- Y. Kobayashi, Y. Li, Y. Wang and D. Wang, Chin. J. Catal., 2013, 34, 2192 CrossRef CAS.
- Y. Ji, M. A. Deimund, Y. Bhawe and M. E. Davis, ACS Catal., 2015, 5, 4456 CrossRef CAS.
- I. L. C. Buurmans, J. Ruiz-Martínez, W. V. Knowles, D. van der Beek, J. A. Bergwerff, E. T. C. Vogt and B. M. Weckhuysen, Nat. Chem., 2011, 3, 862 CrossRef CAS PubMed.
- J. Ruiz-Martínez, I. L. C. Buurmans, W. V. Knowles, D. van der Beek, J. A. Bergwerff, E. T. C. Vogt and B. M. Weckhuysen, Appl. Catal., A, 2012, 419–420, 84 CrossRef.
- T. Remy, J. Cousin Saint Remi, R. Singh, P. A. Webley, G. V. Baron and J. F. M. Denayer, J. Phys. Chem. C, 2011, 115, 8117 CAS.
- D. J. Parrillo, C. Lee, R. J. Gorte, D. White and W. E. Farneth, J. Phys. Chem., 1995, 99, 8745 CrossRef CAS.
- A. K. Ghosh and R. A. Kydd, J. Catal., 1986, 100, 185 CrossRef CAS.
- D. Xi, Q. Sun, J. Xu, M. Cho, H. S. Cho, S. Asahina, Y. Li, F. Deng, O. Terasaki and J. Yu, J. Mater. Chem. A, 2014, 2, 17994 CAS.
- Zeolites and Catalysis, ed. J. Cejka, A. Corma and S. I. Zones, Wiley, 2010, p. 20 Search PubMed.
- M. Kumar, H. Luo, Y. Román-Leshkov and J. D. Rimer, J. Am. Chem. Soc., 2015, 137, 13007 CrossRef CAS PubMed.
- D. W. Fickel, E. D'Addio, J. A. Lauterbach and R. F. Lobo, Appl. Catal., B, 2011, 102, 441 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc00201c |
| This journal is © The Royal Society of Chemistry 2016 |