
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydration-dependent anomalous thermal expansion behaviour in a coordination polymer†
Prem
Lama
,
Lukman O.
Alimi
,
Raj Kumar
Das
and
Leonard J.
Barbour
*
Department of Chemistry and Polymer Science, University of Stellenbosch, Matieland, 7602, Stellenbosch, South Africa. E-mail: ljb@sun.ac.za
First published on 20th January 2016
Abstract
A coordination polymer is shown to possess anomalous anisotropic thermal expansion. Guest water molecules present in the as-synthesised material can be removed upon activation without loss of crystal singularity. The fully dehydrated form shows considerably different thermal expansion behaviour as compared to the hydrate.
Owing to longitudinal vibrational motion most materials expand along all three directions upon heating. This is known as positive thermal expansion (PTE).1 However, in isolated cases the occurrence of other low-energy molecular motions such as rigid unit modes (RUM),2 the invar effect3etc. result in unusual thermal responses such as negative thermal expansion (NTE)4 or zero thermal expansion (ZTE).5 A few geometrically flexible materials have been reported to possess anisotropic thermal expansion as a result of novel types of mechanical responses that include hinge-like6 or stretching–tilting7 motion. A handful of organic and organic–inorganic hybrid materials such as Ag3[Co(CN)6], Prussian blue analogues and methanol monohydrate have been reported to possess extraordinarily anisotropic thermal expansion.8 Being somewhat related to the abovementioned materials, MOFs have attracted substantial attention owing to their novel topologies and various potential applications.9 Additionally, isotropic negative thermal expansion has been reported for several MOF materials, including HKUST-1,10 IRMOFs11 and cyano-bridged MOFs.12
Additional attention has recently been devoted to designing new materials that exhibit anomalous thermal expansion behaviour. However, as yet not much effort has been made to obtain tuneable thermal expansion, with the exception of a few reports of systems that display this highly desirable property.6b,13–16 The phenomenon of tuneable thermal expansion essentially involves invoking changes in weaker interactions or low frequency vibrational modes using external stimuli. For example, Chupas et al. have reported that NTE of Zn(CN)2 can be enhanced by the application of mechanical pressure to the system.13 The flexibility of the system can also be changed using chemical modification either pre-14 or post-synthetically.6b,15,16 In the pre-synthetic approach, isostructural materials with different compositions can be designed, whereas the post-synthetic approach involves modification of framework flexibility either by changing the occupancy or nature of included guests in porous materials.
Herein, we report the modified synthesis of a coordination polymer {[Zn(BTC)(HBPP)]·H2O}n (1·H2O) (Scheme S1, ESI†), which was previously prepared using a different procedure (BTC = 1,3,5-benzenetricarboxylate and HBPP = protonated 1,3-bis(4-pyridyl)propane).17 Single-crystal X-ray diffraction (SCD) analysis at 100 K reveals that 1·H2O crystallises in the non-centrosymmetric orthorhombic space group Pna21. The asymmetric unit consists of one Zn2+ ion, one BTC3− ligand, one HBPP+ ligand and a water molecule (Fig. S1, ESI†). The Zn2+ ion adopts a distorted tetrahedral coordination geometry with three carboxylate groups bound to the metal centre, each in a monodentate coordination mode, and the fourth position is occupied by the pyridyl unit of the HBPP ligand. The remaining pyridyl unit of HBPP is protonated and therefore does not coordinate. The dihedral angles among the BTC units coordinated to the same Zn2+ centres are 12.8(1)°, 34.2(1)° and 36.7(1)° (Fig. S2, ESI†).
Two carboxylate groups of the BTC linkers bridge the metal centers to form approximately hexagonal helices along the crystallographic b axis (Fig. 1). The pitch and diameter of the helices are 7.804(1) Å and 11.262(3) Å, respectively. The metal–metal distances along each helical chain are in the range 8.170(1) to 10.631(1) Å. The individual helical columns are filled by the HBPP+ units and the protonated pyridyl groups are almost parallel to the BTC ligands, (Fig. 1) although there are no significant interactions between them. Each helical column is connected to neighbouring columns via carboxylate groups to generate an overall three dimensional structure (Fig. S3, ESI†). Owing to the presence of both right- and left-handed helices, the framework is achiral (Fig. 2).
 | ||
| Fig. 1 (a) View of an approximately hexagonal helical chain with encapsulated BPPH+ units and (b) perspective view of the helical chain approximately along [010] (BPPH+ units are shown as ball-and-stick models while the remaining components are shown as wire and capped-stick models. All hydrogen atoms except those bonded to nitrogen atoms have been omitted for clarity). | ||
 | ||
| Fig. 2 Perspective view of right- and left-handed (pink and green, respectively) helical chains in 1·H2O (hydrogen atoms and water molecules have been omitted and each helical column is shown as a space filled model). | ||
The overall structure contains small isolated (zero dimensional) voids that are occupied by the water molecules. The protonated pyridyl units and the water molecules form several N–H⋯O, O–H⋯O and C–H⋯O hydrogen bonding interactions (Fig. 3). Interestingly, all of the protonated pyridyl units are oriented in same direction (Fig. 3), consistent with the absence of inversion symmetry of the framework, and the coordination polymer is therefore polar.
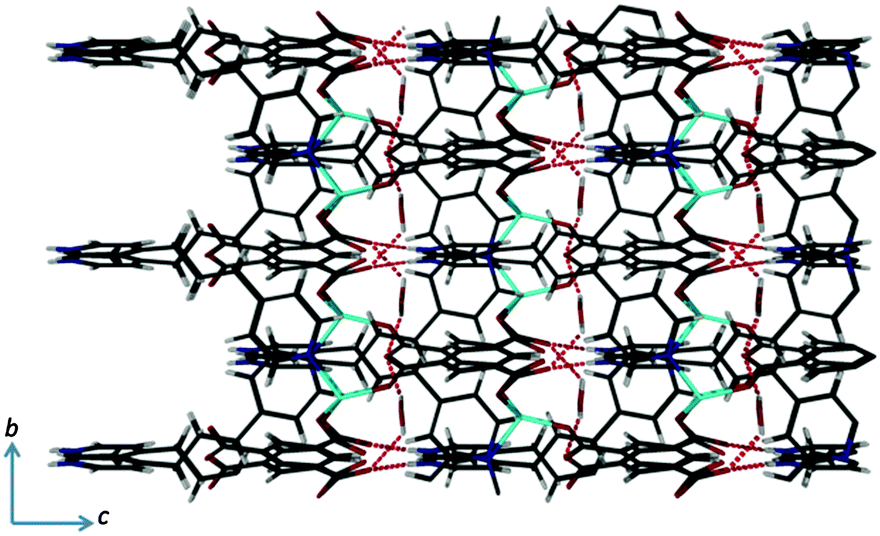 | ||
| Fig. 3 View of the overall packing arrangement of 1·H2O showing the N–H⋯O and O–H⋯O hydrogen bonding interactions (analogous C–H⋯O interactions are omitted for clarity). | ||
The Zn2+ centre forms a pyramidal 3-connected node whereas the BTC units represent 3-connected planar nodes. The topology can thus be described as a 3-connected (10,3)-d (utp) net with extended Schläfli symbol 102·104·104 (Fig. S4, ESI†).
Variable-temperature single-crystal X-ray diffraction (VT-SCD) experiments were carried out with a view to understanding the temperature-dependent structural changes of 1·H2O. Starting at 260 K, the crystal was cooled to 100 K and data were collected at 20 K intervals. Upon cooling the crystallographic b axis contracts significantly while the a and c axes elongate gradually (Fig. 4). The linear thermal expansion coefficients along the three crystallographic axes a, b and c are −6.0(1), 53.0(7) and −7.9(5) MK−1 respectively. Thus 1·H2O shows rare biaxial negative thermal expansion behaviour.8a,b,16b,18 The overall volumetric thermal expansion is 39.0(8) MK−1(Fig. S5, ESI†).
 | ||
| Fig. 4 Variation of unit cell dimensions of 1·H2O (including error bars) with temperature. | ||
The reversibility of the thermal expansion process was verified by first cooling the crystal from 260 K to 100 K and then heating it again to 260 K (260K-R in ESI†). The initial and final unit cell dimensions at 260 K agree within experimental error.
In order to understand the mechanism responsible for the anomalous thermal expansion properties of 1·H2O, we analysed the types of hydrogen bonding interactions and temperature-dependent structural changes. The protonated pyridyl unit of the HBPP+ present in each helical chain forms hydrogen bonding interactions with the carboxylate groups of the neighbouring helices. All of the N–H⋯O hydrogen bonding interactions occur almost exclusively along the crystallographic c axis (Fig. S6, ESI†). Upon cooling, the flexible HBPP+ linkers undergo conformational changes; the bond angle C15⋯C16⋯C17 (θ1) and the non-bonding torsion angle N1⋯C12⋯C18⋯N2 (φ1) decrease from 111.5(3)° to 110.7(2)° and 7.4(5)° to 6.3(4)°, respectively (Table S5, ESI†). Thus the HBPP+ linkers become more bent upon cooling, causing concomitant movement of BTC units and thus resulting in hinge-like motion of helical columns. Hence, the pitch of the helical columns formed by the BTC units decreases from 7.870(1) Å to 7.804(1) Å (Table S6, ESI†) and the diameter of the helices increases from 11.239(3) to 11.262(3) Å (Table S6, ESI†) thereby resulting in PTE along the b direction and NTE along the c direction.
The water molecules also bridge the carboxylate units between two neighbouring helical chains, forming several quite strong O–H⋯O and weak C–H⋯O hydrogen bonding interactions (Fig. S7, ESI†). In this case these interactions act predominantly in the ab plane (Fig. S7, ESI†), thereby transferring a component of the hinge-like motion to the a axis, and consequently causing NTE along this direction too. The N–H⋯O and O–H⋯O hydrogen bonding interactions in 1·H2O are sufficiently strong to preclude any substantial temperature dependent changes in their characteristic geometrical parameters (Tables S7 and S8, ESI†).
Thermogravimetric analysis (TGA) of 1·H2O shows about 3.5% weight loss within the temperature range of 50 to 140 °C, which corresponds to the loss of guest water molecules (calc. 3.7%). The dehydrated framework appears to be stable up to 340 °C (Fig. S8, ESI†). Based on the TGA results, crystals of 1·H2O were activated at 140 °C under dynamic vacuum for 12 hours in order to remove the water guest molecules. Interestingly, the activated crystals retained their crystal singularity and SCD analysis at 100 K yielded the structure of the dehydrated framework 1. TGA of 1 also confirmed the absence of any residual water molecules (Fig. S8, ESI†).
The framework of 1 is isoskeletal19 to that of its hydrated analogue. The N–H⋯O hydrogen bonding interactions remain intact although, of course, the analogous O–H⋯O interactions are absent in 1 owing to the loss of the guest water molecules. The dihedral angles between the BTC units change to 11.8(2)°, 38.3(2)° and 40.2(2)° owing to a slight structural rearrangement upon dehydration.
In order to elucidate the effect of dehydration on the flexibility of the framework, we also investigated the temperature-dependent structural changes in 1. In this case the crystallographic a axis remains practically constant in the temperature range 100 to 310 K, whereas the thermal responses with respect to the crystallographic b and c axes remain quite similar to those of 1·H2O (Fig. 5). The linear thermal expansion coefficients along the crystallographic a, b and c directions are −0.20(7), 47.8(7) and −7.4(7) MK−1, respectively. The dehydrated framework 1 therefore shows a very rare combination of positive, negative and zero thermal expansion behaviour.6,7 In this case, the overall volumetric thermal expansion is 40(1) MK−1 (Fig. S9, ESI†).
 | ||
| Fig. 5 Variation of unit cell dimensions of 1 (including error bars) with temperature. | ||
The thermal response of the BPP unit of 1 is somewhat different from that of the hydrated analogue; the non-bonding torsional angle N1⋯C12⋯C18⋯N2 (φ2) increases from 7.5(8)° to 13.0(5)° whereas the bonding angle C15⋯C16⋯C17 (θ2) now decreases from 112.5(5)° to 109.2(3)° (Table S9, ESI†). The BPP units bend upon cooling, the pitch of the helices decreases from 7.831(1) Å to 7.753(1) Å (Table S10, ESI†) and their diameter increases from 11.105(5) Å to 11.146(4) Å (Table S10, ESI†). This results in similar thermal responses along the b and c directions as compared to the hydrated analogue. However, owing to the absence of O–H⋯O and C–H⋯O hydrogen bonding interactions in the dehydrated framework, the hinge-like motion cannot be transferred to the crystallographic a axis, thereby resulting in zero thermal expansion along that direction. In this case, the N–H⋯O hydrogen bonding interaction remains practically unaffected by temperature variation (Table S11, ESI†). Differential scanning calorimetry (DSC) of both the hydrated and the dehydrated frameworks rules out any possibility of a temperature-induced phase transition – i.e. no distinct thermal events were observed during the thermal cycle in either case (Fig. S10, ESI†).
To date, guest-dependent thermal expansion behaviour has been reported mostly for materials that display NTE purely due to transverse vibrational motion.10,15 For Cd(CN)2,15a ZnPt(CN)615b and HKUST-110 negative thermal expansion behaviour was observed in the guest-free frameworks whereas quite ordinary PTE was observed in the guest-filled systems. In MOF-5, the NTE of the host framework becomes less pronounced under gas pressure as compared to its evacuated form.15c In these instances, PTE of the included guest molecules either dampens or compensates for the NTE of the host framework. The presence of empty channels is necessary for these materials to undergo prominent NTE.
To date there have been very few reports of the thermal expansion behaviour of a host being enhanced upon guest uptake.6b,16 In FMOF-1 the NTE of the host framework is enhanced by N2 pressure16a but gas-loading in the system gradually increases upon cooling, leading to guest-induced phase transitions, as indicated by the two-step breathing process. To the best of our knowledge it is extremely rare to observe guest-induced enhancement of a negative thermal response where the guest occupancy remains constant throughout the thermal cycle.6b,16b We have previously shown how the thermal response of the MOF [Zn(L)2(OH)2]n (L = 4-(1H-naphtho[2,3-d]imidazole-1-yl)benzoic acid) increases upon inclusion of primary alcohols (MeOH, EtOH and nPrOH) but diminishes with inclusion of iPrOH.16b However, such guest dependent behaviour has been attributed to the change in framework flexibility upon desolvation and exchange. Since the guest molecules other than those hydrogen bonded to the [Zn–OH–]n chain could not be modelled, the influence of the guest on the flexibility is not understood. Chen et al. recently reported a similar type of guest-induced enhancement of thermal expansion behaviour6b but in their case the guest molecules were also highly disordered. In the present report the position of the guest water molecule in 1·H2O has been located precisely and the influence of its non-covalent interactions can therefore be rationalised. Consequently, the changes in the weaker interactions and framework dynamics resulting from desolvation can be clearly understood. However, in this case the water molecules occupy a small proportion of the volume and thus the changes in the overall packing arrangement upon dehydration are also very small. Consequently, the changes in the overall thermal response are relatively insignificant compared to those reported by Saha et al.16c
The preparation of porous materials such as 1, together with a detailed elucidation of the guest-dependent changes in their packing arrangements and host–guest interactions may help to establish the design principles for materials having controllable thermal expansion behaviour. Such exploratory studies could lead to several potential single-crystal devices such as thermo-mechanical sensors or actuators.
We are grateful to the National Research Foundation and the Department of Science and Technology (SARCHI Program) for financial support of this work. PL also acknowledges the Claude Leon Foundation for financial support.
Notes and references
- N. W. Ashcroft and N. D. Mermin, Solid State Physics, Holt, Rinehart & Winston, 1976 Search PubMed.
- (a) W. Miller, C. W. Smith, D. S. Mackenzie and K. E. Evans, J. Mater. Sci., 2009, 44, 5441–5451 CrossRef CAS; (b) C. Lind, Materials, 2012, 5, 1125–1154 CrossRef CAS.
- (a) C. E. Guillaume, C. R. Acad. Sci., 1897, 125, 235–238 Search PubMed; (b) C. E. Guillaume, Nature, 1904, 71, 134–139 CrossRef; (c) M. van Schilfgaarde, I. A. Abrikosov and B. Johansson, Nature, 1999, 400, 46–49 CrossRef CAS.
- (a) V. K. Peterson, G. J. Kearly, Y. Wu, A. J. Ramirez-Cuesta, E. Kemner and C. J. Kepert, Angew. Chem., Int. Ed., 2010, 49, 585–588 CrossRef CAS PubMed; (b) A. L. Goodwin and C. J. Kepert, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 140301 CrossRef.
- (a) A. E. Phillips, G. J. Halder, K. W. Chapman, A. L. Goodwin and C. J. Kepert, J. Am. Chem. Soc., 2010, 132, 10–11 CrossRef CAS PubMed; (b) S. Margadonna, K. Prassides and A. N. Fitch, J. Am. Chem. Soc., 2004, 126, 15390–15391 CrossRef CAS PubMed; (c) X. Song, Z. Sun, Q. Huang, M. Rettenmayr, X. Liu, M. Seyring, G. Li, G. Rao and F. Yin, Adv. Mater., 2011, 23, 4690–4694 CrossRef CAS PubMed; (d) Y.-S. Wei, M. Zhang, P.-Q. Liao, R.-B. Lin, T.-Y. Li, G. Shao, J.-P. Zhang and X.-M. Chen, Nat. Commun., 2015, 6, 8348 CrossRef CAS PubMed.
- (a) S. Henke, A. Schneemann and R. A. Fischer, Adv. Funct. Mater., 2013, 23, 5990–5996 CrossRef CAS; (b) H.-L. Zhou, R.-B. Lin, C.-T. He, Y.-B. Zhang, N. Feng, Q. Wang, F. Deng, J.-P. Zhang and X.-M. Chen, Nat. Commun., 2013, 4, 2534 Search PubMed; (c) R. K. Das, H. Aggarwal and L. J. Barbour, Inorg. Chem., 2015, 54, 8171–8173 CrossRef CAS PubMed.
- P. Lama, R. K. Das, V. J. Smith and L. J. Barbour, Chem. Commun., 2014, 50, 6464–6467 RSC.
- (a) D. Das, T. Jacobs and L. J. Barbour, Nat. Mater., 2010, 9, 36–39 CrossRef CAS PubMed; (b) D. Das, T. Jacobs, A. Pietraszko and L. J. Barbour, Chem. Commun., 2011, 47, 6009–6011 RSC; (c) A. D. Fortes, E. Suard and K. S. Knight, Science, 2011, 331, 742–746 CrossRef CAS PubMed; (d) A. L. Goodwin, M. Calleja, M. J. Conterio, M. T. Dove, J. S. O. Evans, D. A. Keen, L. Peters and M. G. Tucker, Science, 2008, 319, 794–797 CrossRef CAS PubMed; (e) K. M. Hutchins, R. H. Groeneman, E. W. Reinheimer, D. C. Swenson and L. R. MacGillivray, Chem. Sci., 2015, 6, 4717–4722 RSC; (f) H.-L. Zhou, Y.-B. Zhang, J.-P. Zhang and X.-M. Chen, Nat. Commun., 2015, 6, 6917 CrossRef CAS PubMed.
- (a) R. Banerjee, H. Furukawa, D. Britt, C. Knobler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 3875–3877 CrossRef CAS PubMed; (b) J. J. Perry IV, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400–1417 RSC; (c) J. M. Ogborn, I. E. Collings, S. A. Moggach, A. L. Thompson and A. L. Goodwin, Chem. Sci., 2012, 3, 3011–3017 RSC; (d) R. K. Das, A. Aijaz, M. K. Sharma, P. Lama and P. K. Braradwaj, Chem. – Eur. J., 2012, 18, 6866–6872 CrossRef CAS PubMed; (e) M. Eddaoudi, D. F. Sava, J. F. Eubank, K. Adil and V. Guillerm, Chem. Soc. Rev., 2015, 44, 228–249 RSC; (f) S. Sen, S. Neogi, K. Rissanen and P. K. Bharadwaj, Chem. Commun., 2015, 51, 3173–3176 RSC.
- (a) V. K. Peterson, G. J. Kearly, Y. Wu, A. J. Ramirez-Cuesta, E. Kemner and C. J. Kepert, Angew. Chem., Int. Ed., 2010, 49, 585–588 CrossRef CAS PubMed; (b) Y. Wu, A. Kobayashi, G. J. Halder, V. K. Peterson, K. W. Chapman, N. Lock, P. D. Southon and C. J. Kepert, Angew. Chem., Int. Ed., 2008, 120, 9061–9064 CrossRef.
- (a) N. Lock, Y. Wu, M. Christensen, L. J. Cameron, V. K. Peterson, A. J. Bridgeman, C. J. Kepert and B. B. Iverson, J. Phys. Chem. C, 2010, 114, 16181–16186 CrossRef CAS; (b) D. Dubbeldam, K. S. Walton, D. E. Ellis and R. Q. Snurr, Angew. Chem., Int. Ed., 2007, 46, 4496–4499 CrossRef CAS PubMed.
- (a) A. L. Goodwin and C. J. Kepert, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 140301 CrossRef; (b) J. L. Korcok, M. L. Katz and D. L. Lenznott, J. Am. Chem. Soc., 2009, 131, 4866–4871 CrossRef CAS PubMed.
- K. W. Chapman and P. J. Chupas, J. Am. Chem. Soc., 2007, 129, 10090–10091 CrossRef CAS PubMed.
- (a) A. L. Goodwin, D. A. Keen, M. G. Tuker, M. T. Love. L. Peters and J. S. O. Evans, J. Am. Chem. Soc., 2008, 130, 9660–9661 CrossRef CAS PubMed; (b) S. G. Duyker, V. K. Peterson, G. J. Kearley, A. J. Ramirez-Cuesta and C. J. Kepert, Angew. Chem., Int. Ed., 2013, 52, 5266–5270 CrossRef CAS PubMed; (c) K. Oka, K. Nabetani, C. Sakaguchi, H. Seki, M. Czapski, Y. Shimakawa and M. Azuma, Appl. Phys. Lett., 2013, 103, 061909 CrossRef; (d) S. Bhattacharya, V. G. Saraswatula and B. K. Saha, Cryst. Growth Des., 2013, 13, 3651–3656 CrossRef CAS; (e) K. W. Chapman, P. J. Chupas and C. J. Kepert, J. Am. Chem. Soc., 2006, 128, 7009–7014 CrossRef CAS PubMed.
- (a) A. E. Phillips, A. L. Goodwin, G. J. Halder, P. D. Southon and C. J. Kepert, Angew. Chem., Int. Ed., 2008, 47, 1396–1399 CrossRef CAS PubMed; (b) A. L. Goodwin, K. W. Chapman and C. J. Kepert, J. Am. Chem. Soc., 2005, 127, 17980–17981 CrossRef CAS PubMed; (c) N. Lock, M. Christensen, C. J. Kepert and B. B. Iversen, Chem. Commun., 2013, 49, 789–791 RSC.
- (a) C. Yang, X. Wang and M. A. Omary, Angew. Chem., Int. Ed., 2009, 48, 2500–2505 CrossRef CAS PubMed; (b) I. Grobler, V. J. Smith, P. M. Bhatt, S. A. Herbert and L. J. Barbour, J. Am. Chem. Soc., 2013, 135, 6411–6414 CrossRef CAS PubMed; (c) V. G. Saraswatula and B. K. Saha, Cryst. Growth Des., 2015, 15, 593–601 CrossRef CAS.
- J. Zhang, Y.-B. Chen, S.-M. Chen, Z.-J. Li, J.-K. Cheng and Y.-G. Yao, Inorg. Chem., 2006, 45, 3161–3163 CrossRef CAS PubMed.
- (a) S. J. Hibble, A. M. Chippindale, A. H. Pohl and A. C. Hannon, Angew. Chem., Int. Ed., 2007, 46, 7116–7118 CrossRef CAS PubMed; (b) L. Zhang, X. Kuang, X. Wu, W. Yang and C. Lu, Dalton Trans., 2014, 43, 7146–7152 RSC; (c) Y.-S. Wei, K.-J. Chen, P.-Q. Liao, B.-Y. Zhu, R.-B. Lin, H.-L. Zhou, B.-Y. Wang, W. Xue, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2013, 4, 1539–1546 RSC.
- (a) G. O. Lloyd, J. Alen, M. W. Bredenkamp, E. J. C. de Vries, C. Esterhuysen and L. J. Barbour, Angew. Chem., Int. Ed., 2006, 45, 5354–5358 CrossRef CAS PubMed; (b) J. L. Atwood, L. J. Barbour and A. Jerga, Science, 2002, 296, 2367–2369 CrossRef CAS PubMed; (c) S. Bhattacharya and B. K. Saha, CrystEngComm, 2011, 13, 6941–6944 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedure, DSC analysis, detailed crystallographic information and additional figures. CCDC 1441452–1441470. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc00158k |
| This journal is © The Royal Society of Chemistry 2016 |