
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational design of multifunctional small-molecule prodrugs for simultaneous suppression of cancer cell growth and metastasis in vitro and in vivo†
Haiyang
Xie‡
a,
Xiao
Xu‡
a,
Jianmei
Chen
a,
Lingling
Li
a,
Jianguo
Wang
a,
Tao
Fang
b,
Lin
Zhou
a,
Hangxiang
Wang
*a and
Shusen
Zheng
a
aThe First Affiliated Hospital, School of Medicine, Key Laboratory of Combined Multi-organ Transplantation, Ministry of Public Health, Key Laboratory of Organ Transplantation of Zhejiang Province, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, Zhejiang University, Hangzhou, 310003, P. R. China. E-mail: wanghx@zju.edu.cn
bJinhua People's Hospital, Jinhua, Zhejiang Province 321000, P. R. China
First published on 21st March 2016
Abstract
We present a design concept for multifunctional prodrugs that simultaneously induce apoptosis and suppress cancer cell metastasis in vitro and in vivo. These “all-in-one” prodrug constructs possess therapeutic potential as novel “integrative” platforms for metastatic cancer treatment.
Cancer remains one of the world's most life-threatening diseases. Systemic chemotherapy is a predominant modality in debulking tumor masses and can also be employed in combination with other therapies.1 However, the successful application of this therapy has been greatly hampered by the non-selectivity of cytotoxic agents, resulting in poor therapeutic indexes and lethal toxicities to the adjacent normal cells and tissues. To address this issue, antibody–drug conjugates (ADCs) have emerged as a promising platform for the targeted delivery of highly potent cytotoxic payloads into cancer cells by harnessing the specificity of monoclonal antibodies.2 Despite their encouraging preclinical activity, the drawbacks of using macromolecular antibodies as targeting motifs are obvious, including poor penetration into solid tumors, immunogenicity, the generation of heterogeneous conjugates and the high cost of ADC production.3 Consequently, there is considerable motivation for the development of an alternative targeting methodology. Compared with antibodies, the utilization of smaller ligands (e.g. peptides, synthetic small molecules) could be much more attractive in overcoming the above-mentioned limitations.4 These ligands can be designed against a tumor-specific antigen or a membrane transporter that is highly over-expressed in various types of cancer. Additionally, these ligands can be coupled to cytotoxic drugs to achieve well-defined stoichiometry, location and chemistry of conjugation using conventional organic synthesis. Indeed, pioneering examples, including drug conjugates that use biotin,5–7 folate8–10 and ligands against carbonic anhydrase IX (CAIX),11 have been reported, representing a promising modality for the targeted delivery of cargos.
During the process of tumor evolution, certain tumor cells acquire metastatic capabilities, thereby facilitating their migration to distant tissues and ultimately surviving at these sites.12,13 Metastasis remains the most common lethal outcome of malignancy, accounting for greater than 90% of cancer-associated mortality. However, most systemically administered cytotoxic therapeutics only reduce tumor growth; they are not capable of suppressing the metastasis of tumor cells. Although much information remains uncharacterized regarding the metastatic process, it is increasingly apparent that a variety of cell-surface molecules are involved in the acquired capability for invasion and metastasis, which are also valuable therapeutic targets for drug development.14,15 However, existing single anticancer agents rarely have the ability to simultaneously kill cancer cells and suppress metastasis. Thus, if we can integrate these functionalities (i.e. anti-proliferation, anti-metastasis and tumor-targeting capability) into a single agent, it would be especially attractive for the management of metastatic cancers from the clinical perspective.
With this motivation in mind, we devised a unique design approach to construct multifunctional prodrug conjugates wherein an apoptosis-inducing chemotherapeutic is combined with a tumor-homing and -penetrating motif, the latter of which also serves as an anti-metastatic agent (Fig. 1a). As a proof of concept, we chose a small cyclic peptidic motif, iRGD (CRGDK/RGPD/EC), that exhibits the capacity to penetrate tumors with its payloads.16 Additionally, it was recently reported that the iRGD motif can inhibit cancer cell metastasis by targeting the corresponding receptors, αν integrins and neuropilin-1, both of which are ubiquitously overexpressed on the surface of cancer cells including hepatocellular carcinoma (HCC) cells.16–18 Therefore, the iRGD motif serves not only as a tumor cell targeting ligand but also as an anti-metastasis agent. In addition, this targeting motif is highly hydrophilic, thus conferring the coupled chemotherapeutics aqueous solubility. To explore the scope of this strategy, three water-insoluble chemotherapeutics, 7-ethyl-10-hydroxycamptothecin (SN38), docetaxel (DTX), and maytansinoid DM1, were separately incorporated into this construct via a bioreductively activatable disulfide linker (Fig. 1b). After careful characterization of the formed prodrug conjugates, their general applicability was demonstrated in in vitro and in vivo experiments.
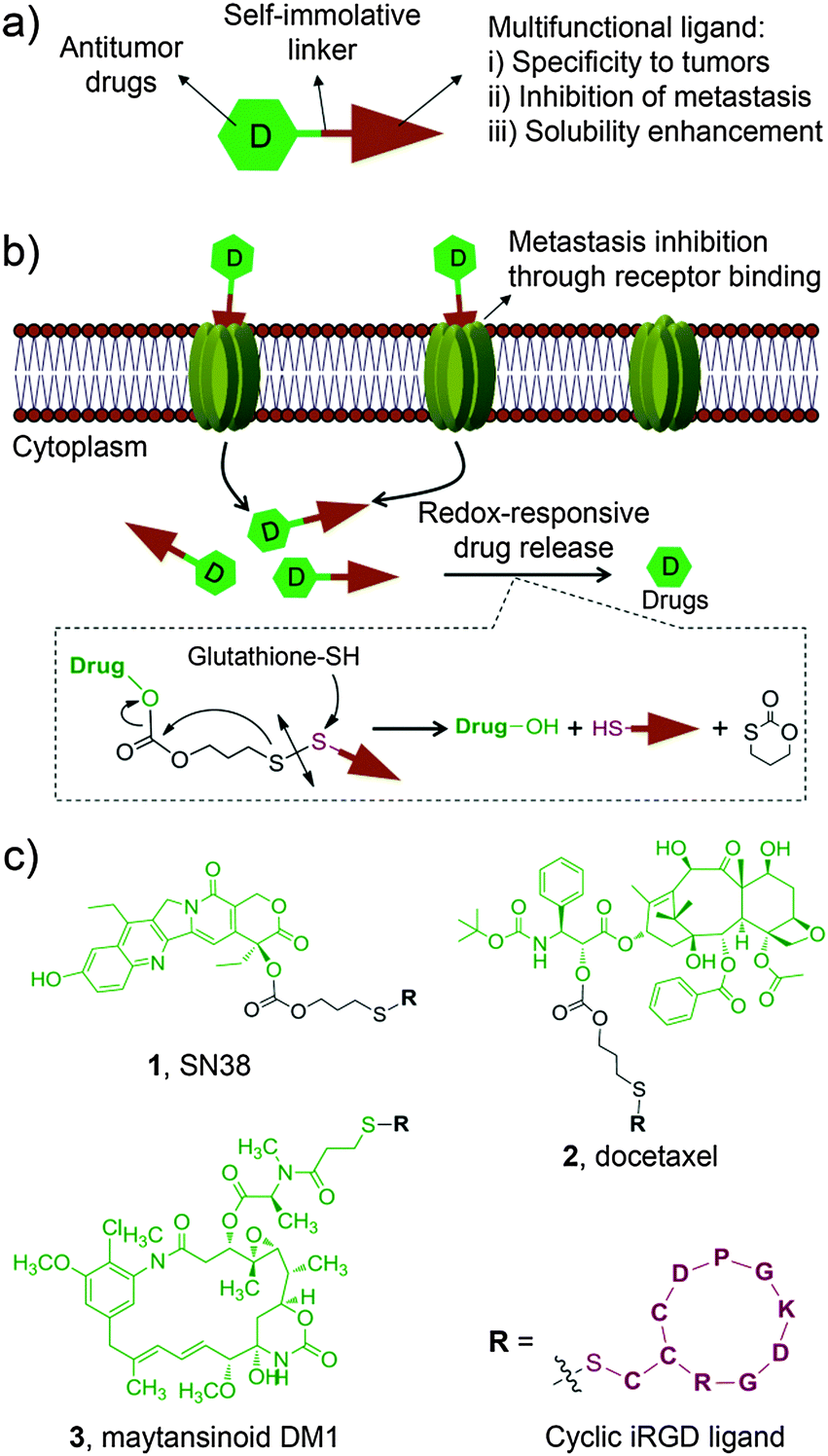 | ||
| Fig. 1 (a) Rational design of multifunctional prodrug conjugates composed of an anticancer chemotherapeutic (SN38, docetaxel or maytansinoid DM1) and a multifunctional tumor-homing and -penetrating peptidic motif via a self-immolative disulfide linker. (b) Tumor cell-specific recognition of prodrug conjugates by cell surface receptors followed by receptor-mediated endocytosis and cleavage of disulfide bonds responsive to intracellular thiols. (c) Molecular structures of synthesized prodrugs 1–3. | ||
The anticancer agents of choice are entirely insoluble in water, thus making them inapplicable for direct clinical use. For instance, to construct water-soluble derivatives of the SN38 molecule, the phenolate group was modified with a bispiperidine moiety to form irinotecan (CPT-11), the only approved prodrug for SN38. However, the hydrolytic release of the active ingredient SN38 requires carboxylesterase-2. This requirement ultimately leads to only 2–8% conversion of the injected dose of CPT-11 in humans,19 implying that alternative chemical strategies should be developed to further improve and fully exploit this compound. Previously, we attempted esterification with lipophilic moieties to reconstruct this agent, thereby enabling SN38-derived prodrugs either to incorporate into the hydrophobic core of amphiphilic block copolymers (e.g. poly(ethylene glycol)-block-poly(D,L-lactic acid))20 or to self-assemble in aqueous media without the use of adjuvant materials.21 We here hypothesized that the hydrophilic iRGD motif possesses the capacity to solubilize the SN38 molecule through covalent chemical conjugation. Moreover, to achieve simultaneous liberation of active drugs in response to reduced glutathione (GSH), which is abundant in mammalian cells, the self-immolative disulfide bond was incorporated as a linker.
For SN38 conjugation, two hydroxyl groups, i.e., 10-OH and 20-OH, could be exploited for chemical modifications. The lactone ring of SN38 can be readily hydrolyzed to an open carboxylate form under biologically relevant conditions, which eliminates its anticancer activity.22 Previous studies indicated that acylation on the 20-OH favors the active lactone form of the prodrugs when circulating in the blood.23 Based on these findings, prodrug 1, in which the iRGD motif was tethered at 20-OH, was initially designed and synthesized as outlined in Scheme S1a (ESI†). Briefly, the phenolic 10-OH was first protected with an acid-labile t-butylcarbonyl (Boc) group by reacting with di-t-butyl dicarbonate to afford compound 7 with an 81% yield. Next, compound 9 bearing a 2-thiopyridyl group was obtained in high yield (52%) using triphosgene. Upon deprotection of the Boc group, derivative 9 can be further converted to the target adduct 1 under mild conditions through substitution with the thiolated iRGD motif. The final prodrug 1 was purified by preparative chromatography for subsequent evaluations. Similarly, we obtained the iRGD-conjugated DTX prodrug 2 with two step reactions. To further explore the feasibility of this approach, we loaded the highly potent cytotoxic compound maytansinoid DM1 to this platform to generate prodrug 3 (Scheme S1b, ESI†). Detailed synthetic procedures, spectroscopic data and purity analysis by high-performance liquid chromatography (HPLC) for 1–3 are provided in the ESI.† All prodrug conjugates 1–3 are highly soluble in saline, thus enabling the systemic administration of therapeutics via intravenous injections. The stabilities of the three conjugates as solids stored at room temperature were also assessed and no hydrolysis was observed for a period extending to months (data not shown).
Having obtained conjugates 1–3, we first verified whether they could spontaneously release therapeutically active compounds upon exposure to intracellular reducing agents, such as GSH, which is present at high concentration (e.g. in a range of 0.5–10 mM) in tumor cells.24 To mimic this condition, prodrug conjugates 1 and 2 were allowed to react with 10 mM of reduced GSH, and the cleavage products were monitored by HPLC. As expected (Fig. S1, ESI†), a burst hydrolytic kinetics for both prodrugs was observed as a consequence of the cleavage of a disulfide bond, releasing approximately 90% of the active drugs within 2 h. The mechanistic study on drug release confirmed that prodrug 1 underwent cyclization by attacking the proximal carbonyl group upon treatment of the reduced GSH, which was evidenced by the production of the metabolite 1,3-oxathian-2-one from HPLC analysis (Fig. 1b and Fig. S2, the details see ESI†). In contrast, after incubating with HEPES buffer (pH 7.4) or the cell culture media containing 20% FBS in the absence of GSH for 2 h, 1 and 2 were fairly stable and showed a nominal cumulative release (less than 18%). This redox-dependent hydrolysis is highly consistent with previous reports,4,25 indicating that this robust linker chemistry could keep the conjugates intact in the bloodstream and concomitantly release the drugs after uptake into the cytosol.
Next, the cytotoxicity using the SN38 conjugate 1 was evaluated with standard MTT assays after 48 or 72 h of drug treatment. In a panel of human tumor cell lines, including human colon carcinoma HT-29 cells, human hepatocellular carcinoma HCC-LM3, BEL-7402, and HepG2 cells, and human lung carcinoma A549 cells, conjugate 1 exhibited high cytotoxicity (Fig. 2 and Fig. S3, Table S1, ESI†). Moreover, this conjugate was approximately 1 to 2 orders of magnitude more effective than clinically used CPT-11 at 48 h incubation, suggesting that the designed construct could bypass the inefficient conversion encountered by CPT-11. In addition, the hydrophilicity imparted by the iRGD motif often impairs the passive cellular uptake of the generated prodrugs; however, we still observed high anti-proliferative activity. This result might contribute to the fact that prodrug 1 can undergo active internalization mediated by the iRGD motif followed by rapid hydrolysis and the spontaneous release of active drugs in cells. As expected, free iRGD alone did not exert anti-proliferative effects in all cancer cells over the concentration range examined (60 μM). We also evaluated the cytotoxicity of iRGD-DTX, 2, against BEL-7402 and HT-29 cells. After 48 h of drug treatment, conjugate 2 also exhibited low IC50 values (77.9 nM for HT-29 and 99.9 nM for BEL-7402, respectively) similar to the results for free DTX (Fig. S4 and Table S2, ESI†). iRGD-DM1, 3, also showed significant anti-proliferative effects in tumor cells (Fig. S5 and Table S3, ESI†). Taken together, the significant in vitro cytotoxicity clearly demonstrated that the iRGD-based platform can bring insoluble anticancer drugs into aqueous solution and effectively release active components by the cleavage of the disulfide linkage in response to intracellular thiols.
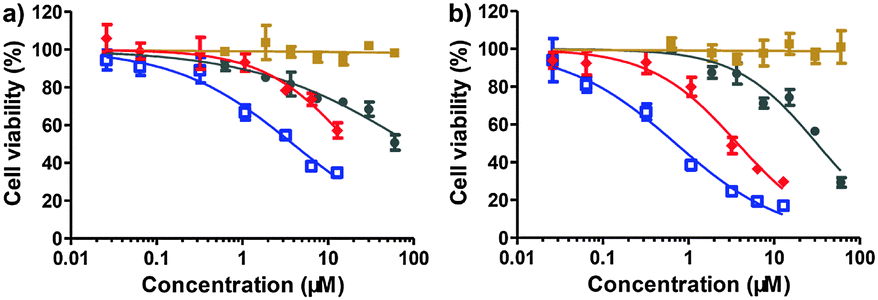 | ||
| Fig. 2 HCC-LM3 cell viability after 48 h (a) or 72 h (b) treatment with CPT-11 (gray), free iRGD (brown), free SN38 (blue), and iRGD-SN38, 1 (red) as measured by standard MTT assay (mean ± SEM). | ||
Metastasis is a major obstacle to anticancer therapies and responsible for most therapeutic failures. Migration and invasion are critical steps in the metastatic cascade.26 It was recently reported that iRGD provides cues to retract cellular protrusions and consequently repels migration of tumor cell populations.17 As we conceive it, in addition to enabling the inhibition of cancer cells through the apoptotic mechanism, the prodrug conjugates bearing the unique iRGD motif could also exert its inhibitory effect on both migration and invasion processes. To verify this hypothesis, we conducted an in vitro motility assay exploiting the iRGD-decorated conjugate 1 as a model agent. The human umbilical vein endothelial cell (HUVEC) involved in tumor vessel formation and invasive HCC-LM3 cells were chosen as in vitro models to evaluate drugs. In wound healing assays, the migration of HUVECs was significantly reduced upon 24 and 48 h treatment of conjugate 1 compared to free SN38 (Fig. 3a and b; for the detailed experimental procedures, see the ESI†); moreover, human HCC-LM3 cancer cells, a highly metastatic cell line, also exhibited similar patterns of wound closure upon drug administration (Fig. S6, ESI†). Compared with free SN38, iRGD-SN38 1 showed a stronger inhibition of migration in HCC-LM3 cells (30.5% vs. 39.1% after 72 h incubation) and HUVECs (29.3% vs. 39.6% and 41.8% vs. 48.9% for 24 and 48 h, respectively) (Fig. 3b).
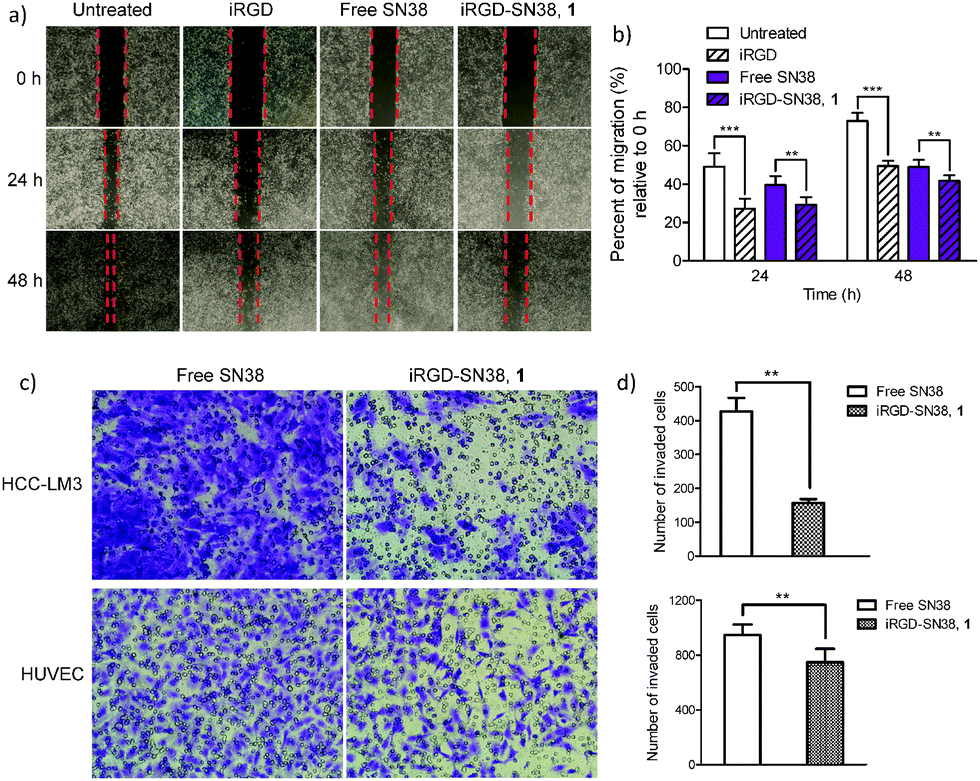 | ||
| Fig. 3 iRGD-decorated prodrug conjugate 1 impaired the metastatic ability of HUVECs and HCC-LM3 cells. (a and b) Wound healing assays indicated that 1 produced a remarkable decrease in the migratory ability of HUVECs. (c and d) Two-chamber Transwell assays revealed that fewer cells invaded through the Transwell filter upon treatment with 1. **p < 0.01, ***p < 0.001. | ||
To further verify the potential for inhibiting invasion, in vitro Transwell assays were conducted. As shown in Fig. 3c and d, conjugate 1 is effective at decreasing the number of invaded cells compared with free SN38 for both HCC-LM3 (427 vs. 157; p < 0.01) and HUVEC cells (946 vs. 623; p < 0.001). These results clearly indicate that iRGD retains its anti-metastatic activity regardless of covalent chemical conjugation (Fig. S7, ESI†).
Coadministration of free iRGD augmented the delivery of sorafenib and doxorubicin in HCC-derived xenograft models, which was demonstrated in a recent report.27 Motivated by this finding, we therefore evaluated the therapeutic effects of prodrug 1 on both tumor inhibition and metastasis. Because HCC-LM3 is a highly invasive cancer cell, which undergoes lung metastases after orthotopic implantation,28 we thus exploit this xenograft-bearing mouse model (see ESI,† for details). Obviously, conjugate 1 suppressed primary tumor growth. At day 25 post-administration, the tumor masses in mice receiving 1 was significantly reduced as compared with mice treated with saline (p < 0.01). In addition, the incidence of lung metastasis of HCC-LM3 was assessed by staining with hematoxylin and eosin. As shown in Fig. 4b and c, the average number of lung metastatic lesions in the 1-treated group was noticeably lower than that of CPT-11 or saline-treated groups (p < 0.01). As a comparison, iRGD alone barely reduced orthotopic tumor masses, but significantly inhibited lung metastases. Collectively, these results strongly supported that the water-soluble platform 1 remained functional in vivo, and that it led to not only the reduction of tumor growth but also the blockade of metastasis.
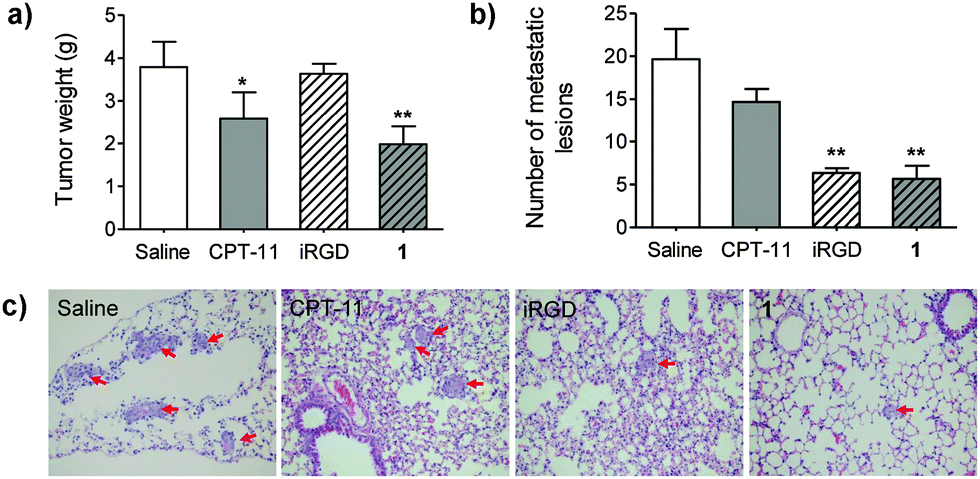 | ||
| Fig. 4 The therapeutic effects of 1 on in vivo tumor growth and metastasis in the orthotopic implantation BALB/c nude mouse model of HCC-LM3. (a) Tumor masses at day 25 after treatment. Mice received five intravenous injection of 6 mg kg−1 CPT-11, 13 mg kg−1 iRGD and 20 mg kg−1 iRGD-SN38, 1 (5 mg kg−1 SN38 equivalent). Saline was injected as control. (b) The number of metastatic lesions in mice groups at week 7 after implantation (n = 3 for each group). (c) iRGD and 1 suppressed lung metastasis in this model. Metastatic lesions are indicated in red arrows. *p < 0.05. **p < 0.01. | ||
In conclusion, we successfully constructed unique multifunctional prodrug platforms composed of potent cytotoxic agents and a tumor-homing and -penetrating peptidic motif, iRGD. The latter component also serves as an enhancer for the drug's aqueous solubility and as an inhibitor of cancer cell invasion and migration. Our results clearly demonstrated that iRGD-tethered prodrug could function well in HCC xenograft-bearing mice in terms of tumor growth and metastasis-inhibitory effects. In addition, the reductive thiol-responsive release behavior and specificity endowed with the addition of the iRGD motif can be expected to release drugs focally within tumor cells of interest. Remarkably, we have readily adapted the potent cytotoxic payload DM1 to this platform; it is worthwhile to evaluate the in vivo therapeutic value of this conjugate, including anti-proliferation and anti-metastasis activities in future work, as recent studies have demonstrated that DM1 derivatives tethered to tumor antigen-specific small-molecule ligands exhibit potent antitumor effects in mouse models.11,29 Overall, our “all-in-one” prodrug design strategy and the resulting constructs may have therapeutic potential as an “integrative” platform for further evaluation in metastatic cancers.
This work was supported by the National Natural Science Foundation of China (No. 81571799, 21202147, and 81421062), the National S&T Major Project of China (2012ZX10002017), the Major Science and Technology Project of Zhejiang Province (2014C03043-2), and the Natural Science Foundation of Zhejiang Province (No. LY13H160004 and LY13H090013).
Notes and references
- B. A. Chabner and T. G. Roberts, Nat. Rev. Cancer, 2005, 5, 65–72 CrossRef CAS PubMed.
- R. V. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796–3827 CrossRef CAS PubMed.
- B. A. Teicher and R. V. Chari, Clin. Cancer Res., 2011, 17, 6389–6397 CrossRef CAS PubMed.
- N. Krall, J. Scheuermann and D. Neri, Angew. Chem., Int. Ed., 2013, 52, 1384–1402 CrossRef CAS PubMed.
- W. X. Ren, J. Han, S. Uhm, Y. J. Jang, C. Kang, J. H. Kim and J. S. Kim, Chem. Commun., 2015, 51, 10403–10418 RSC.
- S. Park, E. Kim, W. Y. Kim, C. Kang and J. S. Kim, Chem. Commun., 2015, 51, 9343–9345 RSC.
- S. Bhuniya, S. Maiti, E. J. Kim, H. Lee, J. L. Sessler, K. S. Hong and J. S. Kim, Angew. Chem., Int. Ed., 2014, 53, 4469–4474 CrossRef CAS PubMed.
- Z. Yang, J. H. Lee, H. M. Jeon, J. H. Han, N. Park, Y. He, H. Lee, K. S. Hong, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 11657–11662 CrossRef CAS PubMed.
- T. Legigan, J. Clarhaut, I. Tranoy-Opalinski, A. Monvoisin, B. Renoux, M. Thomas, A. Le Pape, S. Lerondel and S. Papot, Angew. Chem., Int. Ed., 2012, 51, 11606–11610 CrossRef CAS PubMed.
- S. Santra, C. Kaittanis, O. J. Santiesteban and J. M. Perez, J. Am. Chem. Soc., 2011, 133, 16680–16688 CrossRef CAS PubMed.
- N. Krall, F. Pretto, W. Decurtins, G. J. Bernardes, C. T. Supuran and D. Neri, Angew. Chem., Int. Ed., 2014, 53, 4231–4235 CrossRef CAS PubMed.
- C. L. Chaffer and R. A. Weinberg, Science, 2011, 331, 1559–1564 CrossRef CAS PubMed.
- N. Sethi and Y. Kang, Nat. Rev. Cancer, 2011, 11, 735–748 CrossRef CAS PubMed.
- L. Seguin, J. S. Desgrosellier, S. M. Weis and D. A. Cheresh, Trends Cell Biol., 2015, 25, 234–240 CrossRef CAS PubMed.
- A. Bagri, M. Tessier-Lavigne and R. J. Watts, Clin. Cancer Res., 2009, 15, 1860–1864 CrossRef CAS PubMed.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, O. M. Girard, D. Hanahan, R. F. Mattrey and E. Ruoslahti, Cancer Cell, 2009, 16, 510–520 CrossRef CAS PubMed.
- K. N. Sugahara, G. B. Braun, T. H. de Mendoza, V. R. Kotamraju, R. P. French, A. M. Lowy, T. Teesalu and E. Ruoslahti, Mol. Cancer Ther., 2015, 14, 120–128 CrossRef CAS PubMed.
- M. Bergé, D. Allanic, P. Bonnin, C. Montrion, J. Richard, M. Suc, J.-F. Boivin, J.-O. Contrerès, B. P. Lockhart, M. Pocard, B. I. Lévy, G. C. Tucker, G. Tobelem and T. Merkulova-Rainon, J. Hepatol., 2011, 55, 866–875 CrossRef PubMed.
- P. D. Senter, K. S. Beam, B. Mixan and A. F. Wahl, Bioconjugate Chem., 2001, 12, 1074–1080 CrossRef CAS PubMed.
- H. Wang, H. Xie, J. Wu, X. Wei, L. Zhou, X. Xu and S. Zheng, Angew. Chem., Int. Ed., 2014, 53, 11532–11537 CrossRef CAS PubMed.
- H. Wang, H. Xie, J. Wang, J. Wu, X. Ma, L. Li, X. Wei, Q. Ling, P. Song, L. Zhou, X. Xu and S. Zheng, Adv. Funct. Mater., 2015, 25, 4956–4965 CrossRef CAS.
- V. Bala, S. Rao, B. J. Boyd and C. A. Prestidge, J. Controlled Release, 2013, 172, 48–61 CrossRef CAS PubMed.
- R. H. Mathijssen, R. J. van Alphen, J. Verweij, W. J. Loos, K. Nooter, G. Stoter and A. Sparreboom, Clin. Cancer Res., 2001, 7, 2182–2194 CAS.
- T. Wang, D. Y. Ng, Y. Wu, J. Thomas, T. TamTran and T. Weil, Chem. Commun., 2014, 50, 1116–1118 RSC.
- Y. Zhang, Q. Yin, J. Yen, J. Li, H. Ying, H. Wang, Y. Hua, E. J. Chaney, S. A. Boppart and J. Cheng, Chem. Commun., 2015, 51, 6948–6951 RSC.
- P. Friedl and K. Wolf, Nat. Rev. Cancer, 2003, 3, 362–374 CrossRef CAS PubMed.
- C. Schmithals, V. Köberle, H. Korkusuz, T. Pleli, B. Kakoschky, E. A. Augusto, A. A. Ibrahim, J. M. Arencibia, V. Vafaizadeh, B. Groner, H.-W. Korf, B. Kronenberger, S. Zeuzem, T. J. Vogl, O. Waidmann and A. Piiper, Cancer Res., 2015, 75, 3147–3154 CrossRef CAS PubMed.
- Y. Li, Z. Tang, S. Ye, B. Liu, Y. Liu, J. Chen and Q. Xue, J. Cancer Res. Clin. Oncol., 2003, 129, 43–51 CrossRef CAS PubMed.
- N. Krall, F. Pretto and D. Neri, Chem. Sci., 2014, 5, 3640–3644 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic procedures and characterization data of all compounds. See DOI: 10.1039/c5cc10367c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |