
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystallinity-dependence of ionic conductivity in the ion pairs of a multi-interactive anion†
Gil Ryeong
Lee
a,
Hiroyoshi
Ohtsu
 a,
Jinyoung
Koo
a,
Yumi
Yakiyama‡
*a,
Moon Jeong
Park
b,
Daishi
Inoue
c,
Daisuke
Hashizume
c and
Masaki
Kawano§
*a
a,
Jinyoung
Koo
a,
Yumi
Yakiyama‡
*a,
Moon Jeong
Park
b,
Daishi
Inoue
c,
Daisuke
Hashizume
c and
Masaki
Kawano§
*a
aThe Division of Advanced Materials Science, Pohang University of Science and Technology (POSTECH), RIST Building 3, 77 Cheongam-Ro, Nam-Gu, Pohang 790-784, South Korea. E-mail: mkawano@postech.ac.kr; Fax: +82 54 279 8739; Tel: +82 54 279 8740
bDepartment of Chemistry, Pohang University of Science and Technology (POSTECH), 77 Cheongam-Ro, Nam-Gu, Pohang 790-784, South Korea
cMaterials Characterization Support Unit, RIKEN Center for Emergent Matter Science (CEMS), 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
First published on 9th February 2016
Abstract
Ammonium and sodium salts (ion pairs) of a multi-interactive tri(4-pyridyl)hexaazaphenalenyl anion (TPHAP−) showed completely different ion-conductive properties depending on the crystal structure. TPHAP columnar crystals showed a high conductivity of 10−3 S cm−1 while retaining their structures even under humid conditions, whereas TPHAP dimer crystals exhibited a conductivity of ∼10−5 S cm−1 with crystallinity deterioration. The main unit structures induced by multi-interactivity realized different water accessibility, which explains the differences in their ion conductivity and stability against humidity.
Intermolecular interactions play critical roles in functional materials, crystal engineering, and biological systems.1–3 In such systems, molecules that have multi-point interactions are important components because they can stabilize meta-stable states4,5 by decreasing a local minimum energy state to trap kinetic states. For this purpose, we designed a multi-interactive molecule, the tri(4-pyridyl)hexaazaphenalenyl anion (TPHAP−) which has multi-point interaction sites composed of nine nitrogen atoms in the same π-plane.6 Using this molecule, we trapped a kinetic coordination network composed of the TPHAP ligand6a and prepared diverse coordination networks from the same crystallization setup by changing only solvent or additives.6b Importantly, the potassium salt KTPHAP shows humidity-dependent conductivity with reversible structural transformation by hydration/dehydration.7 This result indicates that water and K+ strongly influence structural change via interaction with TPHAP−.
Here we report the ion conducting properties of two new TPHAP− salts, ion pairs of NH4TPHAP and NaTPHAP, which were isolated as two kinds of phases; highly water-stable phase A and phase B which can readily deteriorate under high humidity. At 95% RH, the A-phases were more conductive than the B-phases by two orders of magnitude. The existence of different crystalline phases indicates that we can prepare various structures by exploiting the multi-interactive nature of TPHAP−. These properties in both phases can be explained by considering the main structural unit, which is a 1-D column or a dimer of the TPHAP ion pair. Water accessibility strongly affects their water-stable crystallinity and the ion conductivity.
We obtained two crystalline phases of both NH4TPHAP and NaTPHAP with clearly different features: one phase keeps high crystallinity (denoted by NH4 (or Na) TPHAP-A) under humid conditions; the other has a low crystallinity (NH4 (or Na) TPHAP-B). NH4TPHAP-A single crystals were obtained by slow recrystallization from dilute MeOH solution; NH4TPHAP-B single crystals were obtained by quick recrystallization from concentrated MeOH solution. NaTPHAP-A crystalline powder was obtained by vapour diffusion using MeOH solution and ethyl acetate; NaTPHAP-B single crystals were obtained using the same method with more ethyl acetate vapour in a smaller crystallization vial than in the case of the NaTPHAP-A crystal (Fig. S1, ESI†). This sensitivity of crystallization to conditions indicates that TPHAP− can respond to very small change in environment effects by forming completely different structures.5,6
The crystal structures of NH4TPHAP-A and NaTPHAP-A were solved by single-crystal X-ray analysis and by ab initio X-ray powder diffraction (XRPD) analysis (ESI†),8 respectively (Fig. 1). Both phases are stable under air and have the same structural features; owing to the highly symmetrical structure of TPHAP−, they are spread onto a 2-D layer in the hexagonal shape with the formation of pores surrounded by six pyridine rings (Fig. 1a and c, red-dotted line). Both NH4TPHAP-A and NaTPHAP-A form π–π stacking columns at hexaazaphenalene (HAP) skeletons9 along the b- and the c-axes with the distances of 3.39 Å and 3.34 Å, respectively (Fig. 1b and d). The remaining open spaces are occupied by counter cations (NH4+, Na+) and severely-disordered water molecules. Two disordered NH4+ in NH4TPHAP-A loosely bind TPHAP− by hydrogen bonding (H-bonding) interactions; N1A(NH4+)⋯N(pyridine): 2.891(2) Å; N2A(NH4+)⋯N(HAP): 2.990(4)–3.485(4) Å (Fig. S2, ESI†).
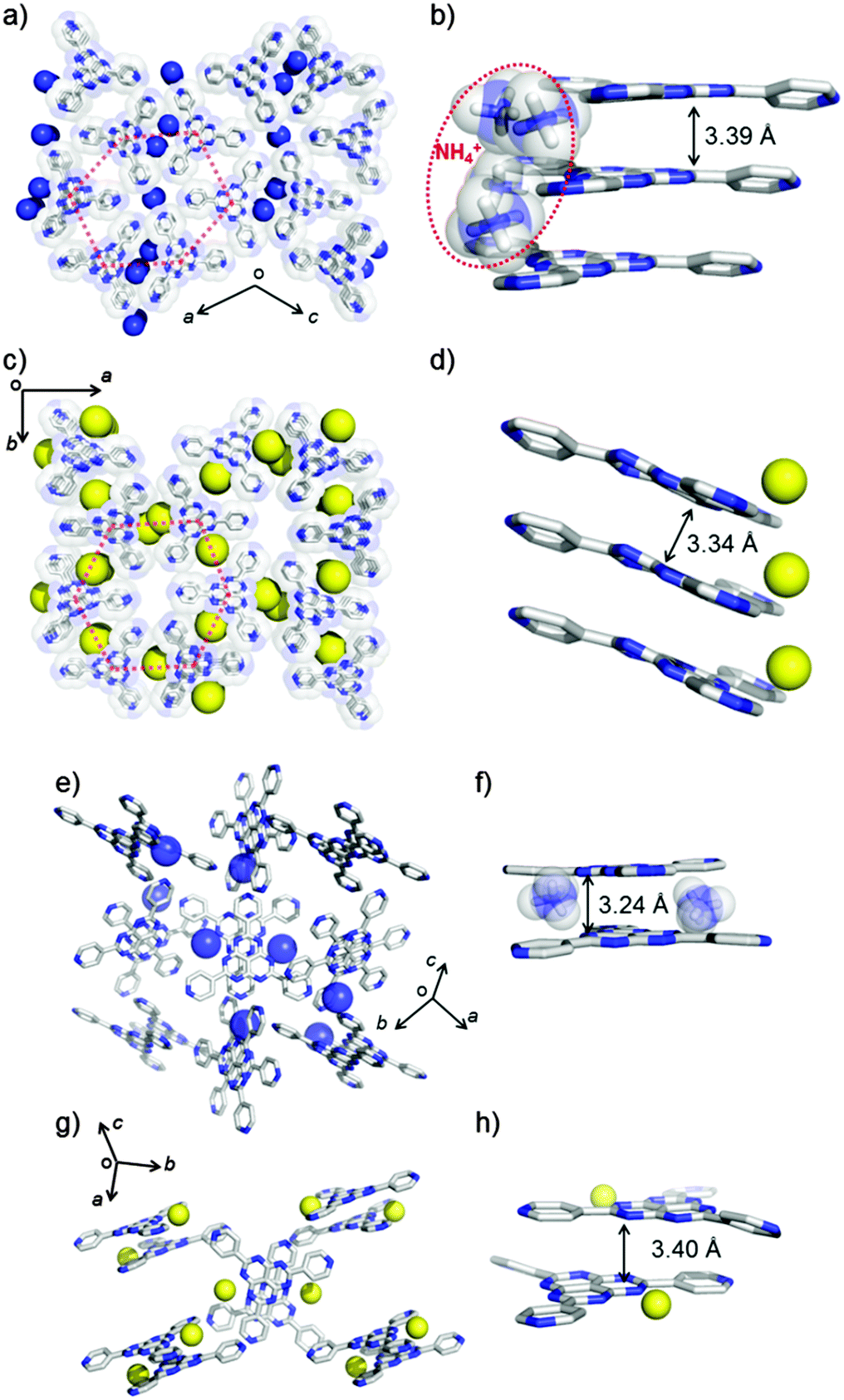 | ||
| Fig. 1 Initial crystal structures. (a) Overview of the NH4TPHAP-A structure; b-axis projection, (b) Infinite π–π stacking of NH4TPHAP-A, (c) overview of the NaTPHAP-A structure; c-axis projection, (d) infinite π–π stacking of NaTPHAP-A, (e) overview of NH4TPHAP-B structure, (f) dimer unit of NH4TPHAP-B, (g) overview of the NaTPHAP-B structure, (h) dimer unit of NaTPHAP-B. Hydrogen and crystal solvents are omitted for clarity. Colour code: C, grey; N, blue; Na+; yellow sphere, NH4+; blue sphere. | ||
Similarly, the structure of NaTPHAP-A has disordered Na+ and loose binding; the Na+⋯N(HAP) distance is in the range 3.0(3)–3.6(2) Å (Fig. S3, ESI†). In contrast, both of the single crystals of NH4TPHAP-B and NaTPHAP-B consist of dimer units of TPHAP− which are bound by counter cations (Fig. 1e–h). In the crystal structure of NH4TPHAP-B, which is the isostructure of KTPHAP, all dimer units bridged by NH4+ are strongly connected by NH4+ (N(NH4+)⋯N(HAP) and the bond distance is in the range 2.987(1)–3.189(1) Å) (Fig. 1e and f and Fig. S4, ESI†).7 This tight binding protects the crystal from disruption by interaction with air and escape of water from the system. In contrast, in single crystals of NaTPHAP-B, the dimers are formed by dipole–dipole interactions and simple π–π stacking between TPHAP−s. Furthermore, interaction between the dimers is too weak to maintain crystallinity. Therefore, its crystallinity drastically decreases in air because of MeOH escape (Fig. S5, ESI†).
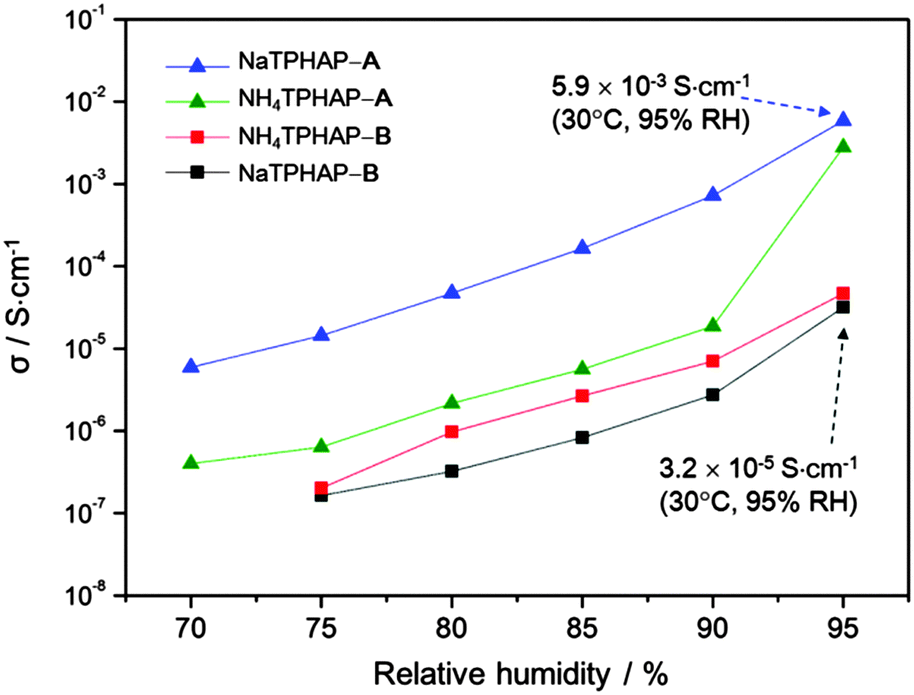 | ||
| Fig. 2 Ionic conductivity σ of different crystalline phases of NaTPHAP and NH4TPHAP measured at 30 °C versus relative humidity. Note that the highest ionic conductivity values in NaTPHAP-A and NaTPHAP-B. (Detailed values are described in Table S1, ESI†). | ||
The ion conductivities of these materials were significantly correlated with the initial structure and atmospheric humidity (Fig. 2). We used ac impedance spectroscopy to measure the conductivity of compressed pellets of each crystal under various relative humidities (RH). The conductivities of all materials increased by more than 2 orders of magnitude as humidity became higher. These observations clearly indicate that water adsorption accelerates ion conduction. In addition, the conductivity measurement under deuterated water conditions (95% RH, 30 °C) gave similar conductivities (NH4TPHAP-A; 3.0 × 10−3 S cm−1, NaTPHAP-A; 9.8 × 10−4 S cm−1, NH4TPHAP-B; 2.2 × 10−5 S cm−1, NaTPHAP-B; 1.3 × 10−5 S cm−1, ESI†) to those which were measured under normal water conditions. These results indicate that the main charge carrier is not proton. Therefore, the possible mechanism of ion conduction in these systems was based on the migration of Na+ and NH4+. Their migration can be accelerated by water adsorption which reduces the interaction between the ions and the framework. A-phases showed drastic conductivity changes from negligible (insulating) at 50% RH to 10−3 S cm−1 at 95% RH. These high conductivities are comparable to cation-exchanged Nafion.10 Na+ ions are less mobile (5.19 × 10−8 m2 s−1 V−1) than NH4+ ions (7.63 × 10−8 m2 s−1 V−1) in water.11 However, NaTPHAP-A showed a higher conductivity (5.9 × 10−3 S cm−1) than NH4TPHAP-A at 95% RH, though the crystal types have similar molecular packing. This is reasonable because larger interaction energy of Na+ with water enabled easier water adsorption than NH4+.12 In addition, the conductivity of NH4TPHAP-A increased abnormally under 95% RH; this result is due to both the stronger stabilization effect by coulombic interaction of NH4+⋯N (HAP or pyridine) (Tables S3 and S4, ESI†) and the weaker interaction energy of NH4+ with water than the Na+ system, which prevented the effective migration of NH4+ within the system. In contrast, TPHAP-dimer-based NH4TPHAP-B and NaTPHAP-B crystals showed lower conductivities ≤3.2 × 10−5 S cm−1 at 95% RH.
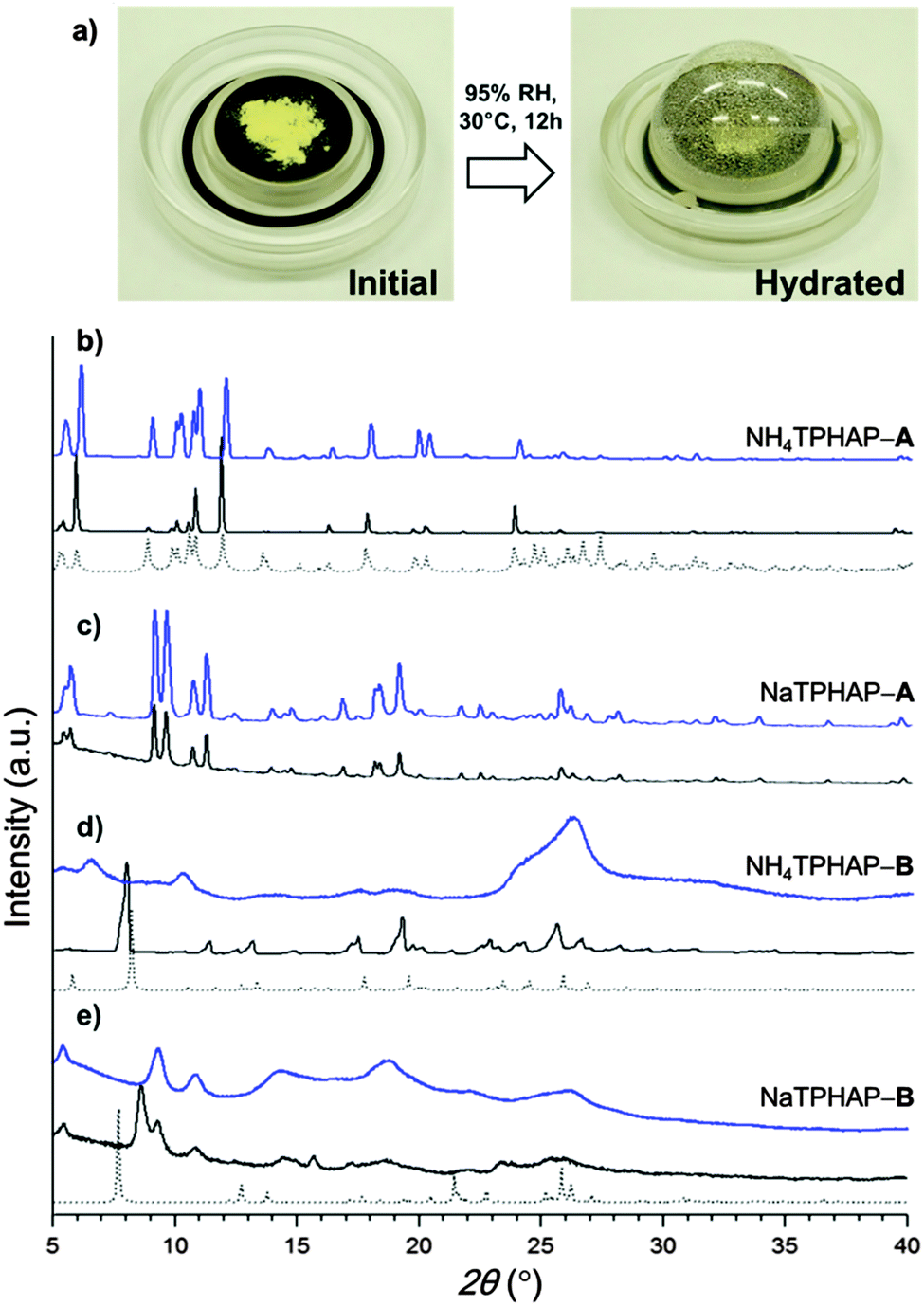 | ||
| Fig. 3 XRPD patterns. Simulated pattern from the single crystal structure (dotted), initial powder (black) and hydrated powder (blue). (a) Experimental setting of XRPD measurement, (b) NH4TPHAP-A, (c) NaTPHAP-A, (d) NH4TPHAP-B, (e) NaTPHAP-B. | ||
We examined XRPD patterns under humid conditions to investigate the hydration effect against the crystal structures and their conductivities (Fig. 3). NH4TPHAP-A and NaTPHAP-A remained crystalline even at 95% RH, as does KTPHAP.7 To our surprise, none of the A-phases showed significant pattern change. In contrast, the crystallinity of NH4TPHAP-B and NaTPHAP-B decayed during hydration at 95% RH and 30 °C for 12 h. These different responses of crystallinity to hydration can be explained by their initial crystal structure, especially the π–π stacking columnar structure or the dimer structure.
A-phases have a stable columnar structure of TPHAP and each column is loosely bound by counter ions; the result is a well-packed structure. This structural feature prevents water solvation which severely degrades the crystallinity. Instead, water can enter only those spaces in the crystals in which the cations are disordered but maintain interactions with TPHAP− ((Na+ or NH4+)⋯N(HAP)) even though their binding energies can be reduced. As a result, at 95% RH, the diffusion rate of the counter cations increases so the ion conductivity becomes high. In contrast, TPHAP dimer-based B-phase crystals include more free space than do the A-phase, and provide more opportunity for water adsorption. Therefore, water adsorption causes the deterioration of initially ordered structures. However, the lower conductivity values of B-phase crystals than those of A-phase ones seem to be inconsistent because water adsorption can increase the diffusion rates of the cations which are linearly related to conductivity.13 The most reasonable explanation for this contradiction is the high stability of the TPHAP− and cation pair. Indeed, the complex can be clearly observed in the CS-ESI-MS spectrum using MeOH.6b This strong coulombic interaction impedes diffusion of the counter cations in water. An additional possibility is suggested by the slight increase of full width at half maximum with no change in intensity of XRPD patterns, indicating formation of finer powder by hydration. In fact, we confirmed the decrease of the particle size using an optical microscope. The observation suggests that a grain boundary effect may be operational: that water adsorption occurs mainly on the crystalline surface, so the grain boundary provides an effective conduction path.
In summary, we exploited the multi-interactive character of TPHAP− to prepare two crystalline phases of NH4TPHAP and of NaTPHAP. The crystallinity and structural features of ion pairs were closely related to their conductivity. The A-phases with infinite π–π stacking structure resisted degradation by high humidity conditions and have high conductivity. However, TPHAP-dimer based B-phases lost their crystallinity by hydration and showed low conductivity. This work emphasizes the importance of molecular interactions in the design of hydrous ionic conducting systems.
The authors acknowledge funding from the Veteran researcher grant (No. 2014R1A2A1A11049978) and the framework of international cooperation program (No. 2014K2A2A4001500) managed by the National Research Foundation of Korea (NRF). The X-ray diffraction study with synchrotron radiation was performed at the Pohang Accelerator Laboratory (Beamline 2D) supported by POSTECH. This work was approved by SPring-8 (Proposals 2014B1022).
Notes and references
-
Supramolecular Chemistry, ed. J. W. Steed and J. L. Atwood, Wiley, UK, 2nd edn, 2009 Search PubMed
.
-
(a) D. Stock, A. G. W. Lesile and J. E. Walker, Science, 1999, 286, 1700–1705 CrossRef CAS PubMed
- G. D. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed
-
(a) M. M. Gromiha and S. Selevaraj, Prog. Biophys. Mol. Biol., 2004, 86, 235–277 CrossRef CAS PubMed
-
(a) M. Kawano, T. Haneda, D. Hashizume, F. Izumi and M. Fujita, Angew. Chem., Int. Ed., 2008, 47, 1269–1271 CrossRef CAS PubMed
-
(a) Y. Yakiyama, A. Ueda, Y. Morita and M. Kawano, Chem. Commun., 2012, 48, 10651–10653 RSC
- Y. Yakiyama, G. R. Lee, S. Y. Kim, Y. Matsushita, Y. Morita, M. J. Park and M. Kawano, Chem. Commun., 2015, 51, 6828–6831 RSC
-
(a) M. Takata, B. Umeda, E. Noshibori, M. Sakata, Y. Saito, M. Ohno and H. Shinohara, Nature, 1995, 377, 46–49 CrossRef CAS
-
(a) S. Suzuki, Y. Morita, K. Fukui, K. Sato, D. Shiomi, T. Takui and K. Nakasuji, Inorg. Chem., 2005, 44, 8197–8199 CrossRef CAS PubMed
- T. Okada, H. Satou, M. Okuno and M. Yuasa, J. Phys. Chem. B, 2002, 106, 1267–1273 CrossRef CAS
-
(a)
Tables of physical and chemical constants, ed. G. W. C. Kaye and T. H. Laby, Longman, London, UK, 1973 Search PubMed
- A. G. Volkov, S. Paula and D. W. Deamer, Bioelectrochem. Bioenerg., 1997, 42, 153–160 CrossRef CAS
- Diffusion constant D can be described as D = σkT/Cz2e2. Here σ is ion conductivity, k is Boltzmann constant, C is carier concentration, z is ion valency and e is elementary charge. See,
Modern Electro Chemistry vol 1: Ionics, ed. J. O. Bockris and A. K. N. Reddy, Springer, 2nd edn, 1998 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the experimental procedures, preparation of sample and ion conductivity results. CCDC 1436930–1436933. For ESI and crystallographic data in CIF or other electronic formation see DOI: 10.1039/c5cc10136k |
| ‡ Present address: Division of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan. |
| § Present address: Department of Chemistry, Graduate School of Science and Engineering, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: E-mail: mkawano@chem.titech.ac.jp. |
| This journal is © The Royal Society of Chemistry 2016 |