
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The interplay of thermally activated delayed fluorescence (TADF) and room temperature organic phosphorescence in sterically-constrained donor–acceptor charge-transfer molecules†
Jonathan S.
Ward‡
a,
Roberto S.
Nobuyasu‡
b,
Andrei S.
Batsanov
a,
Przemyslaw
Data
b,
Andrew P.
Monkman
b,
Fernando B.
Dias
b and
Martin R.
Bryce
*a
aDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
bDepartment of Physics, Durham University, Durham, DH1 3LE, UK
First published on 11th January 2016
Abstract
A series of phenothiazine–dibenzothiophene-S,S-dioxide charge-transfer molecules have been synthesized. Increasing steric restriction around the donor–acceptor bond significantly alters contributions from TADF and phosphorescence. Bulky substituents on the 1-(and 9) position(s) of the phenothiazine result in no TADF in the solid state; instead strong phosphorescence is observed at ambient temperature.
The study of new fluorescent and phosphorescent emitters is central to the development of a wide range of optoelectronic technologies, including organic light-emitting diodes (OLEDs), electrochromic devices, sensors and fluorescence imaging.1 Spin statistics dictate that the efficiency of traditional fluorescent emitters which utilise only singlet excited states for light emission is limited to 25%.2 Phosphorescent emitters based on transition metal complexes, especially third row ions such as Ir(III) and Pt(II), are popular because the heavy metal facilitates spin–orbit coupling and harvesting of both singlet and triplet excitons by enhanced intersystem crossing (ISC). Internal quantum efficiencies (IQEs) approaching 100% can be achieved by this mechanism.3 However, these phosphors suffer from limited stability in practical applications, especially for blue and white emission, and the scarcity and expense of precious metals restricts their commercial applications.
Triplet fusion is an alternative way to convert triplets into emissive singlet states although the IQE in this process is limited to a theoretical maximum of 62.5%.4 Currently, a leading strategy is to exploit thermally activated delayed fluorescence (TADF) to convert triplets to singlet states with a potential IQE of up to 100% using all-organic (metal-free) materials.5 The mechanism of TADF is fundamentally different from traditional fluorescence. TADF uses thermal energy to assist reverse intersystem crossing (RISC) of lower-energy triplet states into emissive singlets. Two main parameters control this mechanism: (i) the energy splitting between the lowest singlet (S1) and triplet (T1) states should be minimised, and (ii) the non-radiative pathways by which the excited singlet and triplet states can decay need to be suppressed in order to achieve high fluorescence quantum yields and long triplet excited state lifetimes.6
Based on the TADF properties of a wide range of donor–acceptor (D–A) molecules,7–15 it is clear that a prerequisite for efficient TADF is the spatial separation of the highest occupied molecular orbital (HOMO) on the electron donor and the lowest unoccupied molecular orbital (LUMO) on the electron acceptor moieties of the emitter molecule. This can be achieved by enforcing a twist between the planes of the D and A moieties to restrict the conjugation leading to an intramolecular charge transfer (ICT) state with a small (ΔES1T1) energy gap.7 For example, tuning of TADF in the green-orange region has been achieved by stabilisation of the ICT state between a phenoxazine donor unit and 2,4,6-triphenyl-1,3,5-triazine acceptor unit separated by a large twist angle.8 A 9,9′-spirobifluorene unit has been used to enforce orthogonality between D and A units in a TADF emitter.9 Systematically engineering the twist angles within a series of D–A structures is, therefore, an important strategy for understanding the parameters which control the overall emission, including TADF, in these types of molecules.
We now report a series of D–A–D and D–A molecules based on phenothiazine donor and dibenzothiophene-S,S-dioxide acceptor units in which the steric bulk of alkyl groups on the donor restricts rotation around the C–N bond linking the D and A units (Fig. 1). Remarkable effects of this functionalization on the structural and photophysical properties of the molecules are observed. Compounds 116a and 616b exhibit efficient TADF in zeonex films whereas the more sterically hindered analogues 3 and 4, which have less conformational flexibility, show strong contributions from phosphorescence, even at RT.
 | ||
| Fig. 1 Structures of D–A–D and D–A molecules. | ||
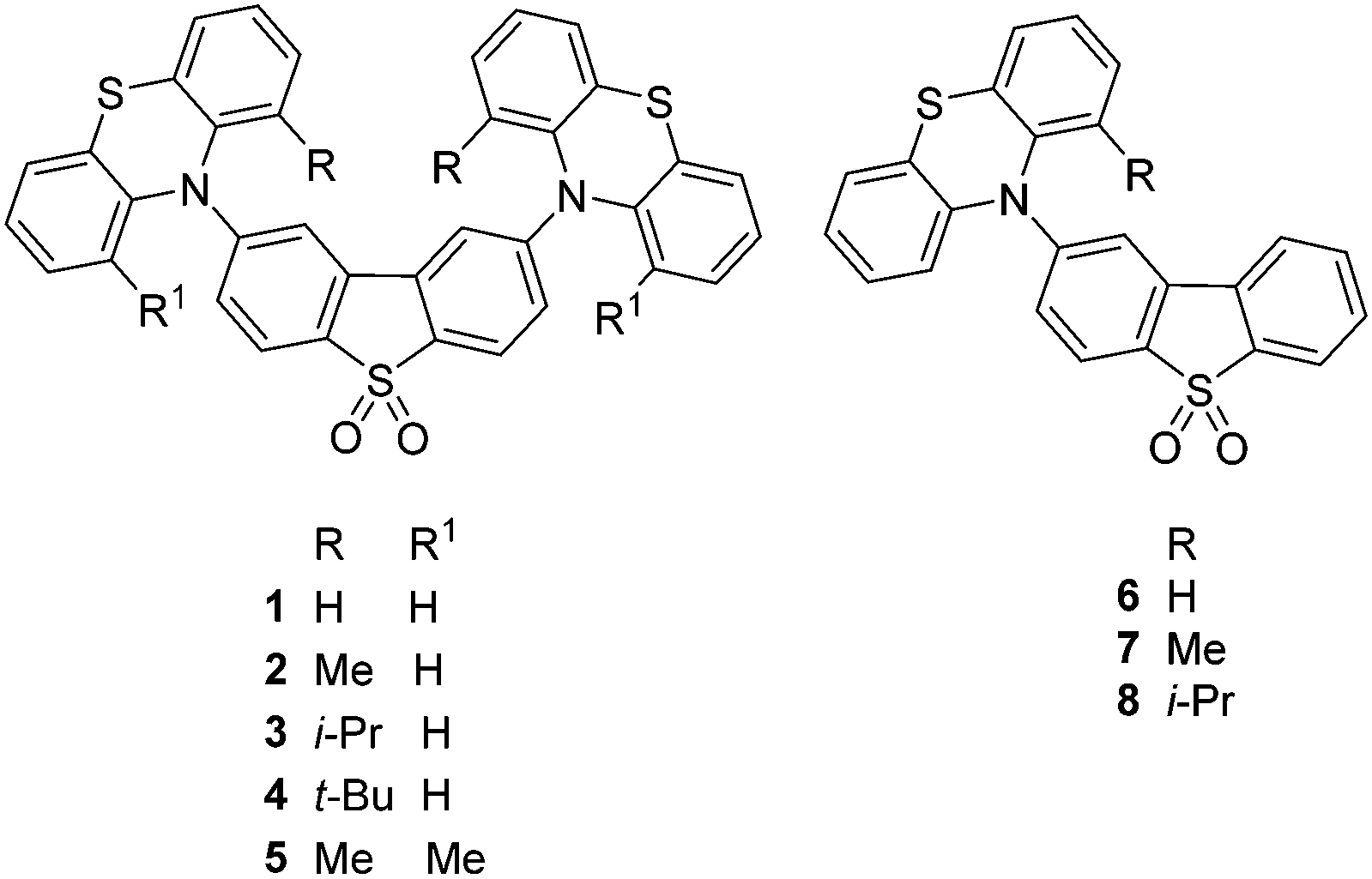
Functionalization of the phenothiazine unit(s) with alkyl substituents at the 1 (and 9) position(s) affords the new compounds 2–5, 7, 8. The synthetic details are reported in the ESI,† S2. The structures of 2·THF, 3, 5·2CH2Cl2 and 7 were determined by single-crystal X-ray diffraction (Fig. 2 and ESI,† S8).
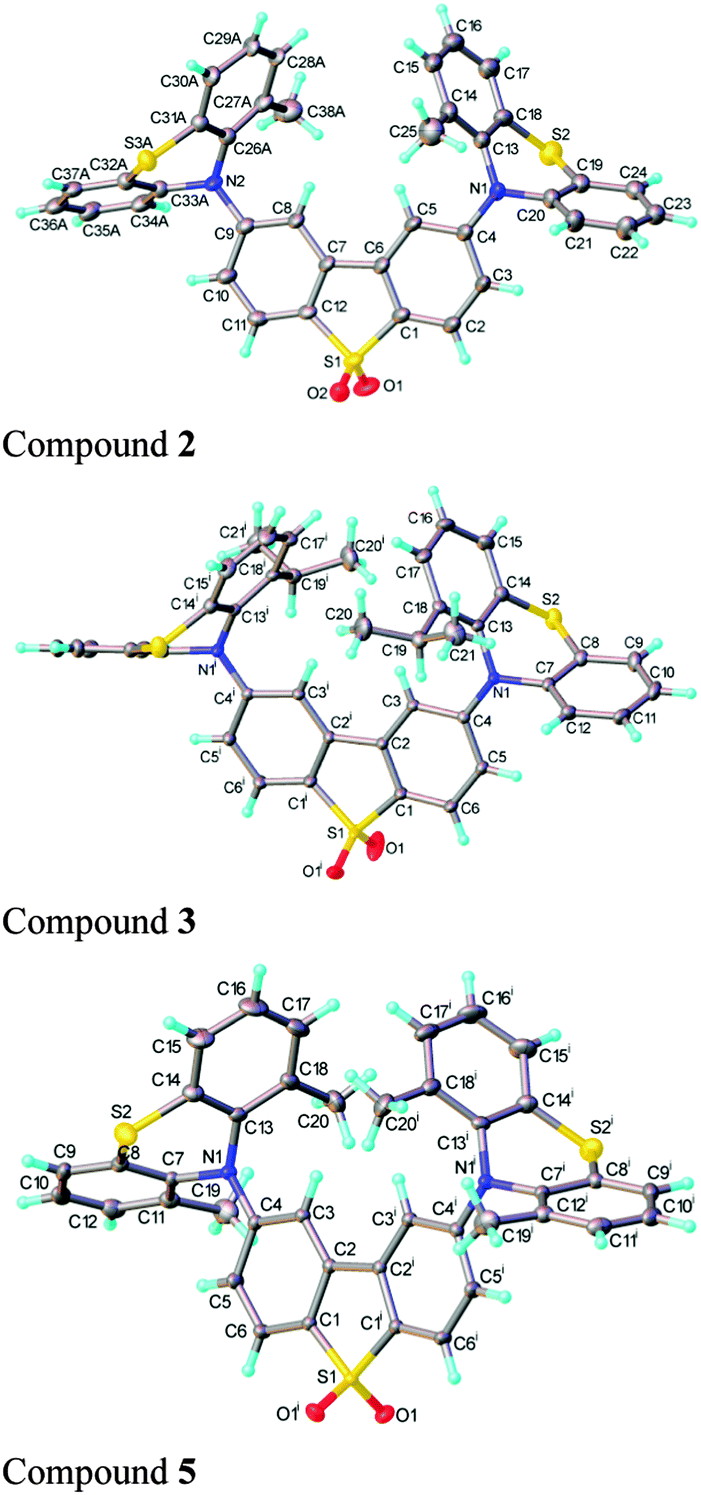 | ||
| Fig. 2 X-ray molecular structures of compounds 2 (major conformer), 3 and 5 displayed with thermal ellipsoids at 50% probability. | ||
The dibenzothiophene moiety in 2 and 5 is practically planar; in 3 it is slightly twisted and in 7 folded, with the two arene rings forming a dihedral angle of 6.9° and 12.5°, respectively. The molecules of 3 and 5 possess a crystallographic two-fold axis, hence the phenothiazine substituents have transoid orientation with respect to the dibenzothiophene plane, i.e. their S atoms lie on opposite sides of this plane. 2 has a similar conformation, although the molecule has no crystallographic symmetry. Interestingly, one of the phenothiazine moieties [at C(9)] is disordered in a 0.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.4 ratio between two conformations, with the methyl substituent on opposite sides of N(2) and the tricyclic system librating around the N(2) as the pivot, to provide room for the methyl group. The phenothiazine moiety is always folded along the N⋯S vector, forming a dihedral angle of 128.7° (3), 135.5° (5) and 135.3° (7). In 2, the ordered phenothiazine moiety is folded by 131.1° and the disordered one by 133.6° or 135.0°. In every case, the N atom is substantially pyramidalised; its lone pair is oriented favourably for conjugation with the dibenzothiophene π-system rather than with the arene rings of phenothiazine itself. Therefore, the N–C(dibenzothiophene) bond is shorter than N–C(phenothiazine), on average by 0.035 Å in 3 and 5 and 0.039 Å in 7 (s.u. 0.001–0.002 Å); the similar difference (0.038 Å) in 2 is not statistically significant due to disorder and high s.u. The packing of 2 and 5 is characterised by stacking (π–π) interactions between phenothiazine arene rings (related via inversion centres and therefore parallel) with interplanar separations of 3.50 and 3.60 Å (intermittently) in 2, or 3.66 Å (uniformly) in 5. These interactions link molecules into infinite chains (ESI,† S8). No stacking is evident in the structures of 3 and 7.
:0.4 ratio between two conformations, with the methyl substituent on opposite sides of N(2) and the tricyclic system librating around the N(2) as the pivot, to provide room for the methyl group. The phenothiazine moiety is always folded along the N⋯S vector, forming a dihedral angle of 128.7° (3), 135.5° (5) and 135.3° (7). In 2, the ordered phenothiazine moiety is folded by 131.1° and the disordered one by 133.6° or 135.0°. In every case, the N atom is substantially pyramidalised; its lone pair is oriented favourably for conjugation with the dibenzothiophene π-system rather than with the arene rings of phenothiazine itself. Therefore, the N–C(dibenzothiophene) bond is shorter than N–C(phenothiazine), on average by 0.035 Å in 3 and 5 and 0.039 Å in 7 (s.u. 0.001–0.002 Å); the similar difference (0.038 Å) in 2 is not statistically significant due to disorder and high s.u. The packing of 2 and 5 is characterised by stacking (π–π) interactions between phenothiazine arene rings (related via inversion centres and therefore parallel) with interplanar separations of 3.50 and 3.60 Å (intermittently) in 2, or 3.66 Å (uniformly) in 5. These interactions link molecules into infinite chains (ESI,† S8). No stacking is evident in the structures of 3 and 7.
The 1H NMR spectra of 1,16a2, 5–8 at 298 K display sharp peaks showing that there is one species in solution at this temperature on the NMR timescale. In contrast, compounds 3 and 4 which have a bulkier iPr or tBu substituent on each phenothiazine ring exhibit restricted rotation. The spectrum of compound 3 at 298 K clearly shows two species in solution: upon heating the peaks coalescence to reveal only one species at 373 K (Fig. 3). 1H NOESY and ROESY NMR experiments strongly suggest that the two species are interconverting with each other (see ESI,† S4). For the t-butyl analogue 4, at 298 K selected peaks are extremely broad and the spectra at 348 K and 373 K are comparable to 3 at 298 K. Rotation in compound 4 is still partially restricted even at 373 K (Fig. 4). 1H NOESY NMR experiments with 4 also demonstrate there is some exchange occurring between the two very broad peaks at 298 K (see ESI,† S4). The presence of these two species is likely to be due to up-up and up-down configurations of the iPr or tBu groups with respect to the acceptor.
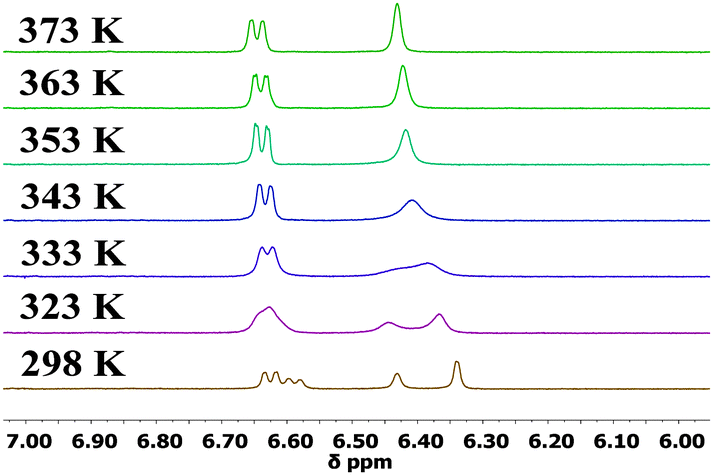 | ||
| Fig. 3 500 MHz variable temperature 1H NMR experiments for 3 from 298–373 K in DMSO-d6. | ||
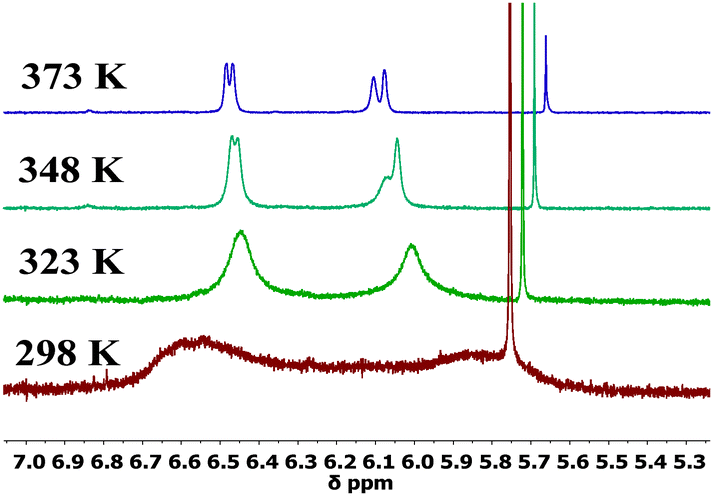 | ||
| Fig. 4 500 MHz variable temperature 1H NMR experiments for 4 from 298–373 K in DMSO-d6. | ||
Photophysical measurements on 2–8 reveal how the increasingly bulky phenothiazine donors affect their optical properties, primarily by restricting rotation around the N–C bond that links the D and A units. Consistent with previous work,10 it has been shown that 1 and 6 emit strongly via a TADF mechanism16 (see ESI,† S6).
The D–A compound 7 has an additional methyl group in the 1-position on phenothiazine compared to 6, and this has a dramatic effect on the emission of this molecule. The same difference is also observed in the D–A–D molecule 2 (Fig. 5).
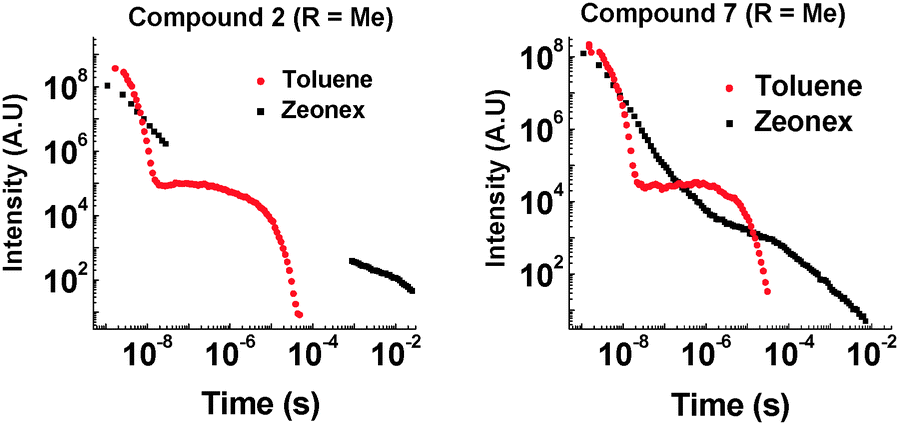 | ||
| Fig. 5 Time-resolved emission decay for 2 and 7 at 290 K (355 nm excitation). | ||
Introduction of the methyl group into molecules 2 and 7 increases the lifetime of the DF emission in toluene solution (compared with molecules 1 and 6, ESI,† S6.3). Temperature and power dependence data confirm that the delayed emission still occurs via a TADF mechanism in solution (Fig. 6).
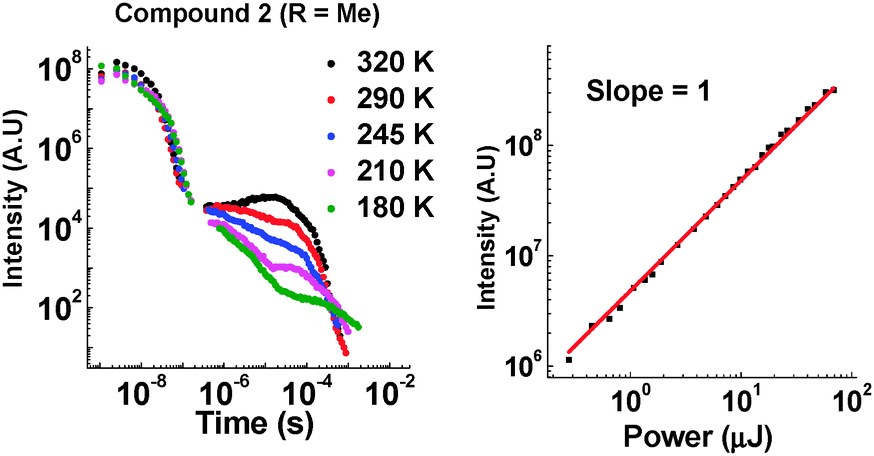 | ||
| Fig. 6 Left: Temperature dependence of emission decay in toluene for 2. Right: Power dependence of DF emission in toluene for 2 (400 ns delay and 50 μs integration time). | ||
There is also a reduction in intensity of the delayed fluorescence on introduction of the methyl group into 2. We propose that the methyl group slows molecular reorganization processes resulting in a slower initial electron transfer step as well as increasing the time taken for the 1CT state to emit. These effects increase the amount of non-radiative decay and direct decay from 1D–1A states, as well as reducing the number of CT states initially created. This is evidenced by the increasing contribution of emission around 400 nm in the steady-state data. The pronounced influence of the side groups in the TADF emission is clearly seen in the case of compound 4, where even in toluene solution no 1CT emission is observed.
The most noticeable difference between unsubstituted 1 and methyl substituted 2 is that in the solid state in a zeonex film, the TADF emission is no longer present. A large time gap with no emission is observed with 2 after fast donor fluorescence decays. It is suggested that the methyl group's ability to restrict reorganization is enhanced in the solid state, resulting in such slow electron transfer that the donor states have all decayed before appreciable electron transfer occurs. The unusual feature in the solid state is that 2 emits significantly more via phosphorescence compared with 1. Quenching experiments of the emission with oxygen show that 2 and 7 have increasing phosphorescent character (Fig. 7). This is consistent with the efficient ISC to the donor triplet state instead of CT formation.
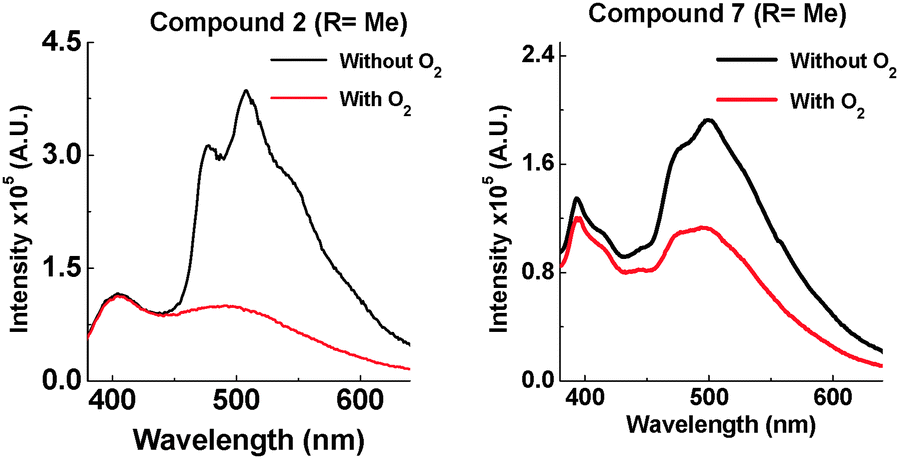 | ||
| Fig. 7 Emission of 2 and 7 at 290 K in zeonex (excited at 355 nm). | ||
D–A–D structure 2 shows more phosphorescence than its D–A analogue 7. More fine structure is observed with 2 and significantly more quenching is observed in the presence of oxygen. This room temperature phosphorescence is unusually strong for an organic molecule,17–20 and the methyl group plays a key role. With increasing steric hindrance the phosphorescence increases even further (Fig. 8). The distinction between TADF and phosphorescence is discussed in more detail in ESI,† S6.8.
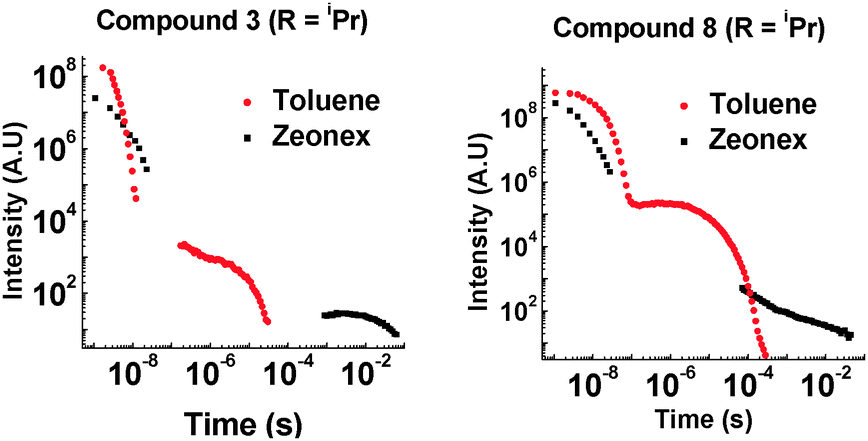 | ||
| Fig. 8 Time-resolved emission decay for 3 and 8 at 290 K (355 nm excitation). | ||
With a bulkier iPr group in the D–A system 8, TADF is still observed in solution (ESI,† S6.5). In zeonex no TADF is observed, but strong phosphorescence is observed at RT. For the D–A–D system 3, the amount of TADF is lowered significantly, even in toluene solution. This correlates with the solution NMR studies showing restricted rotation. With the two iPr systems 3 and 8, the extent of phosphorescence increases significantly (Fig. 9).
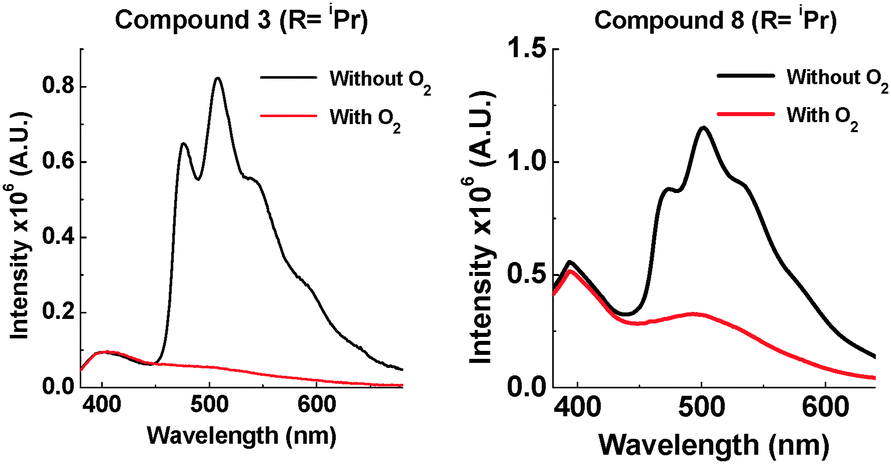 | ||
| Fig. 9 Emission of 3 and 8 at 290 K in zeonex (excited at 355 nm). | ||
The emission of 3 and 8 in zeonex shows even more fine structure compared with 2 and 7 due to the increased ratio of phosphorescence to 1CT fluorescence. D–A–D 3 with two iPr groups, prevents molecular movement more than in the D–A analogue 8 (see Fig. 3 and ESI,† S3). The emission from 3 in the solid state consists almost entirely of phosphorescence as shown by the large decrease in emission on introduction of oxygen. It is unusual for an organic molecule to decay almost completely by phosphorescent pathways. Temperature dependence studies in solid state show that 8 does not emit via a TADF mechanism (ESI,† S6.6). The introduction of these bulkier groups acts like a switch, turning off electron transfer and turning on phosphorescence.
Having observed the drastic change caused by an iPr group, tBu groups were introduced into 4, and two methyl groups were attached onto each phenothiazine in 5. A large contribution from phosphorescence is observed in the solid state of 4 (Fig. 10), and there is also a large contribution of fast fluorescence from the excited S1 state. It is proposed that the motion in 4 in the solid state is so restricted that electron transfer from the 1D–1A states to the 1CT state is prevented. This results in fast fluorescence decay from S1 (1A and 1D) and long lived phosphorescence (3A and 3D) as the main radiative pathways for the excited state to decay. Supporting evidence for extended 1D emission in 4 is seen in time-resolved studies (see ESI,† S6.3). Compound 5 also shows strong phosphorescence at RT, but less 1A and 1D fluorescence than 4, which correlates with the less bulky substituents.
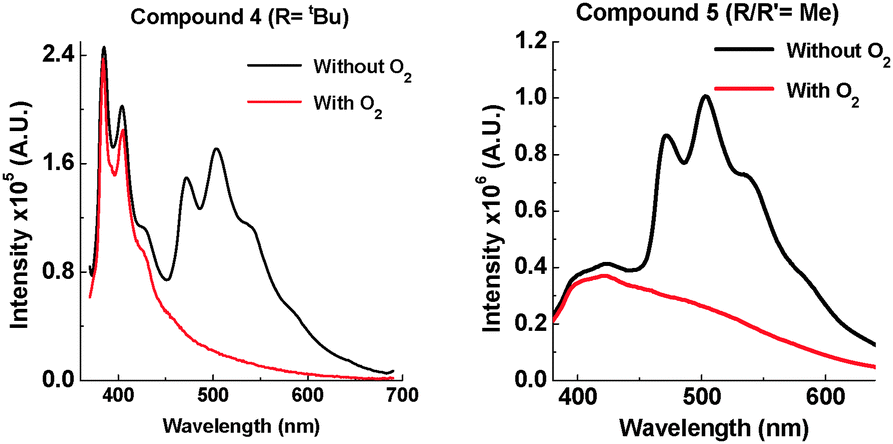 | ||
| Fig. 10 Emission of 4 and 5 at 290 K in zeonex (excited at 355 nm). | ||
In conclusion, suppressing vibrational quenching in a series of D–A–D and D–A molecules strongly activates phenothiazine phosphorescence. By sterically engineering near-orthogonal orientation and restricting rotation between the D and A fragments, in the solid state the main non-radiative deactivation pathway that affects the lowest triplet excited state is thus suppressed and so phosphorescence from phenothiazine is not quenched. This provides valuable mechanistic information on TADF and has led to strongly phosphorescent molecules at room temperature.
Notes and references
- (a) V. W. W. Yam and K. M.-C. Wong, Chem. Commun., 2011, 47, 11579–11592 RSC; (b) Thematic issue, Organic Electronics and Optoelectronics, Chem. Rev., 2007, 107, 923–1386.
- M. A. Aldo, D. F. O'Brien, M. E. Thompson and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 14422–14428 CrossRef.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- (a) D. Y. Kondakov, T. D. Pawlik, T. K. Hatwar and J. P. Spindler, J. Appl. Phys., 2009, 106, 124510 CrossRef; (b) S. M. King, M. Cass, M. Pintani, C. Coward, F. B. Dias, A. P. Monkman and M. Roberts, J. Appl. Phys., 2011, 109, 074502 CrossRef.
- (a) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed; (b) Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931 CrossRef CAS PubMed; (c) K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253 CrossRef CAS; (d) Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706 CrossRef CAS PubMed; (e) C. Adachi, Jpn. J. Appl. Phys., 2014, 53, 060101 CrossRef.
- (a) M. N. Berberan-Santos and J. M. M. Garcia, J. Am. Chem. Soc., 1996, 118, 9391 CrossRef CAS; (b) C. Baleizão and M. N. Berberan-Santos, J. Chem. Phys., 2007, 126, 204510 CrossRef PubMed.
- Q. Zhang, H. Kuwabara, W. J. Potscavage Jr., S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070–18081 CrossRef CAS PubMed.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, Chem. Mater., 2013, 25, 3766–3771 CrossRef CAS.
- T. Nakagawa, S.-Y. Ku, K.-T. Wong and C. Adachi, Chem. Commun., 2012, 48, 9580–9582 RSC.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- P. Chen, L.-P. Wang, W.-Y. Tan, Q.-M. Peng, S.-T. Zhang, X.-H. Zhu and F. Li, ACS Appl. Mater. Interfaces, 2015, 7, 2972–2978 CAS.
- H. Wang, L. Xie, Q. Peng, L. Meng, Y. Wang, Y. Yi and P. Wang, Adv. Mater., 2014, 26, 5198–5204 CrossRef CAS PubMed.
- L. Mei, J. Hu, X. Cao, F. Wang, C. Zheng, Y. Tao, X. Zhang and W. Huang, Chem. Commun., 2015, 51, 13024–13027 RSC.
- S. Xu, T. Liu, Y. Mu, Y. F. Wang, Z. Chi, C. C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 874–878 CrossRef CAS PubMed.
- M. J. Leitl, V. A. Krylova, P. I. Djurovich, M. E. Thompson and H. Yersin, J. Am. Chem. Soc., 2014, 136, 16032–16038 CrossRef CAS PubMed.
- (a) F. B. Dias, J. Santos, D. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, manuscript submitted; (b) R. S. Nobuyasu, Z. Ren, A. S. Batsanov, P. Data, A. P. Monkman, M. R. Bryce, S. Yan and F. B. Dias, Adv. Opt. Mater., DOI:10.1002/adom.201500689.
- Y. Gong, G. Chen, Q. Peng, W. Z. Yuan, Y. Xie, S. Li, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 6195–6201 CrossRef CAS PubMed.
- S. Reineke, N. Seidler, S. R. Yost, F. Prins, W. A. Tisdale and M. A. Baldo, Appl. Phys. Lett., 2013, 103, 093302 CrossRef.
- J. J. Xu, A. Takai, Y. Kobayashi and M. Takeuchi, Chem. Commun., 2013, 49, 8447–8449 RSC.
- M. Koch, K. Perumal, O. Blacque, J. A. Garg, R. Saiganesh, S. Kabilan, K. K. Balasubramanian and K. Venkatesan, Angew. Chem., Int. Ed., 2014, 53, 6378–6382 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, copies of NMR spectra and CV data, additional photophysics and X-ray crystallography. CCDC 1437574 (2), 1426130 (3), 1426129 (5), 1426128 (7). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc09645f |
| ‡ These authors contributed equally to the experimental work. |
| This journal is © The Royal Society of Chemistry 2016 |