
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric synthesis of cyclopentanes bearing four contiguous stereocenters via an NHC-catalyzed Michael/Michael/esterification domino reaction†
Tao
Shu
a,
Qijian
Ni
a,
Xiaoxiao
Song
a,
Kun
Zhao
a,
Tianyu
Wu
a,
Rakesh
Puttreddy
b,
Kari
Rissanen
b and
Dieter
Enders
*a
aInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, 52074, Aachen, Germany. E-mail: enders@rwth-aachen.de
bDepartment of Chemistry, University of Jyväskylä, 40014 Jyväskylä, Finland
First published on 5th January 2016
Abstract
An NHC-catalyzed Michael/Michael/esterification domino reaction via homoenolate/enolate intermediates for the asymmetric synthesis of tetrasubstituted cyclopentanes bearing four contiguous stereocenters is described. A variety of α,β-unsaturated aldehydes and 2-nitroallylic acetates react well with good domino yields and high stereoselectivities.
Cyclopentane motifs are privileged scaffolds present as characteristic structural features in a large number of bioactive natural products and pharmaceuticals such as for instance aristeromycin,1 travoprost,2 pactamycin3 and peramivir4 (Fig. 1). However, to develop direct and efficient catalytic methods for the stereocontrolled construction of multi-substituted cyclopentanes is still challenging for organic chemists.5
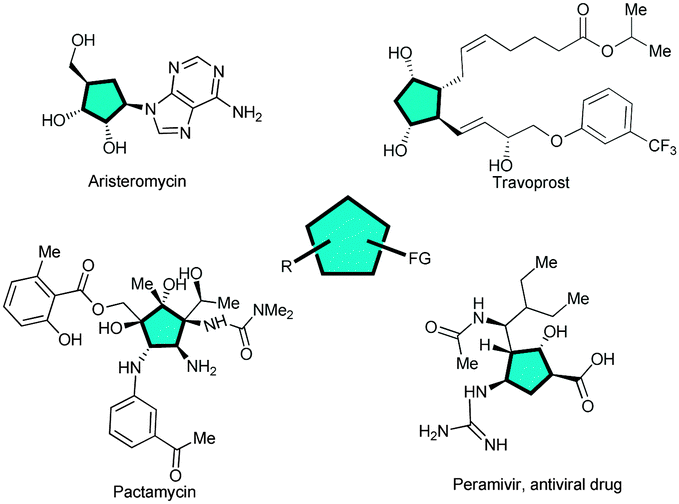 | ||
| Fig. 1 Representative natural products and pharmaceuticals bearing a cyclopentane core. | ||
Nitroalkenes are among the most useful Michael acceptors due to their versatile reactivity and their inherent capacity to undergo further synthetic transformations of the nitro function into other functional groups.6 NHC-catalyzed cascade reactions have emerged as a powerful tool to construct C–C bonds in organic synthesis7 and NHC-catalyzed reactions with nitroalkenes gained quite some interest in recent years.8 The first NHC-catalyzed homoenolate reaction of enals with nitroalkenes to afford anti δ-nitroesters was reported by Nair and co-workers.9a Later the groups of Liu and Rovis reported complementary asymmetric versions of this reaction affording anti and syn δ-nitroesters, respectively.9b–d Very recently, Wang and co-workers have also developed an NHC-catalyzed reaction of enals with nitroalkenes to prepare enantioenriched dihydrocoumarins.8i
Nitroallylic acetates served as versatile dielectrophiles to assemble relatively complex molecules in a domino fashion.10 Seebach and co-workers developed a [3+3] carbocyclization reaction of 2-nitroallylic acetates and enamines to form bicyclic skeletons with multiple stereocenters using the chiral auxiliary concept under stoichiometric conditions.10a An organocatalytic domino reaction of 2-nitroallylic acetates had not been reported until 2009. Tang and Li et al. developed a pyrrolidine-thiourea catalyzed tandem reaction of 2-nitroallylic acetates and cyclic ketones to construct bicyclic [3.3.1] skeletons with four or five stereocenters in a single operation.10b However, to the best of our knowledge, an NHC-catalyzed cascade reaction employing 2-nitroallylic acetates has not been reported yet. Herein, we describe such an NHC organocatalyzed [3+2]-cycloaddition reaction of enals with (E)-2-nitroallylic acetates to afford enantioenriched tetrasubstituted cyclopentanes with four contiguous stereocenters featuring a nitro and an ester group, which can be used for further transformations.
Initially we investigated the reaction of cinnamaldehyde (1a) and (E)-2-nitroallylic acetate 2a as model substrates catalyzed by the NHC catalysts derived from the pre-catalysts A–F in THF/EtOH using one equivalent of NaOAc as a base. The aminoindanol-based triazolium pre-catalyst B provided the desired cyclopentane product 3a in 30% yield and 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :22 e.r., albeit with a low diastereoselectivity (d.r. 3:1) (Table 1).
:22 e.r., albeit with a low diastereoselectivity (d.r. 3:1) (Table 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Solvent | Base | Yield (%)b | d.rc | e.r.d |
| a Reaction conditions: 1a (0.3 mmol), 2a (0.2 mmol), NHC catalyst (10 mol%), base (1.0 equiv.), 24 h at rt. b Yield of isolated compound 3a. c d.r. determined by 1H NMR. d The e.r. values were determined by HPLC on a chiral stationary phase. e The reaction was carried out at −5 °C, 48 h. f The reaction was carried out at −15 °C, 96 h. g The reaction was carried out at −10 °C, 60 h. | ||||||
| 1 | A | THF | NaOAc | <5 | — | — |
| 2 | B | THF | NaOAc | 30 | 3:1 |
78:22 |
| 3 | C | THF | NaOAc | n.r | — | — |
| 4 | D | THF | NaOAc | n.r | — | — |
| 5 | E | THF | NaOAc | 18 | 6.2:1 |
63:37 |
| 6 | F | THF | NaOAc | 26 | 8:1 |
40:60 |
| 7 | B | CHCl3 | NaOAc | 45 | 3.6:1 |
85:15 |
| 8 | B | DME | NaOAc | 36 | 1.7:1 |
72:28 |
| 9 | B | CCl4 | NaOAc | 56 | 1.5:1 |
82:18 |
| 10 | B | CH2Cl2 | NaOAc | 30 | 4.3:1 |
75:25 |
| 11 | B | TBME | NaOAc | 30 | 1.6:1 |
78:22 |
| 12 | B | Toluene | NaOAc | 42 | 1.2:1 |
81:19 |
| 13 | B | CHCl3 | NEt3 | 45 | 3.3:1 |
71:29 |
| 14 | B | CHCl3 | DIPEA | 40 | 3:1 |
75:25 |
| 15 | B | CHCl3 | DABCO | 26 | 5:1 |
70:30 |
| 16 | B | CHCl3 | TMEDA | 32 | 3.7:1 |
69:31 |
| 17 | B | CHCl3 | CsOAc | 30 | 1.8:1 |
68:32 |
| 18 | B | CHCl3 | LiOAc | 36 | 2.6:1 |
74:26 |
| 19e | B | CHCl3 | Cs2CO3 | 20 | 1.3:1 |
87:13 |
| 20e | B | CHCl3 | K3PO4 | 49 | 7.8:1 |
91:9 |
| 21f | B | CHCl3 | K3PO4 | 40 | >20:1 |
93:7 |
| 22g | B | CHCl3 | K3PO4 | 52 | 16:1 |
93:7 |
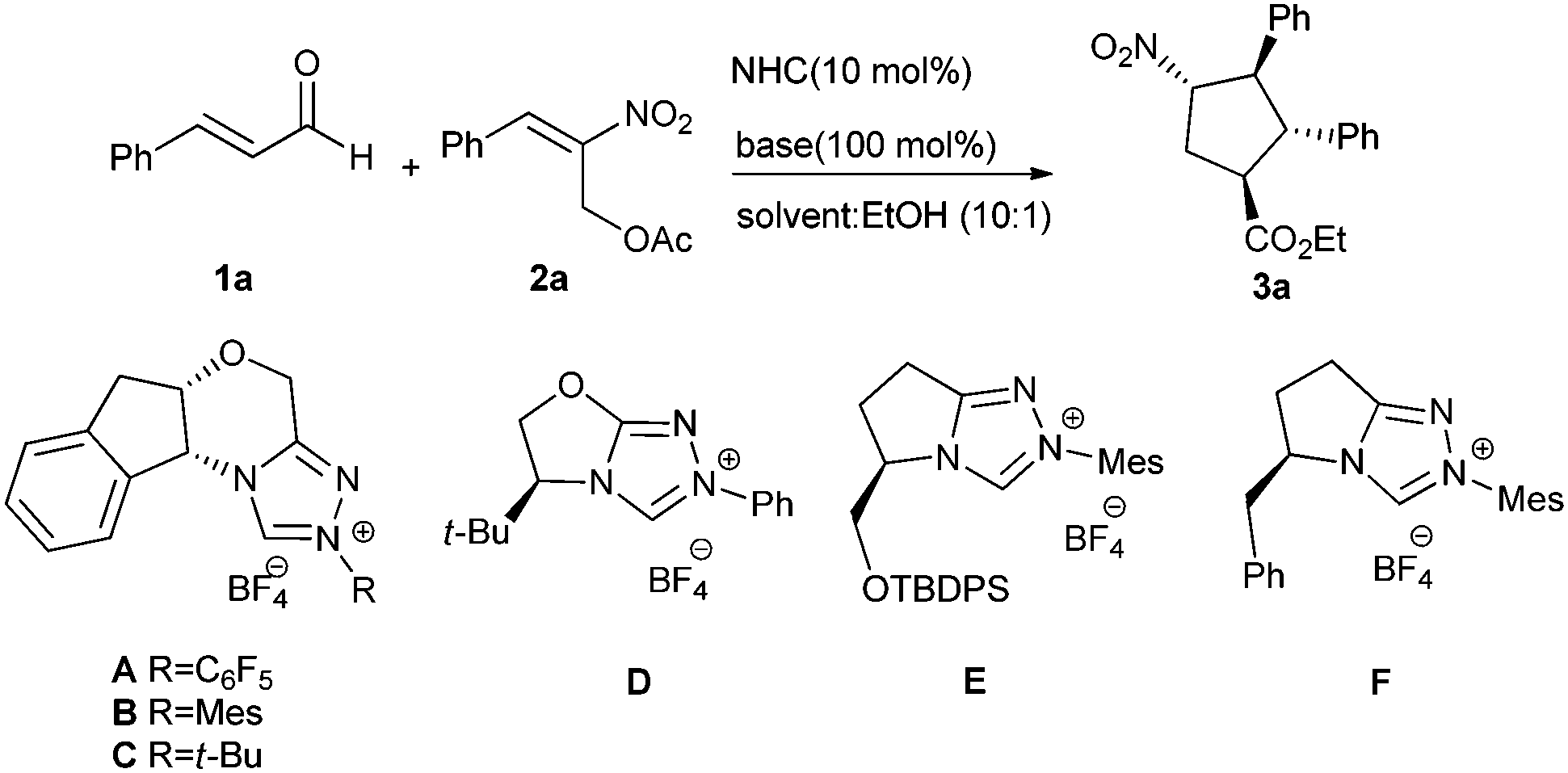
After the screening of the solvents, CHCl3 turned out to be the best solvent, affording 3a in a good domino yield of 45%, a d.r. of 3.6:1 and an e.r. of 85:15 (entry 7). The reaction proceeded well with different bases (entries 13–20). Using strong organic bases such as DBU or DMAP resulted in complex mixtures without any starting material remained and no desired product could be detected. We also screened some Lewis acid additives such as Mg(OtBu)2, Ti(OiPr)4 and Sc(OTf)3 and also MgSO4 as well as 4 Å molecular sieves, but no better result was obtained. Lowering the reaction temperature improved the d.r. and e.r., but the reaction time was extended. The reaction proceeded well at −5 °C (49% yield, 7.8:1 d.r, 91:9 e.r) in 48 h, −15 °C (40% yield, >20:1 d.r., 93:7 e.r.) in 96 h with starting material left and −10 °C (52% yield, 16:1 d.r., 93:7 e.r.) in 60 h. Lowering the loading of the base gave inferior results. Finally, we chose pre-catalyst B, CHCl3:EtOH (10:1), K3PO4 (100 mol%) at −10 °C as the optimized condition for our reaction (entry 22).
With the optimized conditions in hand, we next evaluated the substrate scope with respect to the enals and 2-nitroallylic acetates. With different substituted cinnamaldehydes, heterocyclic enals or (E)-2-nitroallylic acetates, the reaction proceeded well, affording the desired products in good domino yields (18–55%) and enantiomeric ratios (86:14–98:2) (Scheme 1).
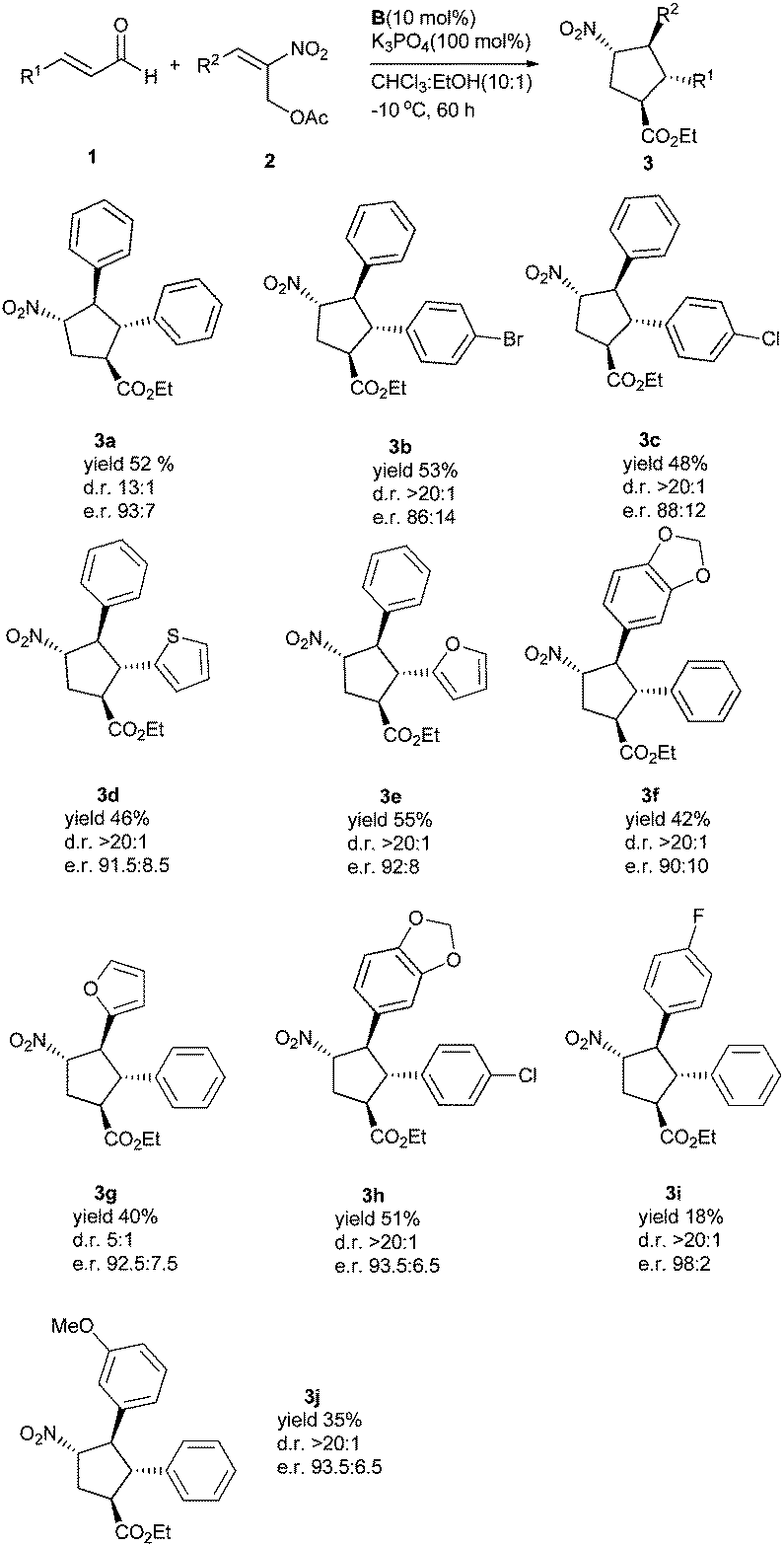 | ||
| Scheme 1 Substrate scope. All reactions were performed on a 0.5 mmol scale. The yields of the isolated products are after column chromatography. The diastereomeric ratios were determined by 1H NMR spectroscopy and the e.r. values by HPLC on a chiral stationary phase. | ||
The absolute configuration was unambiguously determined by X-ray crystal structure analysis of compound 3h and all other cyclopentane products were assigned by analogy (Fig. 2).
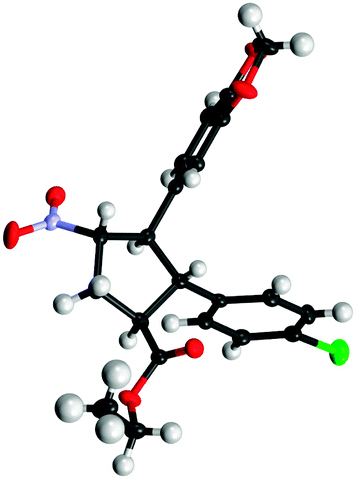 | ||
| Fig. 2 Absolute configuration of 3h determined by X-ray structural analysis.11 | ||
A plausible mechanism for the NHC-catalyzed [3+2] Michael/Michael/esterification cascade is shown in Scheme 2. The reaction proceeds via an extended Breslow intermediate, which as a homoenolate I undergoes a first Michael addition to the nitroallylic acetates 2, followed by the elimination of the acetyl group from the adduct II to generate the second Michael acceptor intermediate III for the intramolecular Michael addition. The resulting acylazolium intermediate IV undergoes an ethanolysis with external ethanol to afford the cyclopentane esters 3 and returns the NHC catalyst for further cycles.
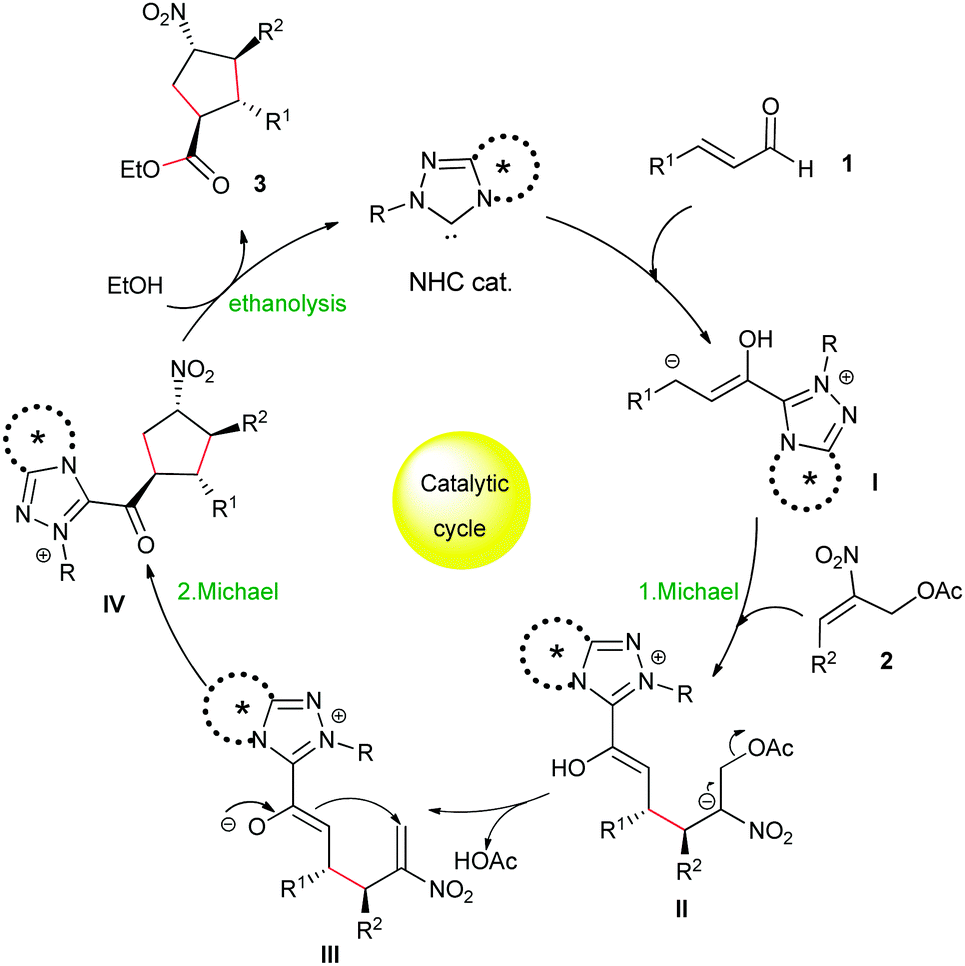 | ||
| Scheme 2 Proposed catalytic cycle. | ||
In conclusion, we have developed a concise protocol for the NHC-catalyzed direct construction of functionalized cyclopentanes bearing four contiguous stereocenters in one single operation with good domino yields and stereoselectivities. A range of functional groups and substituents are tolerated by variation of the enal and nitroallylic acetate substrates. In the novel one-pot protocol two C–C bonds via Michael addition and one C–O bond through a terminating ethanolysis step are formed.
We thank the European Research Council (ERC Advanced Grant 320493 “DOMINOCAT”) for financial support and the BASF SE for the donation of chemicals.
Notes and references
- (a) H. J. Bestmann and D. Roth, Synlett, 1990, 751 CrossRef CAS; (b) S. J. Boyer and J. W. Leahy, J. Org. Chem., 1997, 62, 3976 CrossRef CAS.
- J. T. Whitson, Expert Opin. Pharmacother., 2002, 3, 965 CrossRef CAS PubMed.
- (a) S. Hanessian, R. R. Vakiti, S. Dorich, S. Banerjee, F. Lecomte, J. R. DelValle, J. Zhang and B. Deschênes-Simard, Angew. Chem., Int. Ed., 2011, 50, 3497 CrossRef CAS PubMed; (b) J. T. Malinowski, R. J. Sharpe and J. S. Johnson, Science, 2013, 340, 180 CrossRef PubMed.
- (a) Y. S. Babu, P. Chand, S. Bantia, P. Kotian, A. Dehghani, Y. El-Kattan, T.-H. Lin, T. L. Hutchison, A. J. Elliott, C. D. Parker, S. L. Ananth, L. L. Horn, G. W. Laver and J. A. Montgomery, J. Med. Chem., 2000, 43, 3482 CrossRef CAS PubMed; (b) P. Chand, P. L. Kotian, A. Dehghani, Y. El-Kattan, T.-H. Lin, T. L. Hutchison, Y. S. Babu, S. Bantia, A. J. Elliott and J. A. Montgomery, J. Med. Chem., 2001, 44, 4379 CrossRef CAS PubMed; (c) P. Chand, Y. S. Babu, S. Bantia, S. Rowland, A. Dehghani, P. L. Kotian, T. L. Hutchison, S. Ali, W. Brouillette, Y. El-Kattan and T.-H. Lin, J. Med. Chem., 2004, 47, 1919 CrossRef CAS PubMed.
- (a) T. Hudlicky and J. D. Price, Chem. Rev., 1989, 89, 1467 CrossRef CAS; (b) M. Lautens, W. Klute and W. Tam, Chem. Rev., 1996, 96, 49 CrossRef CAS PubMed; (c) B. Heasley, Eur. J. Org. Chem., 2009, 1477 CrossRef CAS; (d) V. Nair, B. P. Babu, S. Vellalath, V. Varghese, A. E. Raveendran and E. Suresh, Org. Lett., 2009, 11, 2507 CrossRef CAS PubMed; (e) P.-C. Chiang, M. Rommel and J. W. Bode, J. Am. Chem. Soc., 2009, 131, 8714 CrossRef CAS PubMed; (f) J. Kaeobamrung and J. W. Bode, Org. Lett., 2009, 11, 677 CrossRef CAS PubMed; (g) D. T. Cohen, B. Cardinal-David and K. A. Scheidt, Angew. Chem., Int. Ed., 2011, 50, 1678 CrossRef CAS PubMed; (h) G. Liu, M. E. Shirley, K. N. Van, R. L. McFarlin and D. Romo, Nat. Chem., 2013, 5, 1049 CrossRef CAS PubMed; (i) B. T. Parr and H. M. L. Davies, Nat. Commun., 2014, 5, 1 Search PubMed; (j) L.-H. Zou, A. R. Philipps, G. Raabe and D. Enders, Chem. – Eur. J., 2015, 21, 1004 CrossRef CAS PubMed; (k) S. Mukherjee, S. Mondal, A. Patra, R. G. Gonnadeb and A. T. Biju, Chem. Commun., 2015, 51, 9559 RSC.
- For a review, see: O. M. Berner, L. Tedeschi and D. Enders, Eur. J. Org. Chem., 2002, 1877 CrossRef CAS.
- For reviews on NHC-catalyzed domino reactions, see: (a) A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314 CrossRef CAS PubMed; (b) P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2014, 53, 1485 CrossRef CAS PubMed; (c) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed ; For pioneering work on homoenolate intermediates, see: ; (d) C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205 CrossRef CAS PubMed; (e) S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370 CrossRef CAS PubMed ; For reviews on homoenolate intermediates, see: ; (f) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691 RSC; (g) V. Nair, R. S. Menon, A. T. Biju, C. R. Sinu, R. R. Paul, A. Jose and V. Sreekumar, Chem. Soc. Rev., 2011, 40, 5336 RSC; (h) D. T. Cohen and K. A. Scheidt, Chem. Sci., 2012, 3, 53 RSC; (i) P.-C. Chiang and J. W. Bode, TCI MAIL, 2011, 149, 2 Search PubMed; (j) R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040 RSC; (k) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307 CrossRef CAS PubMed.
- For NHC-catalyzed reactions with nitroalkenes, see: (a) A. E. Mattson, A. M. Zuhl, T. E. Reynolds and K. A. Scheidt, J. Am. Chem. Soc., 2006, 128, 4932 CrossRef CAS PubMed; (b) D. A. DiRocco, K. M. Oberg, D. M. Dalton and T. Rovis, J. Am. Chem. Soc., 2009, 131, 10872 CrossRef CAS PubMed; (c) D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 10402 CrossRef CAS PubMed; (d) D. A. DiRocco, E. L. Noey, K. N. Houk and T. Rovis, Angew. Chem., Int. Ed., 2012, 51, 2391 CrossRef CAS PubMed; (e) Q. Ni, H. Zhang, A. Grossmann, C. C. J. Loh, C. Merkens and D. Enders, Angew. Chem., Int. Ed., 2013, 52, 13562 CrossRef CAS PubMed; (f) X.-Y. Chen, L.-H. Sun and S. Ye, Chem. – Eur. J., 2013, 19, 4441 CrossRef CAS PubMed; (g) Y. Du, Y. Wang, X. Li, Y. Shao, G. Li, R. D. Webster and Y. R. Chi, Org. Lett., 2014, 16, 5678 CrossRef CAS PubMed; (h) Q. Zhang, H.-Z. Yu and Y. Fu, Org. Chem. Front., 2014, 1, 614 RSC; (i) Z. Wu, X. Wang, F. Li, J. Wu and J. Wang, Org. Lett., 2015, 17, 3588 CrossRef CAS PubMed.
- For homoenolate additions to nitroalkenes, see: (a) V. Nair, C. R. Sinu, B. P. Babu, V. Varghese, A. Jose and E. Suresh, Org. Lett., 2009, 11, 5570 CrossRef CAS PubMed; (b) B. Maji, L. Ji, S. Wang, S. Vedachalam, R. Ganguly and X.-W. Liu, Angew. Chem., Int. Ed., 2012, 51, 8276 CrossRef CAS PubMed; (c) N. A. White, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2013, 135, 8504 CrossRef CAS PubMed; (d) N. A. White, K. E. Ozboya, D. M. Flanigan and T. Rovis, Asian J. Org. Chem., 2014, 3, 442 CrossRef CAS PubMed.
- For cascade reactions of nitroallylic acetates, see: (a) D. Seebach, M. Missbach, G. Calderari and M. Eberle, J. Am. Chem. Soc., 1990, 112, 7625 CrossRef CAS; (b) C.-L. Cao, Y.-Y. Zhou, J. Zhou, X.-L. Sun, Y. Tang, Y.-X. Li, G.-Y. Li and J. Sun, Chem. – Eur. J., 2009, 15, 11384 CrossRef CAS PubMed; (c) D. K. Nair, S. M. Mobin and I. N. N. Namboothiri, Tetrahedron Lett., 2012, 53, 3349 CrossRef CAS; (d) D. K. Nair, S. M. Mobin and I. N. N. Namboothiri, Org. Lett., 2012, 14, 4580 CrossRef CAS PubMed; (e) L. F. Yeh, S. Anwar and K. Chen, Tetrahedron, 2012, 68, 7317 CrossRef CAS; (f) W.-Y. Huang, Y.-C. Chen and K. Chen, Chem. – Asian J., 2012, 7, 688 CrossRef CAS PubMed; (g) T. Kumar, S. M. Mobin and I. N. N. Namboothiri, Tetrahedron, 2013, 69, 4964 CrossRef CAS; (h) S. Anwar, W.-Y. Huang, C.-H. Chen, Y.-S. Cheng and K. Chen, Chem. – Eur. J., 2013, 19, 4344 CrossRef CAS PubMed; (i) D. K. Nair, R. F. S. Menna-Barreto, E. N. da Silva Junior, S. M. Mobin and I. N. N. Namboothiri, Chem. Commun., 2014, 50, 6973 RSC; (j) T. Zhang, N. Shao, H. Zhu, T. Chen, Q. Zheng and H. Zou, Tetrahedron, 2014, 70, 7454 CrossRef CAS.
- CCDC 1437686.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization date (NMR, IR, MS, HPLC). CCDC 1437686. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc09581f |
| This journal is © The Royal Society of Chemistry 2016 |