
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The SQM/COSMO filter: reliable native pose identification based on the quantum-mechanical description of protein–ligand interactions and implicit COSMO solvation†
Adam
Pecina‡
a,
René
Meier‡
b,
Jindřich
Fanfrlík
a,
Martin
Lepšík
a,
Jan
Řezáč
a,
Pavel
Hobza
*ac and
Carsten
Baldauf
*d
aInstitute of Organic Chemistry and Biochemistry (IOCB) and Gilead Sciences and IOCB Research Center, Flemingovo nám. 2, 16610 Prague 6, Czech Republic. E-mail: hobza@uochb.cas.cz
bInstitut für Biochemie, Fakultät für Biowissenschaften, Pharmazie und Psychologie, Universität Leipzig, Brüderstrasse 34, D-04109 Leipzig, Germany
cRegional Centre of Advanced Technologies and Materials, Department of Physical Chemistry, Palacký University, 77146 Olomouc, Czech Republic
dFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, D-14195 Berlin, Germany. E-mail: baldauf@fhi-berlin.mpg.de
First published on 20th January 2016
Abstract
Current virtual screening tools are fast, but reliable scoring is elusive. Here, we present the ‘SQM/COSMO filter’, a novel scoring function featuring a quantitative semiempirical quantum mechanical (SQM) description of all types of noncovalent interactions coupled with implicit COSMO solvation. We show unequivocally that it outperforms eight widely used scoring functions. The accuracy and chemical generality of the SQM/COSMO filter make it a perfect tool for late stages of virtual screening.
Despite the enormous advances in method development for structure-based in silico drug design, reliable predictions of the structures (docking) and affinities (scoring) of protein–ligand (P–L) complexes still remain an unsolved task.1 A plethora of scoring functions (SFs) have been devised by utilising experimental data for regression analyses, by constructing knowledge-based potentials, or based on physical laws.2,3 As none of the SFs is general enough to perform equally strongly for a diverse set of P–L complexes, utilising several SFs at once (consensus scoring) holds promise.4 Regression analysis and knowledge-based approaches to scoring are trained on a set of P–L complexes and rely on variable master equation terms. Their validity is limited to complexes similar to the training set. In principle, this problem has been overcome in physics-based methods. Because of computational cost, preference has been given to molecular mechanics (MM) methods, such as the combination of MM interaction energies with implicit solvation free energy terms (generalised Born, GB, or Poisson–Boltzmann, PB) to estimate affinities.2 Additionally, the wide coverage of organic chemical space in the GAFF (general AMBER force field)5 has made the parameterisation of ligands for MM straightforward. However, an explicit description of quantum mechanical (QM) effects in P–L interactions, such as charge transfer, polarisation, covalent-bond formation or σ-hole bonding, was missing. QM methods, which describe these effects qualitatively better than the energy functions used in MM-based SFs, were thus introduced into computational drug design.6,7 Recent developments in QM methods and algorithms as well as the availability of a powerful computing infrastructure have paved the way to apply them for P–L complexes in numerous setups: linear scaling or efficient parallelisation of semi-empirical QM (SQM) methods,7–10 QM/MM,7,8,11,12 DFT-D3 on truncated P–L complexes13 or various fragmentation methods.11,14 Specifically, AM1, RM1, PM6 or DF–TB SQM methods have been used7–9,12,15 as such or with empirical corrections for dispersion, hydrogen- and halogen-bonding16 to describe the P–L noncovalent interactions. Merz et al. pioneered this area by introducing a QM-based SF (QMScore), a combination of the AM1 SQM method with an empirical dispersion (D) and the PB implicit solvent [eqn (1)].17 The method was useful for describing metalloprotein–ligand binding, but further corrections were needed, especially for a quantitative treatment of dispersion and hydrogen bonding.10
| Score = ΔEint + ΔΔGsolv + ΔG′wconf − TΔS | (1) |
Our approach is systematic. Using accurate calculations in small model systems as a benchmark, we developed corrections for SQM methods that provide reliable and accurate description of a wide range of noncovalent interactions including dispersion, hydrogen- and halogen-bonding.16 Coupled with the PM6 SQM method,18 the resulting PM6-D3H4X approach is applicable to a wide chemical space and does not require any system-specific parameterisation. We use it here to calculate the ΔEint term. Subsequently, we compared MM-based (PB or GB) and QM-based (COSMO19 or SMD) implicit solvent models and found the latter group to be more accurate.20 These are therefore used for the ΔΔGsolv term. These two dominant terms, ΔEint and ΔΔGsolv, are at the heart of our SQM-based SF.15 We have demonstrated its generality in various noncovalent P–L complexes, such as aldose reductase or carbonic anhydrase and moreover extended it to treat covalent inhibitor binding (ref. 15, 21 and 22).
In this work, we adapt our SQM-based SF to make it usable in virtual screening on the basis of our previous experience. By taking the two dominant terms only, ΔEint and ΔΔGsolv, we define the ‘SQM/COSMO filter’ energy. Its performance is tested here against eight widely used SFs. GlideScore XP (GlideXP),23 PLANTS PLP (PLP),24 AutoDock Vina (Vina),25 Chemscore (CS),26 Goldscore (GS)27 and ChemPLP24 are empirical, regression-based functions which use different terms to describe vdW contacts, lipophilic surface coverage, hydrogen bonding, ligand strain, and desolvation. The Astex Statistical Potential (ASP)28 is a knowledge-based potential. The classical physics-based AMBER/GB SF combines the ff03-GAFF MM force fields with the GB implicit solvent.5,29
The goal is ‘cognate docking’,30i.e. the ability to identify sharply the known native X-ray P–L binding pose from a set of decoy structures generated by docking (Fig. 1). To understand our results in detail, we have not opted for treating them in a statistical manner31 as in the pose decoy test sets available.32 Instead we cautiously selected four unrelated difficult-to-handle P–L systems, which comply with strict criteria for the selection of crystallographic structures for docking (details in the ESI†).33 These systems are acetylcholine esterase (AChE, PDB: 1E66),34 TNF-α converting enzyme (TACE, PDB: 3B92),35 aldose reductase (AR, PDB: 2IKJ)36 and HIV-1 protease (HIV PR; PDB: 1NH0).37 For the latter, the protonation of the active site is inferred from ultra-high resolution X-ray crystallography. Based on these P–L crystal structures, we have created a set of non-redundant poses (2865 in total) by docking with four popular docking programs (Glide, PLANTS, AutoDock Vina and GOLD) coupled to seven widely used SFs23–28 (Fig. 1, Table S2, ESI†).
 | ||
| Fig. 1 The ligand poses generated by the four docking programs. Ligand poses are color-coded by RMSD. | ||
All the poses were re-scored by all nine SFs. For the seven regression- and knowledge-based SFs, we used the recommended protocols. For the two physics-based SFs, only hydrogen atoms and close contacts were relaxed by the AMBER/GB method. RMSD values of the poses relative to the crystal were measured (details in S1.6, ESI†). The scores were normalised and are shown relative to the score of the crystal pose.
The identification of the X-ray pose as the minimum-free-energy structure is an unambiguous criterion for the performance of any SF. The ideal behaviour of such a score vs. RMSD curve (Fig. 2) is characterised by the positive values of energies for the decoy poses. Small deviations (negative energies for very small RMSD values) are acceptable and might be explained by inaccuracies of the crystal structure. These conditions are met by the SQM/COSMO filter, unlike the other SFs (Fig. 2). The numbers of false-positive solutions as well as the maximum RMSD (RMSDmax) from the X-ray pose within a defined interval of the normalised score quantify the virtually ideal behaviour of the SQM/COSMO filter in comparison to the other SFs.
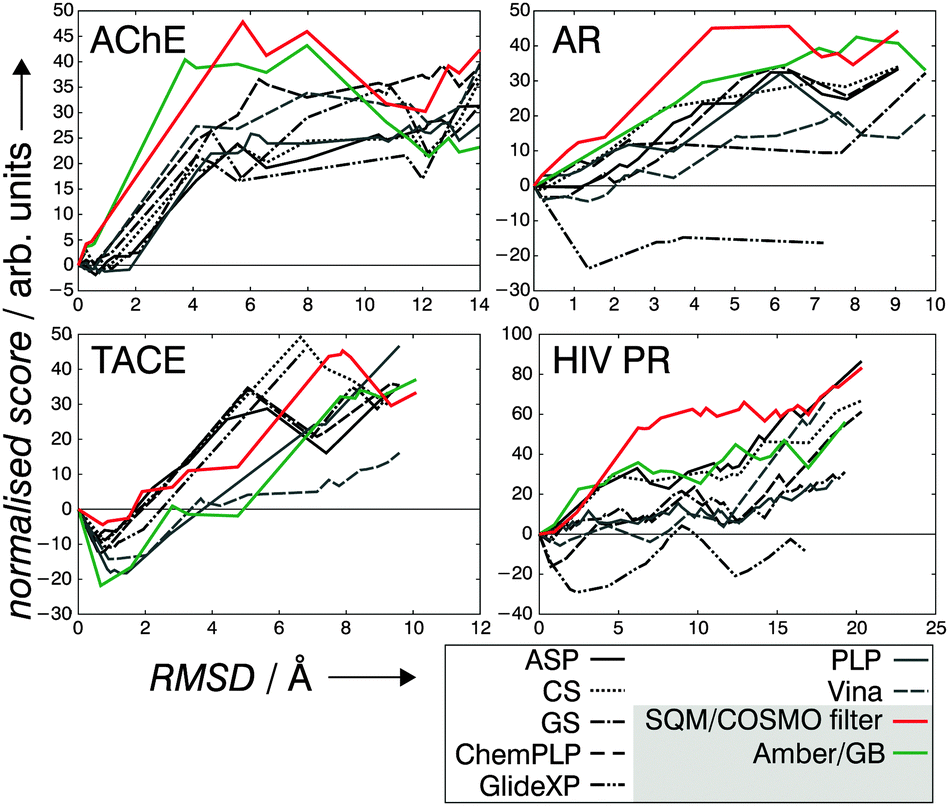 | ||
| Fig. 2 The plots of normalised scores against RMSD values for all four P–L systems. | ||
The number of false-positives is lowest for the SQM/COSMO filter, even zero for three P–L systems (Table 1). CS and ASP perform slightly worse. AMBER/GB performs satisfyingly well for three systems but yields 171 false-positives for TACE. For AChE, all the SFs perform satisfyingly well. For AR and HIV PR, GlideXP generates the highest number of false-positive solutions and also shape-wise the free energy landscape looks ill-defined (Fig. 2). In the case of AR, a plateau of negative relative scores is observed for GlideXP. The hardest case is the TACE metalloprotein. Here, all the SFs produce false-positive solutions but to a different extent. The SQM/COSMO filter performs best, followed by CS. This example in particular shows the strength of an electronic-structure theory description of P–L binding. The presence of the metal cation in this protein and the associated charge-transfer effects between the ligand and the cation are not adequately described by classical force-fields or statistical potentials, but they are well represented by the SQM/COSMO filter.
| Scoring function | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| SQM/COSMO | AMBER/GB | Glide | PLANTS | AutoDock | Gold | ||||
| XP | PLP | Vina | ASP | CS | GS | ChemPLP | |||
| AChE | 0 | 0 | 4 | 12 | 0 | 2 | 3 | 0 | 0 |
| AR | 0 | 1 | 67 | 0 | 10 | 1 | 0 | 1 | 0 |
| TACE | 39 | 171 | 181 | 294 | 63 | 56 | 49 | 78 | 111 |
| HIV PR | 0 | 0 | 98 | 0 | 7 | 0 | 2 | 1 | 8 |
| Total | 39 | 172 | 350 | 306 | 80 | 59 | 54 | 80 | 119 |
The second criterion, RMSDmax, is shown for the interval of the normalised relative scores below 5 (Table 2). The SQM/COSMO filter shows the lowest RMSDmax of 0.88 Å on average. CS follows with 1.28 Å on average. ASP and AMBER/GB satisfy the conditions of an averaged RMSDmax up to 2 Å. AMBER/GB, however, fails in the difficult case of TACE with RMSDmax of 4.76 Å. Analogous analyses at greater intervals have revealed a similar ordering of the SFs (Table S4, ESI†).
| Scoring function | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| SQM/COSMO | AMBER/GB | Glide | PLANTS | AutoDock | Gold | ||||
| XP | PLP | Vina | ASP | CS | GS | ChemPLP | |||
| Maximal RMSD within a window of 5 of the normalised score | |||||||||
| AchE | 0.47 | 0.57 | 2.13 | 0.78 | 0.78 | 1.78 | 1.43 | 1.14 | 0.78 |
| AR | 0.19 | 0.19 | 7.54 | 1.14 | 3.54 | 2.32 | 1.15 | 2.21 | 1.49 |
| TACE | 1.91 | 4.76 | 3.02 | 2.91 | 7.13 | 2.01 | 1.54 | 2.44 | 2.40 |
| HIV PR | 0.94 | 0.94 | 17.26 | 12.60 | 11.62 | 1.00 | 1.01 | 12.60 | 11.62 |
| Average | 0.88 | 1.62 | 7.49 | 4.61 | 5.77 | 1.78 | 1.28 | 4.60 | 4.55 |
The SQM/COSMO filter enables us not only to recognise the correct binding pose (RMSD below 2 Å) but also to go beyond this limit and evaluate even small changes in the geometry of the ligand binding.
The price for such a high accuracy is the increased computational time requirements. The SQM/COSMO filter is ca. 100-times slower than the statistics- and knowledge-based SFs and about 10-times slower than the classical physics-based AMBER/GB. However, compared to the standard SQM-based SF, it is ca. 100-times faster. The speed can be further enhanced by parallelisation.
To summarise, we have pushed the limits of the accuracy of SFs to judge the energetics of P–L noncovalent interactions. Based on our development and the extensive experience with SQM-based scoring functions,3,21 the SQM/COSMO filter has been introduced. It features two dominant terms to describe P–L interaction, namely the ΔEint term at the PM6-D3H4X level for gas-phase noncovalent interactions and the ΔΔGsolv term at the COSMO level for implicit solvation. We showed previously that both these methods are very accurate at a reasonable speed.16,20 The SQM/COSMO energy is calculated in four unrelated P–L complexes. The SQM/COSMO filter is compared to eight widely used SFs, which are statistics-, knowledge- or force-field-based. The SQM/COSMO scheme exhibits a superior performance as judged by two criteria, the number of false positives and RMSDmax. In contrast to standard SFs, no fitting against data sets has been involved. Furthermore, it offers generality and comparability across the chemical space and no system-specific parameterisations have to be performed. The time requirements allow for calculations of thousands of docking poses as we have demonstrated in this pilot study. We propose the SQM/COSMO filter as a tool for accurate medium-throughput refinement in later stages of virtual screening or as a reference method for judging the performance of other scoring functions. The proof of concept that reliable QM calculations can now be performed for tens of thousands of large biochemical entities opens a way to progress in closely related disciplines such as materials design.
This work was supported by research projects RVO 61388963 awarded to the Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic. We acknowledge the financial support of the Czech Science Foundation (grant number P208/12/G016). The authors acknowledge the support by the project L01305 of the Ministry of Education, Youth and Sports of the Czech Republic. The computations were performed at the Center for Information Services and High Performance Computing (ZIH) at TU Dresden.
References
- G. L. Warren, C. W. Andrews, A. M. Capelli, B. Clarke, J. LaLonde, M. H. Lambert, M. Lindvall, N. Nevis, S. F. Semus and S. Senger, et al. , J. Med. Chem., 2006, 49, 5912 CrossRef CAS PubMed; A. R. Leach, B. K. Shoichet and C. E. Peishoff, J. Med. Chem., 2006, 49, 5851 CrossRef PubMed.
- H. Gohlke and G. Klebe, Angew. Chem., Int. Ed., 2002, 41, 2644 CrossRef CAS.
- R. Meier, M. Pippel, F. Brandt, W. Sippl and C. Baldauf, J. Chem. Inf. Model., 2010, 50, 879 CrossRef CAS PubMed.
- P. S. Charifson, J. J. Corkery, M. A. Murcko and W. P. Walters, J. Med. Chem., 1999, 42, 5100 CrossRef CAS PubMed; R. Wang and S. J. Wang, J. Chem. Inf. Comput. Sci., 2001, 41, 1422 CrossRef PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157 CrossRef CAS PubMed.
- K. Raha, M. B. Peters, B. Wang, N. Yu, A. M. Wollacott, L. M. Westerhoff and K. M. Merz, Drug Discovery Today, 2007, 12, 725 CrossRef CAS PubMed; M. Xu and M. A. Lill, Drug Discovery Today: Technol., 2013, 10, 411 CrossRef PubMed.
- D. Mucs and R. A. Bryce, Expert Opin. Drug Discovery, 2013, 8, 263 CrossRef CAS PubMed.
- S. A. Hayik, R. Dunbrack and K. M. Merz, J. Chem. Theory Comput., 2010, 6, 3079 CrossRef CAS PubMed.
- M. Hennemann and T. Clark, J. Mol. Model., 2014, 20, 2331 CrossRef PubMed.
- H. S. Muddana and M. K. Gilson, J. Chem. Theory Comput., 2012, 8, 2023 CrossRef CAS PubMed; P. Mikulskis, S. Genheden, K. Wichmann and U. Ryde, J. Comput. Chem., 2012, 33, 1179 Search PubMed.
- P. Soderhjelm, J. Kongsted and U. Ryde, J. Chem. Theory Comput., 2010, 6, 1726 CrossRef PubMed.
- K. Wichapong, A. Rohe, C. Platzer, I. Slynko, F. Erdmann, M. Schmidt and W. Sippl, J. Chem. Inf. Model., 2014, 54, 881 CrossRef CAS PubMed; P. Chaskar, V. Zoete and U. F. Röhrig, J. Chem. Inf. Model., 2014, 54, 3137 CrossRef PubMed; S. K. Burger, D. C. Thompson and P. W. Ayers, J. Chem. Inf. Model., 2011, 51, 93 CrossRef PubMed.
- J. Antony, S. Grimme, D. G. Liakos and F. Neese, J. Phys. Chem. A, 2011, 115, 11210 CrossRef CAS PubMed.
- M. S. Gordon, D. G. Fedorov, S. R. Pruitt and L. V. Slipchenko, Chem. Rev., 2012, 112, 632 CrossRef CAS PubMed; J. Antony and S. Grimme, J. Comput. Chem., 2012, 33, 1730 CrossRef PubMed.
- M. Lepšík, J. Řezáč, M. Kolář, A. Pecina, P. Hobza and J. Fanfrlík, ChemPlusChem, 2013, 78, 921 CrossRef.
- J. Řezáč, J. Fanfrlík, D. Salahub and P. Hobza, J. Chem. Theory Comput., 2009, 5, 1749 CrossRef PubMed; J. Řezáč and P. Hobza, Chem. Phys. Lett., 2011, 506, 286 CrossRef; J. Řezáč and P. Hobza, J. Chem. Theory Comput., 2012, 8, 141 CrossRef PubMed.
- K. Raha and K. M. Merz, J. Am. Chem. Soc., 2004, 126, 1020 CrossRef CAS PubMed; K. Raha and K. M. Merz, J. Med. Chem., 2005, 48, 4558 CrossRef PubMed.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 RSC.
- M. Kolář, J. Fanfrlík, M. Lepšík, F. Forti, F. J. Luque and P. Hobza, J. Phys. Chem. B, 2013, 117, 5950 CrossRef PubMed.
- J. Fanfrlík, A. K. Bronowska, J. Řezáč, O. Přenosil, J. Konvalinka and P. Hobza, J. Phys. Chem. B, 2010, 114, 12666 CrossRef CAS PubMed; P. Dobeš, J. Řezáč, J. Fanfrlík, M. Otyepka and P. Hobza, J. Phys. Chem. B, 2011, 115, 8581 CrossRef PubMed; A. Pecina, O. Přenosil, J. Fanfrlík, J. Řezáč, J. Granatier, P. Hobza and M. Lepšík, Collect. Czech. Chem. Commun., 2011, 76, 457 CrossRef; A. Pecina, M. Lepšík, J. Řezáč, J. Brynda, P. Mader, P. Řezáčová, P. Hobza and J. Fanfrlík, J. Phys. Chem. B, 2013, 117, 16096 CrossRef PubMed; J. Fanfrlík, M. Kolář, M. Kamlar, D. Hurn, F. X. Ruiz, A. Cousido-Siah, A. Mitschler, J. Řezáč, E. Munusamy and M. Lepšík, et al. , ACS Chem. Biol., 2013, 8, 2484 CrossRef PubMed; J. Fanfrlík, F. X. Ruiz, A. Kadlčíková, J. Řezáč, A. Cousido-Siah, A. Mitschler, S. Haldar, M. Lepšík, M. H. Kolář and P. Majer, et al. , ACS Chem. Biol., 2015, 10, 1637 CrossRef PubMed.
- J. Fanfrlík, P. S. Brahmkshatriya, J. Řezáč, A. Jílková, M. Horn, M. Mareš, P. Hobza and M. Lepšík, J. Phys. Chem. B, 2013, 117, 14973 Search PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177 CrossRef CAS PubMed.
- O. Korb, T. Stützle and T. E. Exner, J. Chem. Inf. Model., 2009, 49, 84 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455 CAS.
- M. D. Eldridge, C. W. Murray, T. R. Auton, G. V. Paolini and R. P. Mee, J. Comput.-Aided Mol. Des., 1997, 11, 425 CrossRef CAS PubMed.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727 CrossRef CAS PubMed.
- W. T. M. Mooij and M. L. Verdonk, Proteins, 2005, 61, 272 CrossRef CAS PubMed.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo and R. T. Lee, J. Comput. Chem., 2003, 24, 1999 CrossRef CAS PubMed; V. Tsui and D. A. Case, Biopolymers, 2001, 56, 275 CrossRef.
- A. Nicholls and A. N. Jain, J. Comput.-Aided Mol. Des., 2008, 22, 133 CrossRef PubMed.
- B. Liu, S. Wang and X. Wang, Sci. Rep., 2015, 50, 15479 CrossRef PubMed.
- E. Perola, W. P. Walters and P. S. Charifson, Proteins, 2004, 56, 235 CrossRef CAS PubMed; J. W. M. Nissink, C. Murray, M. Hartshorn, M. L. Verdonk, J. C. Cole and R. Taylor, Proteins, 2002, 49, 457 CrossRef PubMed; P. Ferrara, H. Gohlke, D. J. Price, G. Klebe and C. L. Brooks, J. Med. Chem., 2004, 47, 3032 CrossRef PubMed.
- G. Klebe, Drug Discovery Today, 2006, 11, 580 CrossRef CAS PubMed.
- H. Dvir, D. M. Wong, M. Harel, X. Barril, M. Orozco, F. J. Luque, D. Munoz-Torrero, P. Camps, T. L. Rosenberry and I. Silman, et al. , Biochemistry, 2002, 41, 2970 CrossRef CAS PubMed.
- U. K. Bandarage, T. Wang, J. H. Come, E. Perola, Y. Wei and B. G. Rao, Bioorg. Med. Chem. Lett., 2008, 18, 44 CrossRef CAS PubMed.
- H. Steuber, A. Heine and G. Klebe, J. Mol. Biol., 2007, 368, 618 CrossRef CAS PubMed.
- J. Brynda, P. Řezáčová, M. Fábry, M. Hořejší, R. Štouračová, J. Sedláček, M. Souček, M. Hradílek, M. Lepšík and J. Konvalinka, J. Med. Chem., 2004, 47, 2030 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc09499b |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |