
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper mediated decarboxylative direct C–H arylation of heteroarenes with benzoic acids†
Tuhin
Patra
,
Sudip
Nandi
,
Santosh K.
Sahoo
and
Debabrata
Maiti
*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai-400076, India. E-mail: dmaiti@chem.iitb.ac.in
First published on 17th November 2015
Abstract
Decarboxylative coupling reactions to date require a stoichiometric oxidant (such as copper and silver salts) for decarboxylation purposes along with a metal catalyst (e.g. palladium) for cross-coupling. In this communication, an economic and sustainable approach by using a simple copper salt was developed in the presence of molecular oxygen as the sole oxidant. A wide range of 5-membered heteroarenes undergo aryl–heteroaryl cross-coupling with electron deficient aryl carboxylic acids.
Five membered heterocycles are recognized as important structural motifs in pharmaceuticals, agrochemicals, natural products, functional organic materials and dye industries (Fig. 1).1 In this context, the synthetic importance of azole derivatives has ensured the continuous interest of chemists to find effective methods for regiospecific formation of aryl–heteroaryl bonds.2
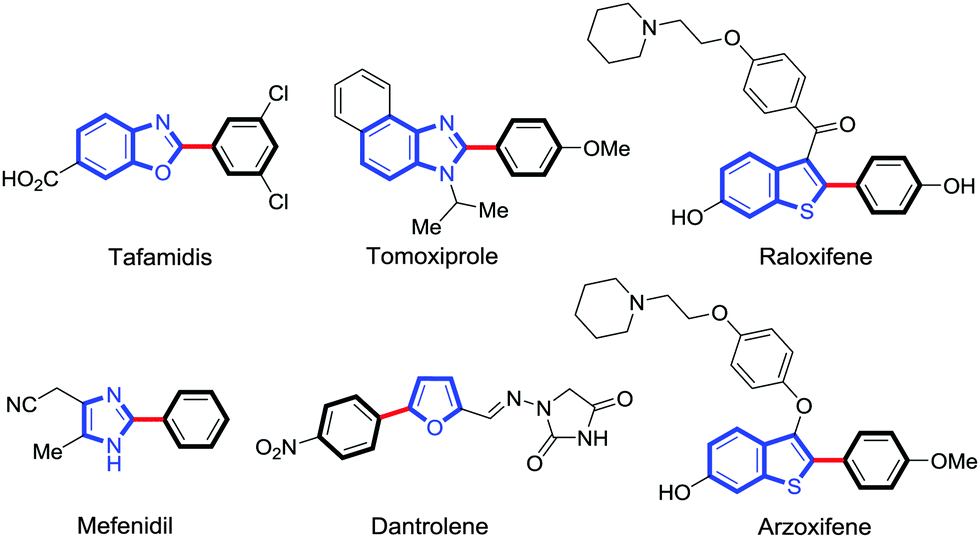 | ||
| Fig. 1 Few relevant aryl-heterocyclic cores in drug molecules. | ||
Traditional cross-coupling methods generally require expensive heavy transition metals, and pre-activated organometallic coupling partners and as a result, these methods often produce toxic and stoichiometric side-products.3 Alternatively, carboxylic acids are an exciting choice in place of traditional organometallic counterparts since they are widely available, inexpensive and easy to store and handle.4 Most importantly, transition metal catalyzed decarboxylation of aromatic carboxylic acids can provide the same aryl–metal intermediates by loss of CO2.5
Over recent years, benzoic acid derivatives have been popularized mainly by Goossen6 and others7 as alternative coupling partners with aryl halides and triflates.8 Consequently, direct C–H arylation reactions have been a prominent field of research since such transformations are capable of streamlining organic synthesis and minimizing wasteful byproducts.9 Therefore, a decarboxylative C–H bond functionalization that combines these two newly emerging approaches, that is, decarboxylation and direct C–H bond functionalization, holds a great potential for new bond forming strategies in synthesis. Interestingly, few methods have already been reported using this decarboxylative direct arylation approach. In these cases either Cu or Ag salts are used in stoichiometric amounts for decarboxylation purposes.10 Additionally, a Pd catalyst was often employed for cross-coupling.11,12 Employment of an economic, greener first row transition metal, for example copper, to play the dual role of decarboxylation followed by direct C–H arylation in the presence of molecular oxygen is yet to be explored (Scheme 1).
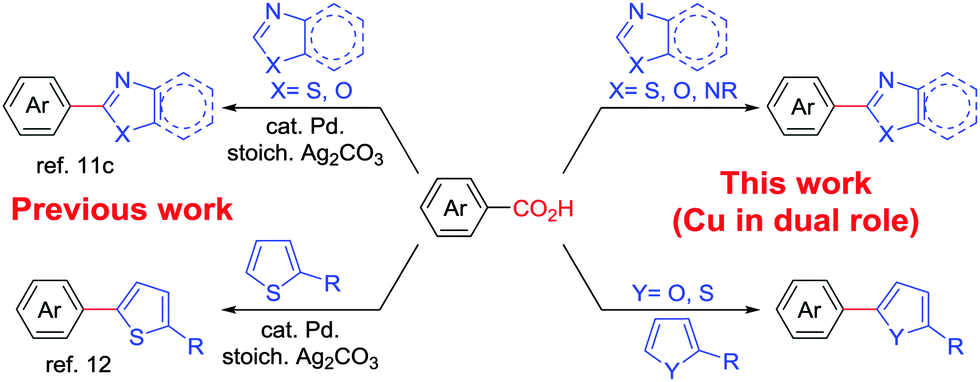 | ||
| Scheme 1 Decarboxylative C–H arylation of heteroarenes. | ||
Copper mediated methods for protodecarboxylation are well studied in the literature.13,14 Additionally, copper is also used broadly in different C–H functionalization reactions.15 Surprisingly, a simple copper catalyzed/mediated method for decarboxylative direct C–H arylation is underdeveloped despite its practical importance and high demand in the present context. Herein we report the first copper mediated C–H arylation of benzoic acid derivatives with different heteroarenes (Scheme 1) using molecular O2.
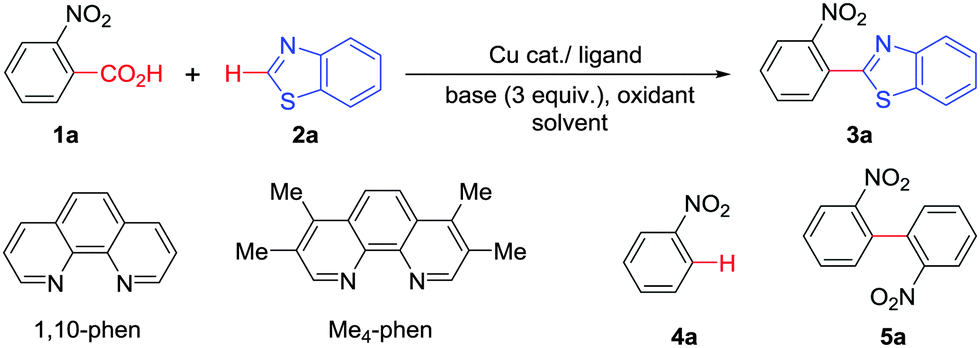
At the outset, we hypothesized that benzoic acid derivatives in the presence of a suitable copper salt can effectively produce direct C–H arylation products with 5-membered heteroarenes in a regioselective manner due to the intrinsic electronic bias among different C–H bonds. To test this presumption, 2-nitrobenzoic acid was employed with benzothiazole in the presence of CuCl2/1,10-phen and a base in different solvents. During optimization studies, we realized that choice of solvent plays a critical role in obtaining the desired product. Polar aprotic solvents (DMSO, DMF) gave the expected compound in trace amounts along with undesired 2,2′-dinitro-1,1′-biphenyl (5a) as the major product. On the other hand, polar protic solvents (EtOH, t-BuOH) resulted in a protodecarboxylative product, nitrobenzene (4a).
Less polar solvents (toluene, xylene) provided the desired coupling product in 15–20% yield along with varying amounts of 4a and 5a.16 Detailed evaluation of the model reaction system by varying different copper salts and ligands revealed that CuBr along with simple 1,10-phenanthroline can promote the desired reaction (Table 1, entries 1–6). Choice of base is also crucial since in the absence of base no cross-coupled product was obtained, and weak bases were found to produce better results (entries 8 and 9).16 Introduction of an oxidant, especially molecular oxygen improved the yield significantly. Upon careful control it was found that addition of 25 mol% of di-tert-butylperoxide (DTBP) in an oxygen atmosphere can improve the yield further to 63% (entries 10–13). Increase in temperature beyond 140 °C promoted the formation of 5a significantly by suppressing desired product formation.16 Control experiments confirmed that copper is responsible for decarboxylation as well as direct C–H arylation (entry 15) in the presence of molecular oxygen.16
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Cu] | Ligand | Base | [O] | 3a (%) |
| a Reaction conditions: 1 (0.6 mmol), Cu salt (30 mol%), ligand (60 mol%), base (3 equiv.), 2 (0.2 mmol) in toluene (1 mL) at 130 °C for 24 h. Yields were determined by gas chromatography using n-decane as the internal standard. b 25 mol% DTBP was used. c 4 Å MS (30 mg). d 140 °C. e Cu salt (15 mol%), ligand (30 mol%). | |||||
| 1 | CuCl2 | 1,10-Phen | K2CO3 | Air | 16 |
| 2 | CuBr2 | 1,10-Phen | K2CO3 | Air | 20 |
| 3 | CuBr | 1,10-Phen | K2CO3 | Air | 27 |
| 4 | CuBr | bipy | K2CO3 | Air | 16 |
| 5 | CuBr | TMEDA | K2CO3 | Air | 14 |
| 6 | CuBr | Me4-phen | K2CO3 | Air | 25 |
| 7 | CuBr | — | K2CO3 | Air | <1 |
| 8 | CuBr | 1,10-Phen | KHCO3 | Air | 12 |
| 9 | CuBr | 1,10-Phen | KF | Air | 24 |
| 10 | CuBr | 1,10-Phen | K2CO3 | K2S2O8 | 20 |
| 11 | CuBr | 1,10-Phen | K2CO3 | DTBP | 28 |
| 12 | CuBr | 1,10-Phen | K2CO3 | O2 | 57 |
| 13b | CuBr | 1,10-Phen | K2CO3 | O2 | 63 |
| 14b,c | CuBr | 1,10-Phen | K2CO3 | O2 | 71 |
| 15 | — | 1,10-Phen | K2CO3 | O2 | — |
| 16 , , | CuBr | 1,10-Phen | K2CO3 | O2 | 76 |
| 17e | CuBr | 1,10-Phen | K2CO3 | O2 | 53 |
Under these optimized conditions, scope of the reaction was investigated with 2-nitrobenzoic acid by varying different 5-membered heterocycles containing 2-heteroatoms. All benzothiazole, benzoxazole and benzimidazole cores were found to provide desired products in good yields (Table 2, entries 3a, 3b and 3c). This observation ruled out the possibility of ring opening of benzothiazole, followed by imine formation and the cyclization pathway, as observed in earlier cases to deliver only benzothiazole coupled products.17 The N-protected imidazole compounds also furnished the decarboxylated coupling product in synthetically useful yield (entry 3d). Chloro-substituted 2-nitrobenzoic acid was found to be effective to deliver the heteroarylated product with benzoxazole (entry 3e). During exploration of scope for substrates we realized that this current method is highly sensitive to the choice of benzoic acid. Mainly, benzoic acids with electron withdrawing groups are prone to easy decarboxylation to provide an aryl–copper intermediate14 and likely to deliver the desired direct C–H arylated product. Accordingly, different fluorosubstituted electron withdrawing benzoic acids were employed to obtain fluorosubstituted heteroarylated coupling products successfully albeit in low yields (entries 3f, 3g, 3h). Please note that decarboxylative coupling reactions are usually very sensitive to the electronic nature of the partners involved.11
|
|
||||
|---|---|---|---|---|
| Entry | Benzoic acid | Heteroarene | Product | Yield (%) |
| a Reaction conditions: 1 (0.6 mmol), CuBr (30 mol%), 1,10-phenanthrolene (60 mol%), K2CO3 (0.6 mmol), 4 Å MS (30 mg), 2 (0.2 mmol), DTBP (25 mol%) in toluene (1 mL) under O2 atmosphere at 140 °C for 24 h. b CuBr (15 mol%), 1,10-phenanthrolene (30 mol%). c CuBr (20 mol%), 1,10-phenanthrolene (40 mol%). | ||||
| 1 |
|
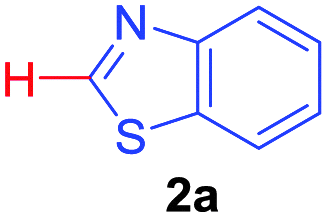
|
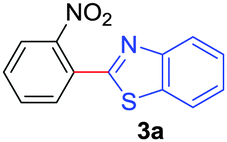
|
76 |
| 53b | ||||
| 2 | 1a |
|
|
71 |
| 51c | ||||
| 3 | 1a |
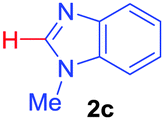
|
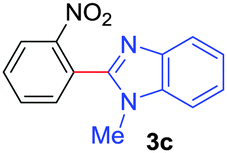
|
74 |
| 43b | ||||
| 56c | ||||
| 4 | 1a |
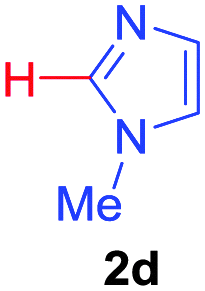
|
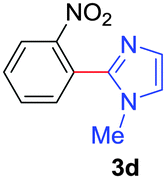
|
60 |
| 47c | ||||
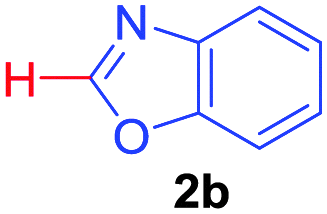
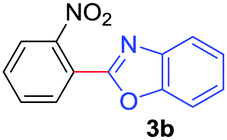
| 5 |
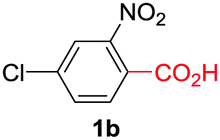
|
2b |
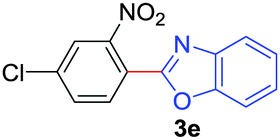
|
78 |
| 49b | ||||
| 6 |
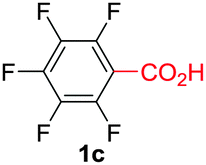
|
2a |
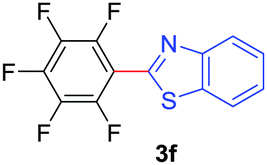
|
32 |
| 7 |
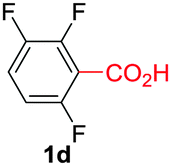
|
2a |
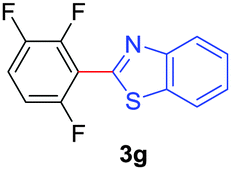
|
36 |
| 23c | ||||
| 8 | 1a | 2b |
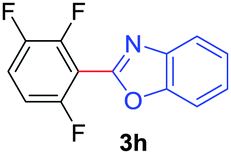
|
29 |
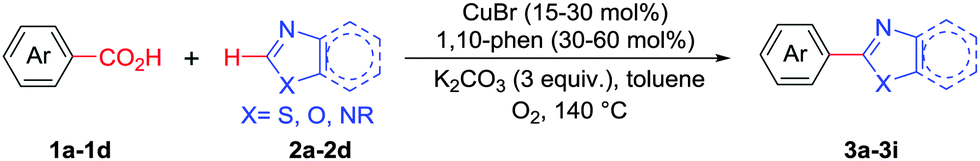
Next, we were intrigued by the possibility of whether the thiophene moiety can be employed in our current methodology or not. Thiophenes are less reactive compared to azole derivatives, and previously stoichiometric silver salts were used with the palladium catalyst for decarboxylative coupling with thiophenes.12 To our delight, benzothiophene delivered the C–H arylated product under the present reaction conditions in synthetically useful yield (Table 3, entry 3i). Next, the scope of different thiophene derivatives was contemplated with 2-nitrobenzoic acid as the model substrate. Aldehyde, cyano and halogen-substitutions were tolerated successfully (entries 3j, 3k, 3l). We were pleased to find that not only thiophene, but the furan moiety can also be used successfully in our present methods (entry 3m). Although this newly described methodology is sensitive to the choice of benzoic acids, it enjoys a wide range of variations in heterocycles.
|
|
||||
|---|---|---|---|---|
| Entry | Benzoid acid | Heteroarene | Product | Yield (%) |
| a Reaction conditions: 1 (0.6 mmol), CuBr (30 mol%), 1,10-phenanthrolene (60 mol%), K2CO3 (0.6 mmol), 4 Å MS (30 mg), 2 (0.2 mmol), DTBP (25 mol%) in toluene (1 mL) under O2 atmosphere at 140 °C for 24 h. b CuBr (15 mol%), 1,10-phenanthrolene (30 mol%). c CuBr (20 mol%), 1,10-phenanthrolene (40 mol%). | ||||
| 1 | 1a |
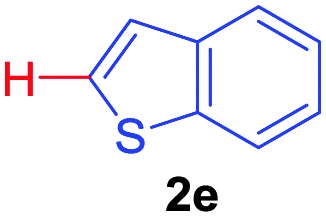
|
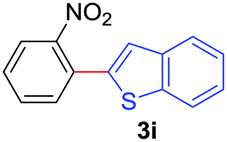
|
83 |
| 54b | ||||
| 2 | 1a |
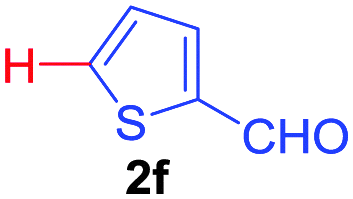
|
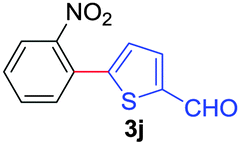
|
68 |
| 44c | ||||
| 3 | 1a |
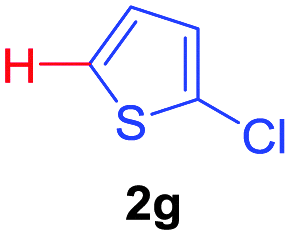
|
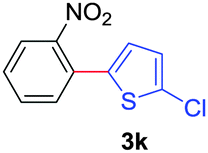
|
73 |
| 46c | ||||
| 4 | 1a |
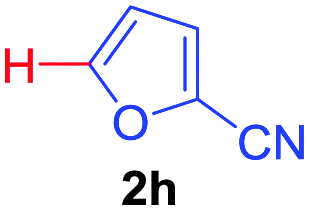
|
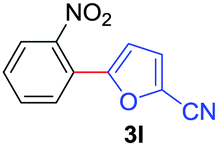
|
76 |
| 49c | ||||
| 5 | 1b | 2g |
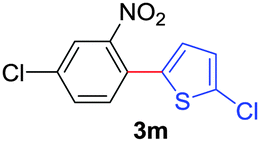
|
70 |
| 57b | ||||
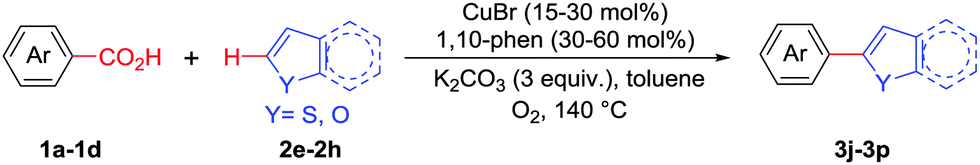
After exploring the scope of substrates, we tried to explicate the mechanistic complexity for this newly developed protocol. Control experiments without heterocycles provided decarboxylated products 4a and 5a, exclusively. Both 4a and 5a were unreactive during their independent reactions with heterocycles. Again, homo-coupling of benzothiazole under the reaction conditions was found to be sluggish (Scheme 2).16 This implies that ligated copper species first forms the copper salt of benzoic acid followed by the extrusion of CO2 to afford an aryl–copper intermediate.14 Additionally, a radical pathway may be disfavoured as no significant drop in product yield was noticed when different radical scavengers were examined.16 In accordance with these observations, along with recent literature reports,18 a plausible mechanistic cycle is proposed. First, ligated copper forms the aryl–copper intermediate via decarboxylation. Then, base assisted metalation of heteroarene with aryl–copper species may lead to either the Cu(I) intermediate (pathway A) or the Cu(II) intermediate (pathway B) by instantaneous oxidation. This Cu(I)/Cu(II) intermediate can undergo reversible sluggish oxidation to generate an aryl–Cu(III) intermediate followed by immediate reductive elimination to provide the desired direct C–H arylated product (Scheme 3). Although the exact role of oxygen is not clear, we believe that it is mainly involved in the formation of aryl–Cu(III) species.19
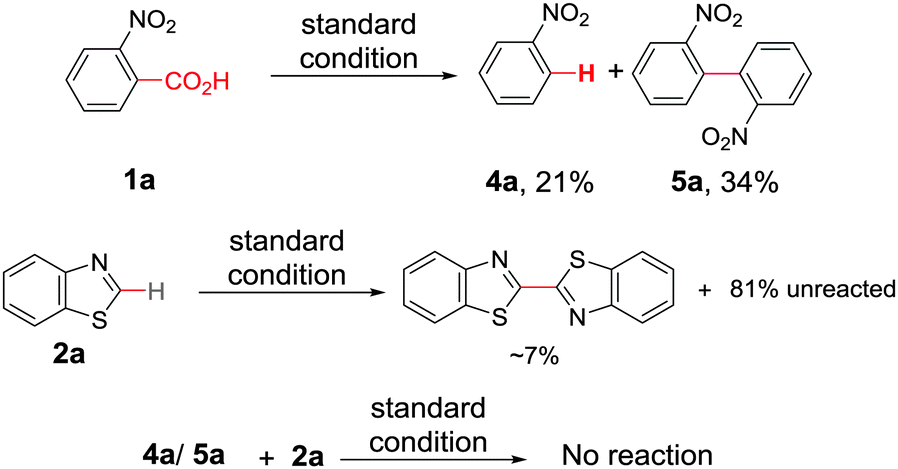 | ||
| Scheme 2 Control experiments. | ||
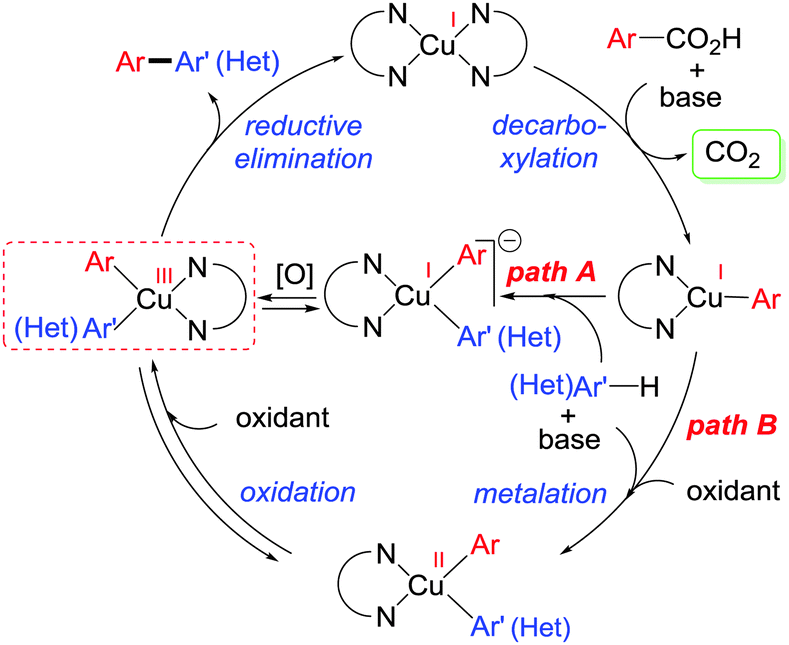 | ||
| Scheme 3 Plausible mechanism. | ||
In summary, we have developed a useful protocol with a simple copper/1,10-phen based system in the presence of molecular oxygen as the sole-oxidant for aryl–heteroaryl cross-coupling employing electron deficient benzoic acids. This method is advantageous because of its wide tolerance towards different heterocycles bearing one or two heteroatoms under relatively mild conditions. Investigation into the mechanistic understanding of the development of direct C–H arylation by copper is currently ongoing in our group.
This activity is supported by SERB, India (EMR/2014/000164). Financial support received from UGC India (T.P.) and IIT-B (S.S.) is gratefully acknowledged.
Notes and references
- (a) R. C. Koehler, D. A. Wilson, M. C. Rogers and R. J. Traystman, J. Pharmacol. Exp. Ther., 1985, 233, 327 CAS; (b) R. E. West Jr, S. M. Williams, H. S. She, N. I. Carruthers, R. W. Egan and M. Motasim Billah, Prostaglandins, 1997, 54, 891 CrossRef CAS; (c) R. Zucchi and S. Ronca-Testoni, Pharmacol. Rev., 1997, 49, 1 CAS; (d) E. Barrett-Connor, Ann. N. Y. Acad. Sci., 2001, 949, 295 CrossRef CAS PubMed; (e) C. R. Overk, K.-W. Peng, R. T. Asghodom, I. Kastrati, D. D. Lantvit, Z. Qin, J. Frasor, J. L. Bolton and G. R. J. Thatcher, ChemMedChem, 2007, 2, 1520 CrossRef CAS PubMed; (f) C. E. Bulawa, S. Connelly, M. DeVit, L. Wang, C. Weigel, J. A. Fleming, J. Packman, E. T. Powers, R. L. Wiseman, T. R. Foss, I. A. Wilson, J. W. Kelly and R. Labaudinière, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9629 CrossRef CAS PubMed.
- (a) A. K. Verma, T. Kesharwani, J. Singh, V. Tandon and R. C. Larock, Angew. Chem., Int. Ed., 2009, 48, 1138 CrossRef PubMed; (b) S. Park, J. Jung and E. J. Cho, Eur. J. Org. Chem., 2014, 4148 CrossRef CAS.
- B. Martín-Matute, K. J. Szabó and T. N. Mitchell, in Metal-Catalyzed Cross-Coupling Reactions and More, ed. A. d. Meijere, S. Brase and M. Oestreich, Wiley-VCH Verlag GmbH & Co. KGaA, 2014, p. 423 Search PubMed.
- M. Nakamura, A. Hajra, K. Endo and E. Nakamura, Angew. Chem., Int. Ed., 2005, 44, 7248 CrossRef CAS PubMed.
- (a) L. J. Goossen, N. Rodríguez and K. Goossen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef CAS PubMed; (b) N. Rodriguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030 RSC; (c) W. I. Dzik, P. P. Lange and L. J. Goossen, Chem. Sci., 2012, 3, 2671 RSC; (d) P. Hu, Y. Shang and W. Su, Angew. Chem., Int. Ed., 2012, 51, 5945 CrossRef CAS PubMed; (e) C. J. Gartshore and D. W. Lupton, Angew. Chem., Int. Ed., 2013, 52, 4113 CrossRef CAS PubMed.
- (a) L. J. Goossen, G. Deng and L. M. Levy, Science, 2006, 313, 662 CrossRef CAS PubMed; (b) L. J. Goossen, N. Rodriguez and C. Linder, J. Am. Chem. Soc., 2008, 130, 15248 CrossRef CAS PubMed; (c) L. J. Goossen, B. Zimmermann and T. Knauber, Angew. Chem., Int. Ed., 2008, 47, 7103 CrossRef CAS PubMed; (d) S. Bhadra, W. I. Dzik and L. J. Goossen, J. Am. Chem. Soc., 2012, 134, 9938 CrossRef CAS PubMed.
- (a) A. Voutchkova, A. Coplin, N. E. Leadbeater and R. H. Crabtree, Chem. Commun., 2008, 6312 RSC; (b) J.-J. Dai, J.-H. Liu, D.-F. Luo and L. Liu, Chem. Commun., 2011, 47, 677 RSC; (c) S. Messaoudi, J.-D. Brion and M. Alami, Org. Lett., 2012, 14, 1496 CrossRef CAS PubMed.
- R. Shang and L. Liu, Sci. China: Chem., 2011, 54, 1670 CrossRef CAS.
- (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed; (b) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed.
- (a) S. Zhao, Y.-J. Liu, S.-Y. Yan, F.-J. Chen, Z.-Z. Zhang and B.-F. Shi, Org. Lett., 2015, 17, 3338 CrossRef CAS PubMed; (b) L. Chen, L. Ju, K. A. Bustin and J. M. Hoover, Chem. Commun., 2015, 51, 15059 RSC.
- (a) J. Cornella, P. Lu and I. Larrosa, Org. Lett., 2009, 11, 5506 CrossRef CAS PubMed; (b) F. Zhang and M. F. Greaney, Angew. Chem., Int. Ed., 2010, 49, 2768 CrossRef CAS PubMed; (c) K. Xie, Z. Yang, X. Zhou, X. Li, S. Wang, Z. Tan, X. An and C.-C. Guo, Org. Lett., 2010, 12, 1564 CrossRef CAS PubMed; (d) J. Zhou, P. Hu, M. Zhang, S. Huang, M. Wang and W. Su, Chem. – Eur. J., 2010, 16, 5876 CrossRef CAS PubMed; (e) H. Zhao, Y. Wei, J. Xu, J. Kan, W. Su and M. Hong, J. Org. Chem., 2011, 76, 882 CrossRef CAS PubMed; (f) S. Seo, M. Slater and M. F. Greaney, Org. Lett., 2012, 14, 2650 CrossRef CAS PubMed; (g) K. Yang, C. Zhang, P. Wang, Y. Zhang and H. Ge, Chem. – Eur. J., 2014, 20, 7241 CrossRef CAS PubMed; (h) G. Shi, C. Shao, S. Pan, J. Yu and Y. Zhang, Org. Lett., 2015, 17, 38 CrossRef CAS PubMed; (i) J. Kan, S. Huang, J. Lin, M. Zhang and W. Su, Angew. Chem., Int. Ed., 2015, 54, 2199 CrossRef CAS PubMed; (j) Y. Zhang, H. Zhao, M. Zhang and W. Su, Angew. Chem., Int. Ed., 2015, 54, 3817 CrossRef CAS PubMed.
- P. Hu, M. Zhang, X. Jie and W. Su, Angew. Chem., Int. Ed., 2012, 51, 227 CrossRef CAS PubMed.
- (a) M. Nilsson, Acta Chem. Scand., 1966, 20, 423 CrossRef CAS; (b) A. Cairncross, J. R. Roland, R. M. Henderson and W. A. Sheppard, J. Am. Chem. Soc., 1970, 92, 3187 CrossRef; (c) T. Cohen and R. A. Schambach, J. Am. Chem. Soc., 1970, 92, 3189 CrossRef CAS; (d) L. J. Goossen, F. Manjolinho, B. A. Khan and N. Rodríguez, J. Org. Chem., 2009, 74, 2620 CrossRef CAS PubMed.
- (a) L. J. Goossen, W. R. Thiel, N. Rodríguez, C. Linder and B. Melzer, Adv. Synth. Catal., 2007, 349, 2241 CrossRef CAS; (b) L. J. Goossen, N. Rodríguez, C. Linder, P. P. Lange and A. Fromm, ChemCatChem, 2010, 2, 430 CrossRef CAS.
- (a) H.-Q. Do and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 12404 CrossRef CAS PubMed; (b) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (c) H.-Q. Do and O. Daugulis, J. Am. Chem. Soc., 2011, 133, 13577 CrossRef CAS PubMed; (d) S. Guin, T. Ghosh, S. K. Rout, A. Banerjee and B. K. Patel, Org. Lett., 2011, 13, 5976 CrossRef CAS PubMed; (e) A. Gogoi, S. Guin, S. K. Rout and B. K. Patel, Org. Lett., 2013, 15, 1802 CrossRef CAS PubMed; (f) M. Ghosh, S. Mishra, K. Monir and A. Hajra, Org. Biomol. Chem., 2015, 13, 309 RSC.
- See the ESI† for detailed description.
- Q. Song, Q. Feng and M. Zhou, Org. Lett., 2013, 15, 5990 CrossRef CAS PubMed.
- (a) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (b) L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2012, 134, 1298 CrossRef CAS PubMed; (c) A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 9797 CrossRef CAS PubMed.
- (a) A. E. King, L. M. Huffman, A. Casitas, M. Costas, X. Ribas and S. S. Stahl, J. Am. Chem. Soc., 2010, 132, 12068 CrossRef CAS PubMed; (b) A. Casitas and X. Ribas, Chem. Sci., 2013, 4, 2301 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Optimization details, reaction procedure, characterization data and 1H, 13C NMR spectra. See DOI: 10.1039/c5cc08367b |
| This journal is © The Royal Society of Chemistry 2016 |