
Expanding the scope of alkyne-mediated bioconjugations utilizing unnatural amino acids†
Johnathan C.
Maza
,
Zachary M.
Nimmo
and
Douglas D.
Young
*
Department of Chemistry, College of William & Mary, Williamsburg, Virginia 23187, USA. E-mail: dyoung01@wm.edu
First published on 21st October 2015
Abstract
The importance of bioconjugates within the field of chemistry drives the need for novel methodologies for their preparation. Well-defined and stable bioconjugates are easily accessible via the utilization of unnatural amino acids (UAAs). As such, we have synthesized and incorporated two new UAAs into green fluorescent protein, and optimized a novel Cadiot–Chodkiewicz bioconjugation, effectively expanding the toolbox of chemical reactions that can be employed in the preparation of bioconjugates.
Bioconjugates represent a class of molecules in which a biomacromolecule is linked to another molecule, typically a probe, a surface, or a cytotoxic compound.1,2 Protein-based bioconjugates, in which a protein is the biomolecule, represent an ever-expanding field of research. Specifically, protein-based bioconjugates are becoming increasingly popular for “lab-on-a-chip” technologies, in which a diagnostic protein is immobilized on a surface, as well as towards the development of novel cancer therapeutics via the preparation of antibody-drug conjugates.3–5
The methods to generate a bioconjugate range from non-covalent interactions, typically adsorption and encapsulation, to covalent linkages.6 While a covalent attachment is more robust, and less easily disrupted in a biological setting, obtaining a degree of specificity during the synthesis has precluded its widespread application. This is a consequence of often utilizing reactive residues within the protein of interest. However, there are typically multiple nucleophilic residues that can react within the protein.1 The result is a non-specific coupling that can either disrupt normal protein function, result in improper orientation of the protein-bioconjugate, or lead to heterogeneous mixtures of linkages at multiple residues.6 These limitations can be overcome by site-specifically incorporating UAAs with chemical functionalities not found in the canonical amino acids.7,8 In particular, the suppression of the amber (TAG) stop codon via an exogenous amino-acyl tRNA synthetase (aaRS)/tRNA pair allows a high level of control over the position of the UAA.9 These UAA-containing proteins can then be conjugated to other molecules via a bioorthogonal reaction that occurs at physiological conditions (pH = 7, 37 °C) with no chance of cross-reaction with other biomolecules.10,11
Indeed a variety of bioorthogonal reactions have already been developed. These include the copper(I)-catalyzed cycloaddition between azides and terminal alkynes,12–14 the strain-promoted cycloaddition,15–17 the oxime formation,18–21 and more recently, the formation of a conjugated diyne via the copper(I)-catalyzed Glaser–Hay coupling.22 Despite the wide array of bioorthogonal chemistries available, each possessing advantages and disadvantages, the individual requirements of the bioconjugate help dictate which reaction may be best to employ. Due to the increasing application of bioconjugates, the development of novel bioorthogonal reactions (and UAAs with which they can be employed) are at the forefront of the field.
The Cadiot–Chodkiewicz reaction affords a conjugated diyne via the reaction of a terminal alkyne and a halo-alkyne (Fig. 1). In the presence of a copper(I) salt and a monodentate nitrogenous ligand, usually triethylamine (TEA), the reaction proceeds to form a covalent linkage in the form of a conjugated diyne in a relatively chemoselective fashion.23,24 Furthermore, the overall reaction is net redox neutral, as a single copper(I) catalyst goes through a series of oxidative additions and reductive eliminations with the bromoalkyne and terminal alkyne reactant to yield the conjugated diyne.25
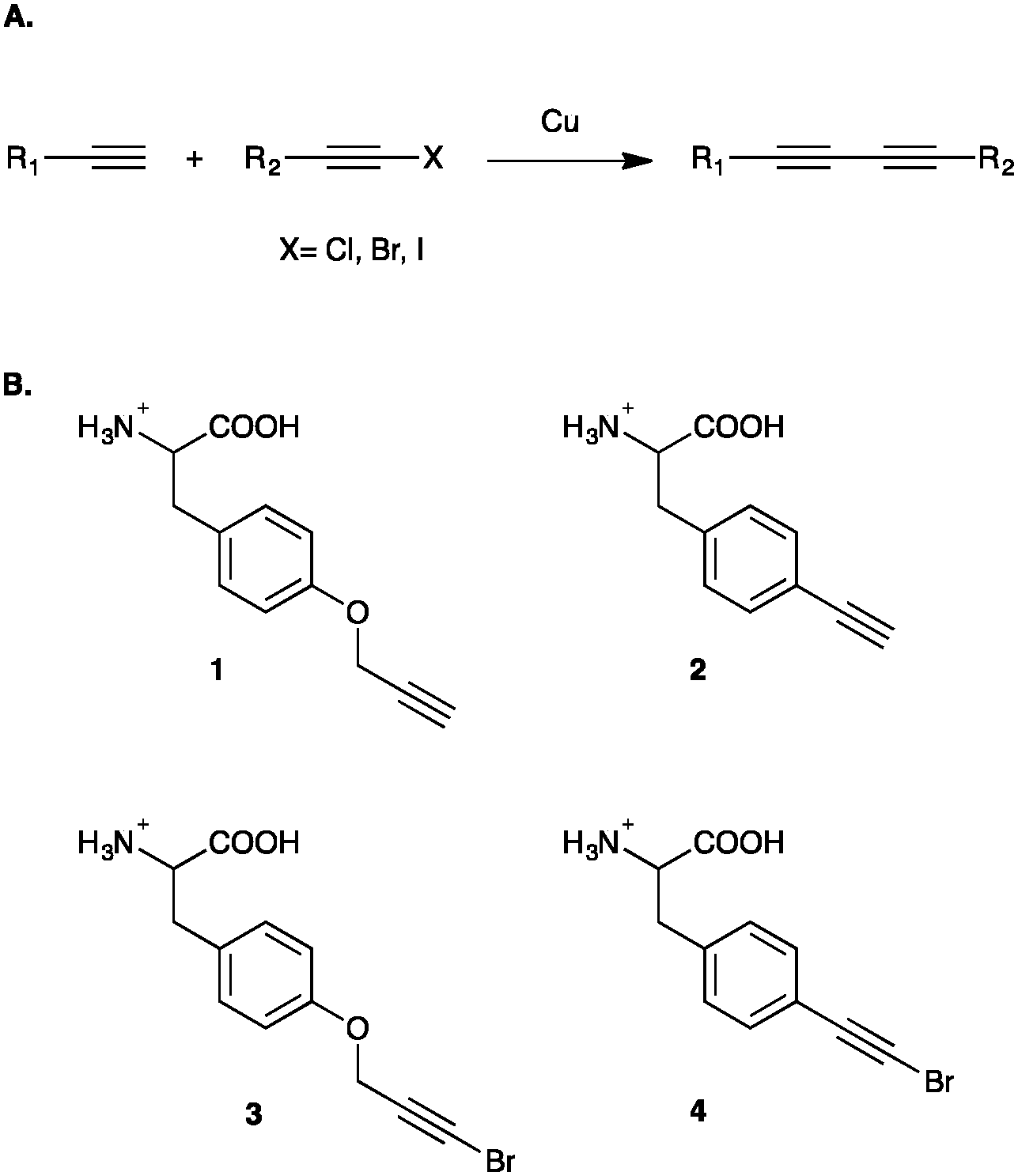 | ||
| Fig. 1 Cadiot–Chodkiewicz reaction and associated UAAs. (A) Standard Cadiot–Chodkiewicz reaction employing a copper(I) salt to couple a terminal alkyne to a haloalkyne, affording an asymmetrical conjugated diyne product. (B) p-Propargyloxyphenylalanine (1, pPrF), p-ethynylphenylalanine (2, pEtF), p-bromopropargyloxyphenylalanine (3, pBrPrF), p-bromoethynylphenylalanine (4, pBrEtF) incorporated into GFP-151 for Glaser–Hay and Cadiot–Chodkiewicz reactions. | ||
The Cadiot–Chodkiewicz reaction has diverse applications to many areas of chemistry. It has been utilized in polymerization reactions, such as in the formation of the backbone of a solid-state polymer crystal, or in the fabrication of polymerized monolayer assemblies.26,27 Additionally, several acetylenic natural products exhibiting valuable biological properties can be obtained via a Cadiot–Chodkiewicz reaction.28
Similar to the Cadiot–Chodkiewicz reaction, the Glaser–Hay coupling of two terminal alkynes also affords a diyne.29,30 The reaction brings together two terminal alkynes generate a diyne linkage; however, it is not redox neutral and has chemoselectivity issues as nothing differentiates the two terminal alkynes.31 The reaction involves the addition of a bidentate nitrogenous ligand, triethylmethylenediamine (TMEDA),32 which lowers the reaction temperatures and enhances the kinetics of the reaction. Recently, we demonstrated a bioorthogonal variant of the Glaser–Hay reaction that can be conducted in an aqueous setting under physiological conditions.22 Using a terminal alkyne-containing UAA, we were able to demonstrate that the reaction proceeds to completion within approximately 6 h at 4 °C, with near quantitative conjugation. As previously mentioned, the Glaser–Hay coupling of terminal alkynes has a chemoselectivity issue when the terminal alkynes differ, resulting in the formation of unwanted homodimers. However, due to the steric bulk of the protein, we found that the homodimerization of the reaction was mostly inhibited, leading to primarily the desired protein heterodimer product. As such we were able to demonstrate that the Glaser–Hay reaction could be employed as a novel bioorthogonal chemistry, yielding stable conjugates with well-defined geometries. However, the reaction was limited by the oxidative damage of the protein due to the mechanistic cycling of the copper through three different redox states. As a result, reactions proceeding for longer than 6 h, resulted in oxidative damage and protein degradation.
A key component of the Glaser–Hay mechanism is the formation of a copper(II)-hydroxyl intermediate, which has been shown to produce hydroxyl radicals that are deleterious to living systems.33 Because the copper(I) of the Cadiot–Chodkiewicz reaction is not thought to utilize copper(II) intermediates, we reasoned that the chemistry could be employed in a biological context to minimize previously observed oxidative damage. Furthermore, the reaction is highly chemoselective, as the use of a halo-alkyne minimizes the formation of homodimer side products by differentiating the two alkynes. Ultimately, this has the potential to increase the yield of the conjugated protein product. Based on these facts and the limitations of the bioorthogonal Glaser–Hay, we sought to develop a bioorthogonal variant of the Cadiot–Chodkiewicz reaction that could be conducted in an aqueous setting and under physiological conditions.
In order to conduct and optimize a Cadiot–Chodkiewicz bioconjugation, new UAAs harboring a terminal haloalkyne needed to be synthesized and incorporated into a protein. In order to probe UAA-dependent effects on the reaction, aliphatic and aromatic brominated alkynyl UAAs were prepared from the previously reported protected p-propargyloxyphenylalanine (pPrF, 1) and the p-ethynylphenylalanine (pEtF, 2) respectively.34,35 Gratifyingly, the well-established bromination of phenylacetylene using N-bromosuccinamide (NBS) and silver nitrate worked well for the synthesis of both UAAs in moderate yields.36,37 Following deprotection, the final UAAs, p-bromo-propargyloxyphenylalanine (pBrPrF, 3) and p-bromo-ethynylphenylalanine (pBrEtF, 4), were recovered in overall good yields (67% and 34% respectively) (Fig. 1).
With the pBrPrF and pBrEtF in hand, it was imperative to incorporate these UAAs into a model protein. Due to both its fluorescent properties and well-documented prior use in UAA development technologies, green fluorescent protein (GFP) was selected as a model system. Specifically, attempts were made to incorporate newly synthesized UAAs at residue 151 by suppressing the amber stop codon. Furthermore, our previous work immobilizing GFP revealed that this surface exposed site is ideal for UAA placement, as the rigidity of the residue helps orient the bioorthogonal functional handle.38 In lieu of undergoing a tedious aaRS selection process, we hoped to incorporate the brominated UAAs using the previously described promiscuous pCNF aaRS.39,40 The pCNF aaRS was investigated first to incorporate pBrPrF and pBrEtF due to their structural similarity to other UAAs that the pCNF aaRS incorporates.
BL21(DE3) E. coli were co-transformed with a pEVOL–pCNF plasmid and a pET-GFP-TAG151 plasmid and used to initiate an expression culture at OD600 0.1 which was grown to an OD600 0.7. The culture was subsequently centrifuged and the cell pellet was resuspended in 4 mL of LB broth supplemented with antibiotics, IPTG, arabinose, and the presence or absence of a UAA.34 This previously reported expression protocol allowed for the minimization of the amount of UAA employed, and was found to be very effective. After 18–20 h at 37 °C, cells were pelleted and the expressed GFP was purified.
Gratifyingly, the promiscuous pCNF aaRS incorporated both brominated-UAA variants with a higher fidelity than the simple terminal alkyne analogs (Fig. 2). As is to be expected, the smaller pBrEtF UAA, had a higher incorporation than the pBrPrF. We hypothesize that the 1-bromo-alkyne moiety provides a degree of hydrophobic character to the UAA, making the interaction between the amino acid and the hydrophobic binding pocket of the aaRS more favourable.
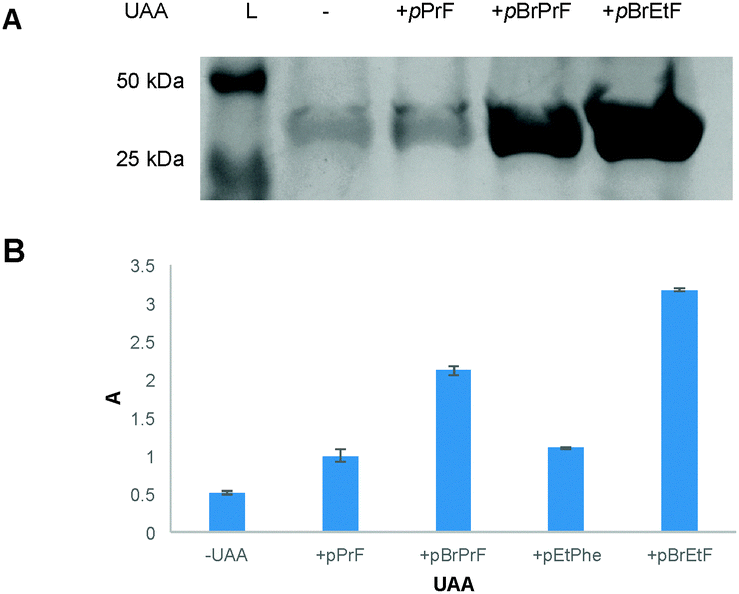 | ||
| Fig. 2 Expression of UAA-containing GFP151 using the promiscuous pCNF aaRS. (A) A stained SDS-PAGE gel indicated successful incorporation of 3 and 4 over background (−). A positive control, 1, was also utilized in an expression. (B) Data for overall incorporation of UAAs via the pCNF aaRS as measured via absorbance at 280 nm on a nanodrop spectrophotometer (ε/1000 = 20, MW = 26.80 kDa). Data obtained were normalized to the pPrF UAA (1) to demonstrate the difference in incorporation efficiency over the previously reported UAA. | ||
With both brominated alkyne UAAs in hand, it was feasible to develop a bioorthogonal Cadiot–Chodkiewicz reaction. Initial studies employed the pBrPrF-containing GFP variant, to mimic previous Glaser–Hay pPrF-GFP studies and provide an effective comparison. The Cadiot–Chodkiewicz reaction was carried out in PBS (pH = 7) using copper iodide and triethylamine (TEA) (both at a final concentration of 5 mM) in the presence of a terminal alkyne-containing fluorophore (AlexaFluor 488 alkyne) at 4 °C for 6 h. The reaction was successful, as fluorescence could be detected on a denatured SDS-PAGE gel only when protein and fluorophore were exposed to the CuI/TEA system (Fig. 3). Even more exciting was the minimal protein degradation relative to the previously reported Glaser–Hay reaction. Also, due to the chemoselective nature of the reaction no protein dimerization was detected, and fluorophore dimerization was minimal and easily removed.
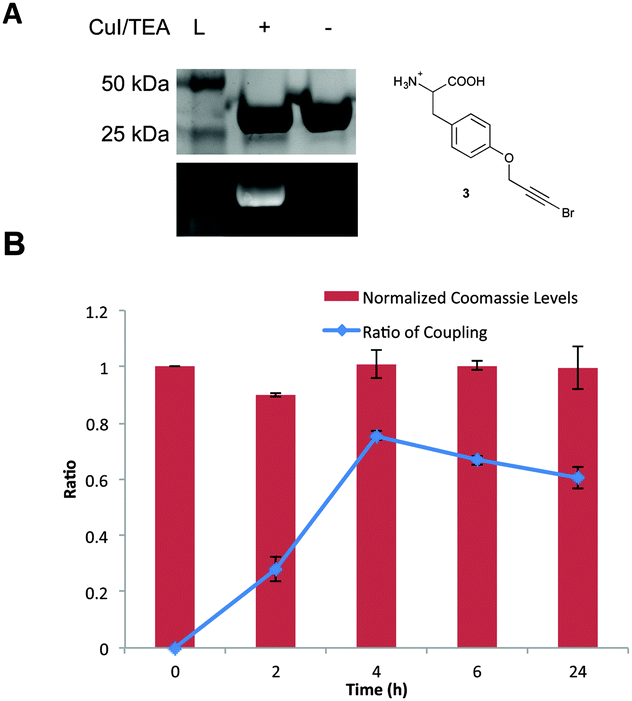 | ||
| Fig. 3 Bioorthogonal Cadiot–Chodkiewicz reaction. (A) The reaction performed on GFP (0.2 mg mL−1) and AlexaFluor 488 (0.2 mM) in the presence of 5 mM of CuI and 5 mM TEA at 4 °C. Fluorescence is only observed in the presence of the CuI/TEA system. (B) Reaction profile at 4 °C over a 24 h time period. Following analysis via SDS-PAGE, fluorescent imaging, and staining with coomassie blue, protein levels were normalized to the 0 time point control. The protein levels indicate minimal to no protein degradation occurs, even at longer time points. Analysis of coupling efficiency was determined by calculating the ratio of fluorescence to coomassie staining for each time point. All reactions were performed in triplicate. | ||
In an attempt to further optimize the reaction, both copper concentrations and temperatures were varied. A 5 mM working Cu(I) concentration was found to be ideal, which represents a marked improvement over the ∼50 mM concentrations required for the Glaser–Hay reaction (see ESI†). Previous reports have indicated that in vivo use of copper-mediated bioorthogonal chemistries required working concentrations of near 0.1 mM of copper(I) salt to minimize cytotoxicity.10,41 Thus, the minimized copper concentrations help bring bioorthogonal conjugated diyne chemistry into the range of in vivo use. These copper concentrations also had no impact on GFP fluorescence as determined by control reactions. Additionally, the optimal temperature profile for the reaction was also investigated. After performing a time course of the reaction at both 37 °C and 4 °C, we were able to determine very little difference between either temperature at early time-points. However, as the reaction was extended to 24 h, greater protein degradation at 37 °C occurred, most likely due to an increase in the rate of disproportionation of the Cu(I) catalyst at this temperature, producing a reactive copper(II) species (Fig. 3 and ESI†). However, for the 4 °C temperature profile the reaction reached approximately 86% completion in 4 h with minimal protein degradation, indicating that the bioorthogonal Cadiot–Chodkiewicz reaction can be performed quickly and in a relatively mild conditions. Extended times and temperatures resulted in higher yields, however were accompanied by protein degradation.
We next sought to explore the effects of an aromatic variant of the pBrPrF. As such, pBrEtF-GFP151 was expressed, and subjected to coupling conditions at 4 °C in the presence of an alkyne fluorophore. Once again a successful conjugation was observed as determined by SDS-PAGE (see ESI†). Only samples exposed to the CuI/TEA system exhibited fluorescence while other controls did not, indicating the fluorescence was not due to non-specific interactions. Interestingly, the use of an aromatic containing bromoalkyne appears to be less effective in the Cadiot–Chodkiewicz reaction than its aliphatic analog.
Next we became interested in exploring how the novel biological Cadiot–Chodkiewicz conjugation compared to our previously described Glaser–Hay reactions. In direct comparison, the Cadiot–Chodkiewicz exhibited far less protein degradation as compared to the Glaser–Hay versions of either an aliphatic (pPrF) or aromatic (pEtF) terminal alkyne containing UAAs. Furthermore, the data indicates that the biological Cadiot–Chodkiewicz reaction proceeds at a faster rate than the Glaser–Hay, with the aliphatic version (pBrPrF) of the coupling reaching completion the fastest in 4 h (Fig. 4). Gratifyingly, these results correlate well with the mechanistic understanding of both reactions, as the Cadiot–Chodkiewicz requires a single copper atom, while the Glaser–Hay necessitates two copper–alkyne conjugates to form the diyne product. Moreover, the Cu(I)/(III) redox couple of the Cadiot–Chodkiewicz reaction most likely aids in the minimized protein oxidation relative to the Glaser–Hay coupling that involves a reactive Cu(II) intermediate. However, it is important to note that the Cadiot–Chodkiewicz requires brominated UAAs, requiring additional synthetic preparation. In comparison to other bioconjugation techniques, the Cadiot–Chodkiewicz reaction may be slower; however, it employs more synthetically accessible UAAs and results in a well-defined linear geometry primed for further reactions. Thus, the selection of the Cadiot–Chodkiewicz reaction may be dependent on the downstream application and available resources.
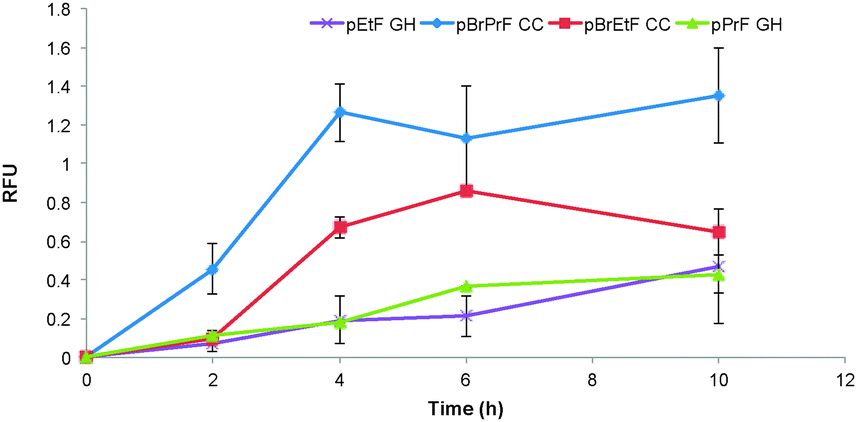 | ||
| Fig. 4 Comparison of alkyne reactions. Reactions were conducted either under the described Cadiot–Chodkiewicz conditions or the Glaser–Hay conditions at 4 °C depending on incorporated UAA. Due to differences in protein levels resulting from oxidative damage, the ratio of fluorophore coupling for each data set was normalized to the 24 hour time point in order to compare coupling trends between reactions. | ||
Overall, we have accomplished the successful application of the Cadiot–Chodkiewicz reaction to a biological context. Furthermore, we have demonstrated that the reaction can be performed with minimal protein oxidation. Finally, we have showed that the Cadiot–Chodkiewicz variant requires less harsh copper(I) concentrations, bringing the reaction near the range for in vivo use. Future work will involve optimization of conditions to increase the compatibility of the reaction with biological systems, and extension of the reaction towards in vivo applications.
Notes and references
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, London, 3rd edn, 1996 Search PubMed.
- K. Lang and J. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- H. Zhu and M. Snyder, Curr. Opin. Chem. Biol., 2003, 7, 55–63 CrossRef CAS PubMed.
- W. Tan, L. Sabet, Y. Li, T. Yu, P. R. Klokkevold, D. T. Wong and C. M. Ho, Biosens. Bioelectron., 2008, 24, 266–271 CrossRef CAS PubMed.
- D. Brady and J. Jordaan, Biotechnol. Lett., 2009, 31, 1639–1650 CrossRef CAS PubMed.
- F. Rusmini, Z. Zhong and J. Feijen, Biomacromolecules, 2007, 8, 1775–1789 CrossRef CAS PubMed.
- C. Liu, P. Schultz, R. Kornberg, C. Raetz, J. Rothman and J. Thorner, Annu. Rev. Biochem., 2010, 79, 413–444 CrossRef CAS PubMed.
- T. S. Young and P. G. Schultz, J. Biol. Chem., 2010, 285, 11039–11044 CrossRef CAS PubMed.
- L. Wang and P. G. Schultz, Angew. Chem., Int. Ed., 2004, 44, 34–66 CrossRef PubMed.
- E. Sletten and C. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- N. J. Agard, J. M. Baskin, J. A. Prescher, A. Lo and C. R. Bertozzi, ACS Chem. Biol., 2006, 1, 644–648 CrossRef CAS PubMed.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192–3193 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- H. Kolb and K. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- J. Baskin and C. Bertozzi, QSAR Comb. Sci., 2007, 26, 1211–1219 CAS.
- J. Kalia and R. Raines, Curr. Org. Chem., 2010, 14, 138–147 CrossRef CAS PubMed.
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS PubMed.
- B. Hutchins, S. Kazane, K. Staflin, J. Forsyth, B. Felding-Habermann, V. Smider and P. Schultz, Chem. Biol., 2011, 18, 299–303 CrossRef CAS PubMed.
- S. A. Kazane, J. Y. Axup, C. H. Kim, M. Ciobanu, E. D. Wold, S. Barluenga, B. A. Hutchins, P. G. Schultz, N. Winssinger and V. V. Smider, J. Am. Chem. Soc., 2013, 135, 340–346 CrossRef CAS PubMed.
- J. S. Lampkowski, J. K. Villa, T. S. Young and D. D. Young, Angew. Chem., Int. Ed., 2015, 54, 9343–9346 CrossRef CAS PubMed.
- W. Chodkiewicz, P. Cadiot and A. Willemart, C. R. Hebd. Seances Acad. Sci., 1957, 245, 2061–2062 CAS.
- J. Montierth, D. DeMario, M. Kurth and N. Schore, Tetrahedron, 1998, 54, 11741–11748 CrossRef CAS.
- J. Berná, S. M. Goldup, A. L. Lee, D. A. Leigh, M. D. Symes, G. Teobaldi and F. Zerbetto, Angew. Chem., Int. Ed., 2008, 47, 4392–4396 CrossRef PubMed.
- M. Mowery and C. Evans, Tetrahedron Lett., 1997, 38, 11–14 CrossRef CAS.
- H. Matsuda, H. Nakanisi, T. Hosomi and M. Kato, Macromolecules, 1988, 21, 1238–1240 CrossRef CAS.
- A. Shun and R. Tykwinski, Angew. Chem., Int. Ed., 2006, 45, 1034–1057 CrossRef CAS PubMed.
- C. Glaser, Ber. Dtsch. Chem. Ges., 1896, 2, 422–424 CrossRef.
- A. Hay, J. Org. Chem., 1962, 27, 3320 CrossRef CAS.
- M. Vilhelmsen, J. Jensen, C. Tortzen and M. Nielsen, Eur. J. Org. Chem., 2013, 701–711 CrossRef CAS.
- A. Hay, J. Polym. Sci., Part A: Polym. Chem., 1962, 58, 581–591 CAS.
- J. Ueda, Y. Shimazu and T. Ozawa, Free Radical Biol. Med., 1995, 18, 929–933 CrossRef CAS PubMed.
- S. I. Lim, Y. Mizuta, A. Takasu, Y. H. Kim and I. Kwon, PLoS One, 2014, 9, e98403 Search PubMed.
- A. Deiters and P. G. Schultz, Bioorg. Med. Chem. Lett., 2005, 15, 1521–1524 CrossRef CAS PubMed.
- X. Nie and G. Wang, J. Org. Chem., 2006, 71, 4734–4741 CrossRef CAS PubMed.
- J. P. Marino and H. N. Nguyen, J. Org. Chem., 2002, 67, 6841–6844 CrossRef CAS PubMed.
- B. K. Raliski, C. A. Howard and D. D. Young, Bioconjugate Chem., 2014, 25, 1916–1920 CrossRef CAS PubMed.
- D. Young, T. Young, M. Jahnz, I. Ahmad, G. Spraggon and P. Schultz, Biochemistry, 2011, 50, 1894–1900 CrossRef CAS PubMed.
- D. Young, S. Jockush, N. Turro and P. Schultz, Bioorg. Med. Chem. Lett., 2011, 21, 7502–7504 CrossRef CAS PubMed.
- A. J. Link, M. K. Vink and D. A. Tirrell, J. Am. Chem. Soc., 2004, 126, 10598–10602 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols, bioconjugation optimization and SDS-PAGE analysis. See DOI: 10.1039/c5cc08287k |
| This journal is © The Royal Society of Chemistry 2016 |