
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence[(ClImDipp)P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) P(Dipp)][GaCl4]: a polarized, cationic diphosphene†
P(Dipp)][GaCl4]: a polarized, cationic diphosphene†
Kai
Schwedtmann
a,
Michael H.
Holthausen
a,
Chris H.
Sala
a,
Felix
Hennersdorf
a,
Roland
Fröhlich
b and
Jan J.
Weigand
*a
aChair of Inorganic Molecular Chemistry, University of Technology Dresden, 01062 Dresden, Germany. E-mail: jan.weigand@tu-dresden.de
bInstitute of Organic Chemistry, Westfälische Wilhelms-Universität Münster, 48149 Münster, Germany
First published on 2nd December 2015
Abstract
The reaction of the neutral diphosphanide [(ClImDipp)P–P(Cl)(Dipp)] (6) (ClImDipp = 4,5-dichloro-1,3-bis(Dipp)-imidazol-2-yl; Dipp = 2,6-di-iso-propylphenyl) with methyl triflate (MeOTf) leads to the formation of cationic diphosphane [(ClImDipp)(Me)P–P(Cl)(Dipp)]+ (8+) in a stereoselective methylation. In contrast, reacting 6 with the Lewis acid GaCl3 yields cationic diphosphene [(ClImDipp)P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P(Dipp)]+ (7+), which is explained by a low P–Cl bond dissociation energy. The significantly polarized PP double bond in 7+ allows for its utilization as an acceptor for nucleophiles – the reaction with Cl− regenerates diphosphanide 6 and the reaction with PMe3 gives cation [(ClImDipp)P–P(PMe3)(Dipp)] (9+). In depth DFT investigation provides detailed insights into the bonding situation of the reported compounds.
P(Dipp)]+ (7+), which is explained by a low P–Cl bond dissociation energy. The significantly polarized PP double bond in 7+ allows for its utilization as an acceptor for nucleophiles – the reaction with Cl− regenerates diphosphanide 6 and the reaction with PMe3 gives cation [(ClImDipp)P–P(PMe3)(Dipp)] (9+). In depth DFT investigation provides detailed insights into the bonding situation of the reported compounds.
A few decades ago, the concept of kinetic stabilization by sterically demanding substituents provided a breakthrough in the field of multiple bonded compounds based on heavier main group elements.1 In 1981 Yoshifuji succeeded in the preparation of the first diphosphene Mes*P
PMes* 1 by introducing the very bulky Mes* substituent (Mes* = 2,4,6-tri-tert-butylphenyl, “super-mesityl”, Fig. 1).2 Only recently, N-heterocyclic carbenes (NHCs) have gathered comparable attention in phosphorus chemistry for their ability to stabilize low-coordinate bonding environments in poly-phosphorus compounds, which can also be explained by thermodynamic stabilization (conjugated π-system, charge delocalization).3 This was shown by Robinson who prepared the neutral (ImDipp)P–P(ImDipp) (ImDipp = 1,3-bis(Dipp)-imidazol-2-yl)4 and explored its further reactivity.5 Later, Bertrand reported on the stepwise oxidation of (ImDipp)P–P(ImDipp) and isolated the dicationic P2 species 22+,6 illustrating that imidazoliumyl-substituents can be used for the stabilization of cations (Fig. 1). The activation of white phosphorus (P4) by carbenes was investigated thoroughly and gave access to extended frameworks of low-coordinate P atoms,4,7 such as the neutral catena P4-species 3.7a,b We reported on the stepwise transformation of P4 by using an electrophilic phosphenium cation and a nucleophilic carbene ClNHCDipp which yielded the linear P3 cation 4+ featuring two terminal imidazoliumyl-substituents.8,9 In a very recent contribution, Grützmacher isolated the cationic disphosphene 5+, via the reaction of (ImDipp)PH with PCl2(Ni-Pr2) in the presence of DABCO (1,4-diazabicyclo[2.2.2]octane) and subsequent chloride abstraction with GaCl3.10
 | ||
| Fig. 1 First reported diphosphene 1 and selected imidazoliumyl functionalized polyphosphorus compounds (22+–5+) (only one representative Lewis structure is presented). | ||
The aforementioned compounds have illustrated the ability of imidazoliumyl-substituents to accept π-electron density from adjacent two-coordinate P atoms which is important for their stability since it significantly lowers the nucleophilicity of the phosphorus moiety. The termination of the Pn (n = 2, 3, 4) chains in 22+–4+ by two imidazoliumyl-groups, however, leads to symmetrical distribution of electron density within the multiple bonded polyphosphorus fragments. We envisioned that a diphosphene, bearing a sterically demanding aryl group and an imidazoliumyl-substituent has a polarized PP double bond and serves well for an interesting reactivity.
Aiming at the synthesis of such a diphosphene, we reacted neutral diphosphanide 6 with distinct electrophiles (GaCl3, MeOTf). This gave rise to cationic diphosphene salt 7[GaCl4] via halide abstraction or cationic diphosphane salt 8[OTf] via stereoselective methylation. Reactions of 7+ with nucleophiles (PMe3, Cl−) demonstrate its remarkable acceptor properties. The reaction of diphosphanide 6 with GaCl3 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry in C6H6 solution resulted in the immediate formation of a red precipitate of 7[GaCl4] (Scheme 1, 76% yield). This compound constitutes a rare example of an unsymmetrically substituted, cationic diphosphene.10,11 Its 31P{1H} NMR spectrum shows an AX spin system. The observed large 1J(PP) coupling constant (1J(PAPX) = −577.9 Hz) is characteristic for a PP double bond.12 The A part, assigned to the imidazoliumyl-substituted P atom, is shifted to higher field (δ(PA) = 398.1 ppm). On the other hand, the X part, assigned to the Dipp-substituted P atom, is at remarkable low field (δ(PX) = 605.8 ppm) compared to (E)-configured diphosphenes (e.g. Mes*PPMes* δ(P) = 492 ppm).12 This implies a significant polarization of the PP double bond, which can be indicated by resonance structure 7b+ (Scheme 1). An inverse polarization was observed previously by 31P NMR spectroscopy for π-donor substituted diphosphenes (e.g. Mes*–P1P2–(Ni-Pr2): δ(P1) = 276 ppm, δ(P2) = 447 ppm,135+: (ImDipp)–P1P2–(Ni-Pr2)+: δ(P2) = 158 ppm, δ(P1) = 492 ppm).10 Cation 7+ is bright, red-colored and the UV/vis spectrum of 7[GaCl4] reveals two absorptions at 490 nm (ε = 429 cm2 mol−1) and 349 nm (ε = 4995 cm2 mol−1). The first absorption is assigned to a symmetry forbidden n(P) → π*(PP) transition and the second to the symmetry allowed π(P–P) → π*(PP) transition.12 The molecular structure of 7[GaCl4] is depicted in Fig. 2 and confirms the (E)-configuration (C28–P2–P1–C1: 179.9(2)°). The PP bond length (P1–P2: 2.038(1) Å) is typical for diphosphenes.10,12 The C–P–P angle involving the imidazoliumyl-substituent (C1–P1–P2: 105.0(1)°) is larger than that involving the Dipp-substituent (C28–P2–P1 95.1(1)°). This might be a result of a higher degree of π-bonding interactions involving the more electron withdrawing imidazoliumyl-group, or the large steric demand of the imidazoliumyl-substituent.14 Interestingly, the reaction of 6 with MeOTf does not yield MeCl and 7[OTf]. Instead, addition of MeOTf to a solution of 6 in benzene gave a yellowish reaction mixture from which 8[OTf] was conveniently obtained in good yields via addition of n-hexane and isolation of the formed precipitate (77% yield). The 31P{1H} NMR spectrum of the reaction mixture shows the prominent resonances of an AX spin system. These are assigned to diastereomeric 8+ (δ(PA) = −15.1 ppm, δ(PX) = 75.1 ppm, 1J(PAPX) = −218.2 Hz) which mainly comprises a pair of enantiomers with (R,S)- and (S,R)-configuration. Thus, the relatively small absolute value of the 1J(PP) coupling constants in (R,S)- and (S,R)-configuration is attributed to an anti-periplanar arrangement of the lone pairs of electrons.15 In contrast, the relatively large absolute value observed for the (S,S)- and (R,R)-configuration is attributed to the gauche arrangement.15 Similar observations were reported for meso- and rac-1,2-bis(trifluoromethyl)-diphosphane where the trans dispositions of electronegative CF3 groups determines the favoured rotamers.16 The A part is assigned to the Me-substituted P atom on the basis of the observed 2J(PH) coupling constant (8.7 Hz). A second AX spin system of low intensity (<5%) is assigned to the second diastereomer of 8+ (δ(PA) = −10.5 ppm, δ(PX) = 86.4 ppm, 1J(PAPX) = −354.8 Hz) comprised of a pair of enantiomers with (R,R)- and (S,S)-configuration. The significantly different values of 1J(PP) coupling constants observed for the (R,S)/(S,R) and (R,R)/(S,S) pairs of enantiomers are explained by the relative arrangement of the lone pairs of electrons in 8+. Most likely, a trans-conformation of the sterically demanding imidazoliumyl- and Dipp-substituent represents the most stable rotamer of 8+.15 The high stereoselectivity of the methylation of 6 might be a consequence of significant differences in the steric demand of the substituents at both, the di-coordinate P atom and the adjacent chiral P atom. Thus, the favoured approach of the electrophile (E+) to the di-coordinate P atom occurs from the less crowded side as illustrated in Fig. 3. The molecular structure of 8[OTf] confirms the (R,S)- and (S,R)-configuration of the major isomer (Fig. 4). The anti-periplanar conformation of the imidazoliumyl- and Dipp-substituents as observed in solution is also present in the solid state (C1–P1–P2–C29: 152.8(1)°). The formation of 8[OTf] in the reaction of 6 with MeOTf indicates that the di-coordinate P atom exhibits the most nucleophilic properties. Therefore, it is reasonable to assume that the halide abstraction from 6 by GaCl3, which yields 7[GaCl4], proceeds via coordination of the electrophile to the di-coordinate P atom, followed by 1,2-elimination of GaCl4−. Additionally, resonance structure 7b+ indicates that this compound is a suitable acceptor for nucleophiles due to the polarization of the diphosphene moiety by the adjacent imidazoliumyl-substituent. The acceptor properties of 7+ were further investigated by DFT calculations (Fig. 5). The polarization of the PP double bond is expressed by a higher contribution of Pa (57.4% vs. 42.6% for Pb) to the π-type orbital and a donor acceptor interaction with the adjacent π* CN orbital of 18.5 kcal mol−1, indicating a stabilization of the positive charge by delocalization. Analysis of the natural charges of 7+ showed that Pb carries the highest charge in the molecule of +0.44e compared to +0.24e on Pa, making Pb the preferred reaction site for nucleophiles. This reactivity was elucidated by the reaction of 7[GaCl4] with suitable nucleophiles – a Cl− source and PMe3 (Scheme 2). The addition of [n-Bu4N][Cl] to a solution of 7[GaCl4] in o-C6H4F2 results in the immediate color change from red to yellow, associated with diphosphanide 6. 31P NMR investigation of the reaction mixture revealed quantitative regeneration to 6via adduct formation of the cationic diphosphene with the chloride anion. The previously unknown molecular structure of 68 was determined by means of X-ray single crystal structure determination in the course of this study (Fig. 4). The P–C bond length of compound 6 involving the imidazoliumyl-substituent (P2–C13: 1.803(2) Å) is longer than related bond distances in (ImMes)PPh (P–C: 1.763(6) Å)17 or 2 (P–C: 1.750(2) Å).4 In addition, the P–P bond length (P1–P2: 2.1327(9) Å) is shorter than the typical P–P single bond distance observed for diphosphanes (2.22 Å)18 but significantly longer than a typical PP double bond length observed for diphosphenes (2.00 Å).12 This can be explained by the donation of electron density from the p-type lone pairs of electrons on P2 into the lobe of the σ*-orbital of the adjacent P–Cl bond (vide infra). This is supported by a longer P–Cl bond length (P1–Cl1: 2.1586(8) Å) than those typically observed for chloro-substituted diphosphanes (2.10 Å).19 Collectively, the structural data indicates that the interaction of chloride in 6 is significantly weaker than a covalent P–Cl bond in a chlorophosphane. This is supported by computations which show a significant donor acceptor interaction of 22.5 kcal mol−1 of the p-type lone pair of Pa and the antibonding σ* Pb–Cl orbital (Fig. 5). A comparison of the energy profiles of the P–Cl bond dissociation of 6me (all iso-propyl moieties are substituted by methyl groups, r0 = 2.131 Å) and Ph2PCl (r0 = 2.104 Å) yielded a dissociation energy of 34.1 kcal mol−1 and a force constant of 154.4 N m−1 for 6meversus 88.2 kcal mol−1 and 218 N m−1 for Ph2PCl (Fig. S4.1, ESI†). This result further proves the weaker P–Cl bond in 6 compared to regular chlorophosphanes.
:1 stoichiometry in C6H6 solution resulted in the immediate formation of a red precipitate of 7[GaCl4] (Scheme 1, 76% yield). This compound constitutes a rare example of an unsymmetrically substituted, cationic diphosphene.10,11 Its 31P{1H} NMR spectrum shows an AX spin system. The observed large 1J(PP) coupling constant (1J(PAPX) = −577.9 Hz) is characteristic for a PP double bond.12 The A part, assigned to the imidazoliumyl-substituted P atom, is shifted to higher field (δ(PA) = 398.1 ppm). On the other hand, the X part, assigned to the Dipp-substituted P atom, is at remarkable low field (δ(PX) = 605.8 ppm) compared to (E)-configured diphosphenes (e.g. Mes*PPMes* δ(P) = 492 ppm).12 This implies a significant polarization of the PP double bond, which can be indicated by resonance structure 7b+ (Scheme 1). An inverse polarization was observed previously by 31P NMR spectroscopy for π-donor substituted diphosphenes (e.g. Mes*–P1P2–(Ni-Pr2): δ(P1) = 276 ppm, δ(P2) = 447 ppm,135+: (ImDipp)–P1P2–(Ni-Pr2)+: δ(P2) = 158 ppm, δ(P1) = 492 ppm).10 Cation 7+ is bright, red-colored and the UV/vis spectrum of 7[GaCl4] reveals two absorptions at 490 nm (ε = 429 cm2 mol−1) and 349 nm (ε = 4995 cm2 mol−1). The first absorption is assigned to a symmetry forbidden n(P) → π*(PP) transition and the second to the symmetry allowed π(P–P) → π*(PP) transition.12 The molecular structure of 7[GaCl4] is depicted in Fig. 2 and confirms the (E)-configuration (C28–P2–P1–C1: 179.9(2)°). The PP bond length (P1–P2: 2.038(1) Å) is typical for diphosphenes.10,12 The C–P–P angle involving the imidazoliumyl-substituent (C1–P1–P2: 105.0(1)°) is larger than that involving the Dipp-substituent (C28–P2–P1 95.1(1)°). This might be a result of a higher degree of π-bonding interactions involving the more electron withdrawing imidazoliumyl-group, or the large steric demand of the imidazoliumyl-substituent.14 Interestingly, the reaction of 6 with MeOTf does not yield MeCl and 7[OTf]. Instead, addition of MeOTf to a solution of 6 in benzene gave a yellowish reaction mixture from which 8[OTf] was conveniently obtained in good yields via addition of n-hexane and isolation of the formed precipitate (77% yield). The 31P{1H} NMR spectrum of the reaction mixture shows the prominent resonances of an AX spin system. These are assigned to diastereomeric 8+ (δ(PA) = −15.1 ppm, δ(PX) = 75.1 ppm, 1J(PAPX) = −218.2 Hz) which mainly comprises a pair of enantiomers with (R,S)- and (S,R)-configuration. Thus, the relatively small absolute value of the 1J(PP) coupling constants in (R,S)- and (S,R)-configuration is attributed to an anti-periplanar arrangement of the lone pairs of electrons.15 In contrast, the relatively large absolute value observed for the (S,S)- and (R,R)-configuration is attributed to the gauche arrangement.15 Similar observations were reported for meso- and rac-1,2-bis(trifluoromethyl)-diphosphane where the trans dispositions of electronegative CF3 groups determines the favoured rotamers.16 The A part is assigned to the Me-substituted P atom on the basis of the observed 2J(PH) coupling constant (8.7 Hz). A second AX spin system of low intensity (<5%) is assigned to the second diastereomer of 8+ (δ(PA) = −10.5 ppm, δ(PX) = 86.4 ppm, 1J(PAPX) = −354.8 Hz) comprised of a pair of enantiomers with (R,R)- and (S,S)-configuration. The significantly different values of 1J(PP) coupling constants observed for the (R,S)/(S,R) and (R,R)/(S,S) pairs of enantiomers are explained by the relative arrangement of the lone pairs of electrons in 8+. Most likely, a trans-conformation of the sterically demanding imidazoliumyl- and Dipp-substituent represents the most stable rotamer of 8+.15 The high stereoselectivity of the methylation of 6 might be a consequence of significant differences in the steric demand of the substituents at both, the di-coordinate P atom and the adjacent chiral P atom. Thus, the favoured approach of the electrophile (E+) to the di-coordinate P atom occurs from the less crowded side as illustrated in Fig. 3. The molecular structure of 8[OTf] confirms the (R,S)- and (S,R)-configuration of the major isomer (Fig. 4). The anti-periplanar conformation of the imidazoliumyl- and Dipp-substituents as observed in solution is also present in the solid state (C1–P1–P2–C29: 152.8(1)°). The formation of 8[OTf] in the reaction of 6 with MeOTf indicates that the di-coordinate P atom exhibits the most nucleophilic properties. Therefore, it is reasonable to assume that the halide abstraction from 6 by GaCl3, which yields 7[GaCl4], proceeds via coordination of the electrophile to the di-coordinate P atom, followed by 1,2-elimination of GaCl4−. Additionally, resonance structure 7b+ indicates that this compound is a suitable acceptor for nucleophiles due to the polarization of the diphosphene moiety by the adjacent imidazoliumyl-substituent. The acceptor properties of 7+ were further investigated by DFT calculations (Fig. 5). The polarization of the PP double bond is expressed by a higher contribution of Pa (57.4% vs. 42.6% for Pb) to the π-type orbital and a donor acceptor interaction with the adjacent π* CN orbital of 18.5 kcal mol−1, indicating a stabilization of the positive charge by delocalization. Analysis of the natural charges of 7+ showed that Pb carries the highest charge in the molecule of +0.44e compared to +0.24e on Pa, making Pb the preferred reaction site for nucleophiles. This reactivity was elucidated by the reaction of 7[GaCl4] with suitable nucleophiles – a Cl− source and PMe3 (Scheme 2). The addition of [n-Bu4N][Cl] to a solution of 7[GaCl4] in o-C6H4F2 results in the immediate color change from red to yellow, associated with diphosphanide 6. 31P NMR investigation of the reaction mixture revealed quantitative regeneration to 6via adduct formation of the cationic diphosphene with the chloride anion. The previously unknown molecular structure of 68 was determined by means of X-ray single crystal structure determination in the course of this study (Fig. 4). The P–C bond length of compound 6 involving the imidazoliumyl-substituent (P2–C13: 1.803(2) Å) is longer than related bond distances in (ImMes)PPh (P–C: 1.763(6) Å)17 or 2 (P–C: 1.750(2) Å).4 In addition, the P–P bond length (P1–P2: 2.1327(9) Å) is shorter than the typical P–P single bond distance observed for diphosphanes (2.22 Å)18 but significantly longer than a typical PP double bond length observed for diphosphenes (2.00 Å).12 This can be explained by the donation of electron density from the p-type lone pairs of electrons on P2 into the lobe of the σ*-orbital of the adjacent P–Cl bond (vide infra). This is supported by a longer P–Cl bond length (P1–Cl1: 2.1586(8) Å) than those typically observed for chloro-substituted diphosphanes (2.10 Å).19 Collectively, the structural data indicates that the interaction of chloride in 6 is significantly weaker than a covalent P–Cl bond in a chlorophosphane. This is supported by computations which show a significant donor acceptor interaction of 22.5 kcal mol−1 of the p-type lone pair of Pa and the antibonding σ* Pb–Cl orbital (Fig. 5). A comparison of the energy profiles of the P–Cl bond dissociation of 6me (all iso-propyl moieties are substituted by methyl groups, r0 = 2.131 Å) and Ph2PCl (r0 = 2.104 Å) yielded a dissociation energy of 34.1 kcal mol−1 and a force constant of 154.4 N m−1 for 6meversus 88.2 kcal mol−1 and 218 N m−1 for Ph2PCl (Fig. S4.1, ESI†). This result further proves the weaker P–Cl bond in 6 compared to regular chlorophosphanes.
 | ||
| Scheme 1 Synthetic route to 7[GaCl4] and 8[OTf] (comprised of a pair of enantiomers, only the (R,S) enantiomer is shown) and resonance structures 7a+ and 7b+. | ||
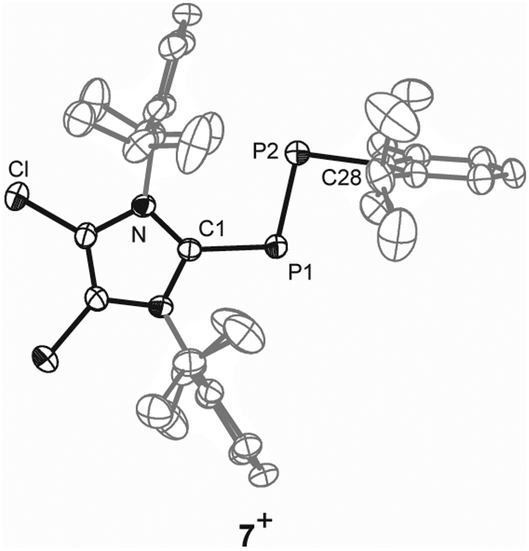 | ||
| Fig. 2 Molecular structure of cation 7+ in 7[GaCl4]·C6H5F (anions, hydrogen atoms and solvate molecules are omitted for clarity and thermal ellipsoids are displayed at 50% probability); selected experimental bond lengths [Å] and angles [°]: C1–P1 1.834(2), P1–P2 2.038(1), P2–C28 1.854(4), C1–P1–P2 105.0(1), C28–P2–P1 95.1(1). | ||
 | ||
| Fig. 3 Molecular structure of 6 (M06-2X/def2svp) showing the favoured and disfavoured approach of electrophiles. Hydrogen atoms are omitted for clarity. | ||
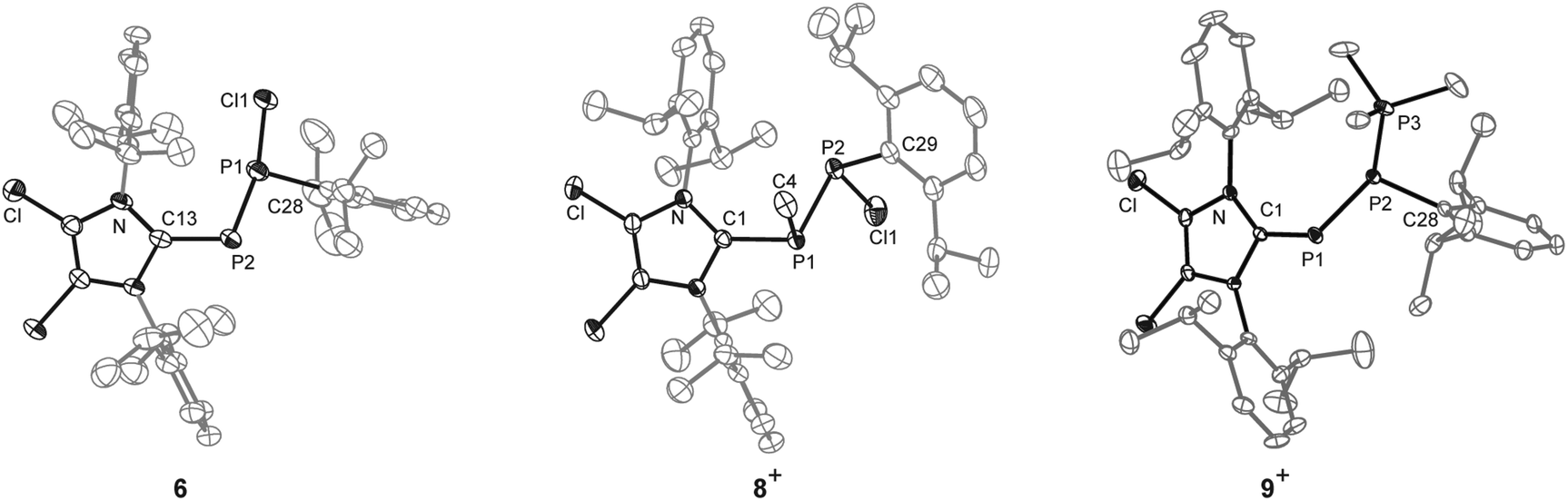 | ||
| Fig. 4 Molecular structure of 6·(Et2O)·(n-hexane), 8+ in 8[OTf]·2(1,2-C6H4F2), 9+ in 9[GaCl4] (hydrogen atoms and solvent molecules are omitted for clarity and thermal ellipsoids are displayed at 50% probability); selected experimental bond lengths in Å and angles in °; 6: C1–P1 1.865(3), P1–P2 2.1327(9), P1–Cl1 2.1586(8), C1–P1–P2 95.83(8), C13–P2–P1 102.2(1). 8+: C1–P1 1.842(2), P1–P2 2.2250(9), P2–C29 1.841(2), P2–Cl1 2.0673(9); 9+: C1–P1 1.799(3), P1–P2 2.151(1), P2–P3 2.208(1), P1⋯P3 3.478(1), C1–P1–P2 100.5(1), P1–P2–P3 105.8(1). | ||
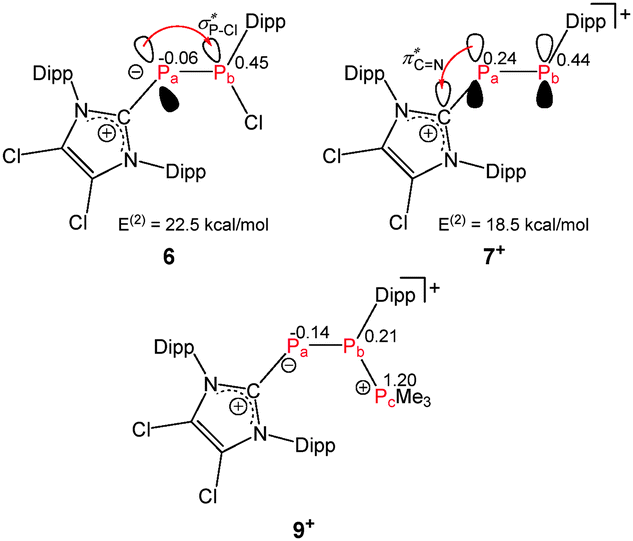 | ||
| Fig. 5 Natural charges and selected secondary interactions of NBO analysis on DFT optimized structures of 6, 7+ and 9+. | ||
 | ||
| Scheme 2 Synthetic route to 6 and 9[GaCl4]. | ||
The addition of PMe3 to a red solution of 7[GaCl4] in o-C6H4F2 again leads to an immediate color change to yellow. The phosphane adduct 9[GaCl4] was isolated in high yields from the reaction mixture (96%, Scheme 2). Its 31P{1H} NMR spectrum shows an AMX spin system. The A part of the spin system (δ(PA) = −108.1 ppm) is assigned to the di-coordinate P atom and the pronounced high field shift indicates an inverse polarized phosphaalkene (or a phosphanide) moiety.20 The resonance at lowest field (δ(PX) = 11.2 ppm) is assigned to the tetra-coordinate P atom (compare [Ph2P–PMe3]+: δ(PMe) = 15 ppm)21 while the M part (δ(PM) = −62.1 ppm) corresponds to the tri-coordinate P atom.22 As the 1J(PP) couplings are of expected magnitude (1J(PAPM) = −322.1 Hz, 1J(PMPX) = −343.3 Hz) a comparatively large 2J(PP) coupling (2J(PAPX) = 72.8 Hz) might indicate a through space interaction between the di- and tetra-coordinate P centers. The connectivity of 9+ was confirmed by X-ray single crystal structure determination (Fig. 4). Its P–C bond length involving the imidazoliumyl-moiety is in the typical range of inverse polarized phosphaalkenes (C1–P1: 1.799(3) Å) and is comparable to 6. Two distinct P–P bond lengths are observed in 9+ (P1–P2: 2.151(1) Å, P2–P3: 2.208(1) Å) and that one involving the di-coordinate P atom is significantly shorter than a typically P–P single bond (P–P: 2.22 Å).18 The distance between both terminal P atoms (P1⋯P3: 3.478(1) Å) is well within the sum of the van der Waals radii of the respective atoms (∑rvdW(P,P) = 3.80 Å),23 which might explain the observed large 2J(PP) coupling between both atoms. It is important to note that polyphosphorus compounds featuring di-, tri- and tetra-coordinate P atoms in one molecule are very rare24 and, to the best of our knowledge, 9+ is the first example of a phosphane environment that bridges a phosphanide and a phosphonium moiety.
In summary, we have studied reactions of neutral diphospanide 6 with selected electrophiles (GaCl3, MeOTf). They proceed either via stereoselective methylation yielding cationic diphosphane 8+ or halide abstraction giving the remarkable cationic diphosphene 7+. The latter features a sterically demanding aryl group and a cationic, π-electron accepting imidazoliumyl-substituent. The substitution pattern in 7+ causes a significant polarization of the PP double bond. This allows for its utilization as an acceptor towards chloride or PMe3 as nucleophiles and the corresponding adducts 6 and 9+ were obtained. The utilization of polarized PP double bonded cations as Lewis acids is expected to provide new avenues in diphosphene chemistry, which is subject of ongoing studies in our laboratories.
This work was supported by the Fonds der Chemischen Industrie (FCI, scholarships for M. H. H. and F. H.), the German Science Foundation (DFG, WE 4621/2-1) and the ERC (SynPhos 307616). We thank the Center for Information Services and High Performance Computing (ZIH) for the generous allocation of computation time.
References
- P. Power, J. Chem. Soc., Dalton Trans., 1998, 2939–2951 RSC ; and references reported therein.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587–4589 CrossRef CAS.
- (a) Y. Wang and G. H. Robinson, Inorg. Chem., 2014, 53, 11815–11832 CrossRef CAS PubMed; (b) M. H. Holthausen and J. J. Weigand, Chem. Rev., 2014, 43, 6639–6657 RSC.
- (a) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970–14971 CrossRef CAS PubMed; (b) F. D. Henne, E.-M. Schnöckelborg, K.-O. Feldmann, J. Grunenberg, R. Wolf and J. J. Weigand, Organometallics, 2013, 32, 6674–6680 CrossRef CAS ; for related species (CAAC)PP(CAAC) (CAAC = cyclic alkyl amino carbene) see: O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 5530–5533 Search PubMed.
- (a) Y. Wang, Y. Xie, M. Y. Abraham, P. Wei, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, Chem. Commun., 2011, 47, 9224–9226 RSC; (b) Y. Wang, Y. Xie, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2013, 135, 19139–19142 CrossRef CAS PubMed.
- O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Nat. Chem., 2010, 2, 369–373 CrossRef CAS PubMed.
- (a) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 7052–7055 CrossRef CAS PubMed; (b) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2007, 129, 14180–14181 CrossRef CAS PubMed; (c) D. Holschumacher, T. Bannenberger, K. Ibrom, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2010, 39, 10590–10592 RSC.
- M. H. Holthausen, S. K. Surmiak, P. Jerabek, G. Frenking and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 11078–11082 CrossRef CAS PubMed.
- Grützmacher and co-workers reported on a similar derivative of 4+. M. Tondreau, Z. Benkö, J. R. Harmer and H-J. Grützmacher, Chem. Sci., 2014, 5, 1545–1554 RSC.
- A. Beil, R. J. Gillard Jr. and H.-J. Grützmacher, Dalton Trans., 2015 10.1039/c5dt03014e.
- M. Sanchez, V. Romanenko, M.-R. Mazieres, A. Gudima and L. Markowski, Tetrahedron Lett., 1991, 32, 2775–2778 CrossRef CAS.
- L. Weber, Chem. Rev., 1992, 92, 1839–1905 CrossRef CAS.
- L. N. Markovski, V. D. Romanenko, E. O. Klebanski and S. V. Iksanova, Zh. Obshch. Khim., 1985, 55, 1867–1874 Search PubMed.
- T. Busch, W. W. Schoeller, E. Niecke, M. Nieger and H. Westermann, Inorg. Chem., 1989, 28, 4334–4340 CrossRef CAS.
- A. A. M. Ali and R. K. Harris, J. Chem. Soc., Dalton Trans., 1983, 583–587 RSC.
- J.-P. Albrand, J.-B. Robert and H. Goldwhite, Tetrahedron Lett., 1976, 17, 949–952 CrossRef.
- A. J. Arduengo III, J. C. Calabrese, A. H. Cowley, H. V. Rasika Dias, J. R. Goerlich, W. J. Marshall and B. Riegel, Inorg. Chem., 1997, 36, 2151–2158 CrossRef.
- K. F. Tebbe, Z. Anorg. Allg. Chem., 1980, 468, 202–212 CrossRef CAS.
- (a) M. Nieger, E. Niecke, J. Tirree, Private Communication, 2002, CCDC 178050; (b) L. Heuer, D. Schomburg and R. Schmutzler, Phosphorus, Sulfur Silicon Relat. Elem., 1989, 45, 217–222 CrossRef CAS.
- L. Weber, Eur. J. Inorg. Chem., 2000, 2425–2441 CrossRef CAS.
- N. Burford, P. J. Ragogna, R. McDonald and M. J. Ferguson, J. Am. Chem. Soc., 2003, 125, 14404–14410 CrossRef CAS PubMed.
- A. Schmidtpeter, S. Lochschmidt, K. Karaghiosoff and W. S. Sheldrick, J. Chem. Soc., Chem. Commun., 1985, 1447–1448 RSC.
- A. F. Hollemann, E. Wiberg and N. Wiberg, Lehrbuch der Anorganischen Chemie, ed. W. d Gruyter, Berlin, New York, 102nd edn, 2007, appendix IV4 Search PubMed.
- K.-O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 7545–7549 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For detailed experimental procedures and characterization details of new compounds, NMR spectra, crystallographic details and computational data. CCDC 1429021–1429024. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc08248j |
| This journal is © The Royal Society of Chemistry 2016 |