
Stimuli-responsive polymersomes for cancer therapy
Thavasyappan
Thambi
,
Jae Hyung
Park
and
Doo Sung
Lee
*
School of Chemical Engineering, Sungkyunkwan University, Suwon 440-746, Republic of Korea. E-mail: dslee@skku.edu; Fax: +82-31-292-8790; Tel: +82-31-290-7282
First published on 12th October 2015
Abstract
Cancer is the leading cause of mortality and remains a major challenge for modern chemotherapy. Recent advances in cancer therapy have made a modest impact on patient survival. Nanomedicine represents an innovative field with significant potential to improve cancer treatment. Nanomedicine utilizes numerous nanoconstructs, including polymersomes, micelles, and drug conjugates, to deliver therapeutic agents at the target site of interest. In particular, polymeric vesicles, also known as polymersomes, are self-assembled amphiphilic polymers in which an aqueous compartment is enclosed by a thick bilayer membrane. Unlike liposomes, polymersomes consist of high-molecular-weight amphiphilic polymer analogues. Since polymersomes are prepared using synthetic amphiphilic polymers, the bilayer membrane thickness can be readily altered by tuning the molecular weight of hydrophobic blocks. As a consequence, the polymersomes prepared from high-molecular-weight amphiphiles strengthen their membranes, making them inherently more stable than liposomes. The intriguing aggregation of polymersomes offers numerous advantages, including stability, tunable membrane properties, and the capability of encapsulating hydrophilic and hydrophobic agents. Owing to these properties, polymersomes are attractive candidates for various applications such as drug delivery, gene therapy, and tissue engineering. Although these properties have placed polymersomes at the forefront of drug delivery applications, to attain an enhanced therapeutic effect polymersomes are supposed to rapidly release the drug at the target site. To fulfill this requirement, stimuli-responsive polymersomes that respond to various internal or external stimuli have been developed. This review focuses on recently developed stimuli-responsive polymersomes and their potential application in cancer therapy.
 Thavasyappan Thambi | Thavasyappan Thambi was born in the Indian State of Tamil Nadu in 1983. He received his B.Sc. degree (2003) in Chemistry from Bharathiar University and his M.Sc. degree (2005) in Chemistry from the University of Madras. Then, he was appointed as a Scientist in Biocon, India, where he worked on synthesis of biologically active molecules. After the 3.5 years industrial experience, he moved to Sungkyunkwan University to receive his Ph.D. degree in 2013, working in the field of microenvironment-specific polymeric nanoparticles for imaging and therapy of intractable diseases. He is currently working as a postdoctoral fellow in Sungkyunkwan University with Prof. Doo Sung Lee, and his research focuses on the development of stimuli-responsive injectable polymeric materials for sustained delivery of therapeutic agents. |
 Jae Hyung Park | Jae Hyung Park received his Ph.D. degree in Materials Science and Engineering from Gwangju Institute of Science and Technology in 2002. Thereafter, he carried out a two-year postdoctoral research at Korea Institute of Science and Technology. He then joined the School of Pharmacy, Purdue University, as an associate postdoctoral researcher in 2004. He became an Assistant Professor at the Department of Chemical Engineering, Kyung Hee University in 2011. Since then, he has been an Associate Professor at the School of Chemical Engineering, Sungkyunkwan University. Thus far, he has published over 100 peer-reviewed papers and 5 book chapters. |
 Doo Sung Lee | Doo Sung Lee received his Ph.D. at Korea Advanced Institute of Science and Technology, South Korea, in 1984. Currently he is a full professor at the School of Chemical Engineering, Sungkyunkwan University, and the Director of the Theranostic Macromolecules Research Center (TMRC) funded by NRF of Korea. He also served as a member of the National Academy of Engineering of Korea and Korean Academy of Science and Technology in 2007 and 2011, respectively. In 2011, he was elected as the President of the Polymer Society of Korea. He has published over 150 articles in peer-reviewed journals, which have been cited more than 7000 times so far. His research interests include functionalized injectable hydrogels for drug delivery, cell delivery and tissue engineering, and biodegradable polymeric nanoparticles for controlled drug/protein delivery and imaging. |
1. Introduction
The development of smart nanocarriers is an active subject of recent studies.1 The self-assembly of amphiphilic copolymers in aqueous solutions can be utilized to form a range of nanocarriers of varying morphologies, including spherical micelles, worm-like micelles, and polymersomes.2–4 Among the variety of nanocarriers, polymersomes are artificial vesicles that have received considerable attention due to their entangled architecture.5 Polymersomes, composed of amphiphilic polymers, are spherical nanostructures in which an aqueous core is enclosed by a hydrophobic membrane. The membrane thickness of polymersomes can be elegantly tailored by altering the hydrophobic ratio of the copolymers.6 Polymersomes are generally prepared using high-molecular-weight amphiphilic block copolymers, and their stability and toughness are superior to those of their lipid counterpart, liposomes. Owing to the unique properties of polymersomes, their critical aggregation concentrations are low, which allows these particles to be retained longer in the bloodstream. The unique structural feature of polymersomes allows the encapsulation of hydrophobic and hydrophilic agents, such as anticancer drugs, proteins, and genes.7 It is known that polymersomes with an optimal size can alter the tissue distribution of anticancer drugs.8 In addition, the surface modification of polymersomes with target-specific ligands can facilitate the cellular uptake of anticancer drugs by binding to receptors that are overexpressed on cancer cells.9 Additionally, poly(ethylene glycol) (PEG) conjugation, known as the PEGylation of polymersomes, or the preparation of PEG-containing polymersomes is a widely used strategy to prevent the adsorption of blood protein components.10,11 The hydrophilic and neutral nature of PEG in polymersomes reduces protein opsonization, which makes polymersomes invisible to phagocytic and dendritic cells. As a consequence, systemically administered polymersomes can passively accumulate at the tumor site via the enhanced permeation and retention (EPR) effect.10,12 Given this unique structure, polymersomes could efficiently protect encapsulated agents from hydrolytic or enzymatic degradation without the loss of biological activity. It is important that systemically administered polymersomes should trigger the drug after it arrives at the target site.13,14 Thus, nanocarriers should be stable in the bloodstream and rapidly release their payload after arriving at the target site of action. To fulfill this contradictory requirement, smart polymersomes, also known as stimuli-responsive polymersomes, have been developed. Stimuli-responsive polymersomes are able to circulate in the bloodstream for long periods and maintain their nanostructures; however, they are disassembled at the target site. Fig. 1 graphically represents the state-of-the-art of polymersomes and their tumor-targeting characteristics.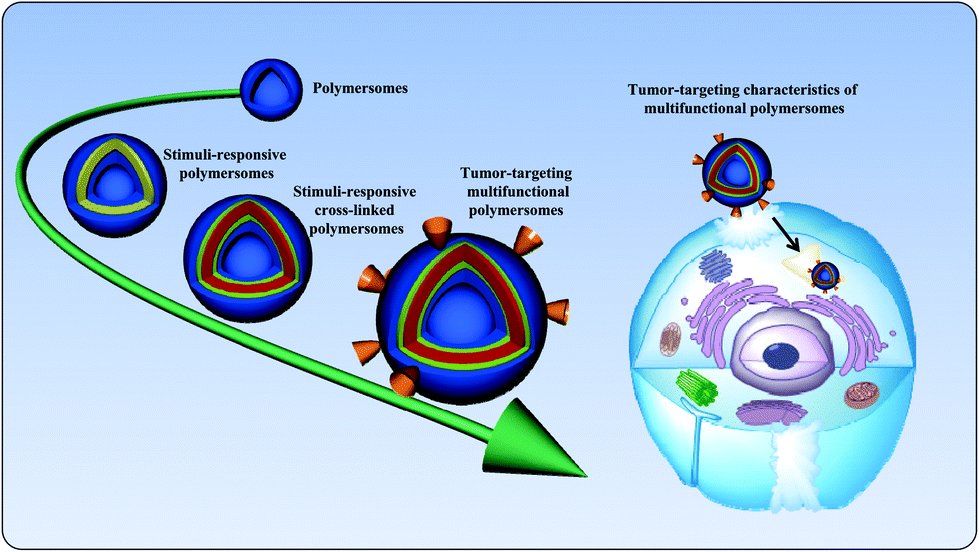 | ||
| Fig. 1 Schematic illustration of the state-of-the-art of polymersomes and their targeting characteristics in cancer therapy. | ||
2. Stimuli-responsive polymersomes
Traditional nanocarriers, which lack stimuli-responsive linkers, release anticancer drugs in a sustained manner via a diffusion-controlled mechanism. The utilization of such nanocarriers may not endow tumor tissues with cytotoxic local concentrations of anticancer drugs. All drugs are characterized by a plasma level above which the drug is toxic and below which it is ineffective. Thus, nanocarriers primarily serve to lower the systemic level of the drug while maintaining the desired therapeutic range after a single dose.15 Therefore, numerous stimuli-responsive polymersomes that respond to physical stimuli (e.g., temperature, ultrasound, light, electrochemical, and magnetic), chemical stimuli (e.g., pH, redox, and ionic) or biological stimuli (e.g., enzymes, inflammation and glucose) have been developed to improve the therapeutic efficacy of anticancer drugs (Fig. 2).16–20 Among the various types of stimuli-responsive carriers, pH-, temperature-, and redox-responsive nanocarriers have been widely used for various biomedical applications, including cancer therapy. In this review, we focused on recently developed chemical and biological stimuli-responsive polymersomes for cancer therapy as well as other representative physical stimuli-responsive polymersomes that might be applicable in cancer therapy in the near future.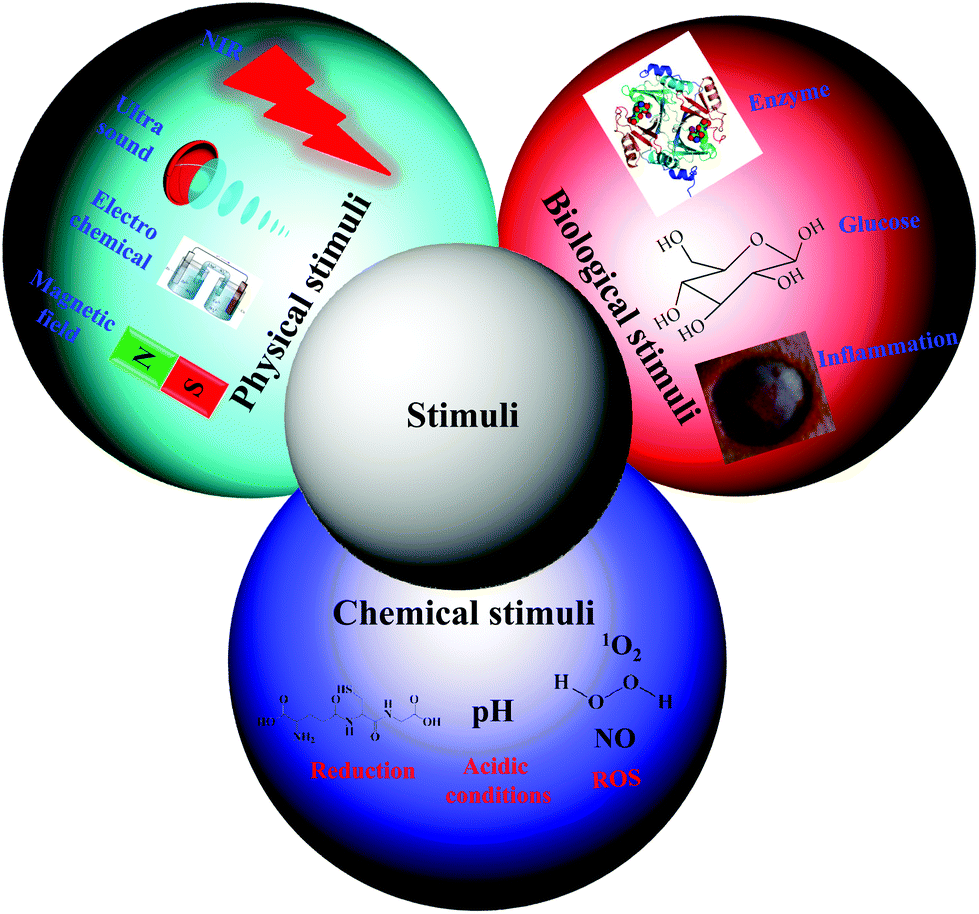 | ||
| Fig. 2 Schematic illustration of various types of stimuli and their representative examples. | ||
3. Bioreducible polymersomes
Recently, bioreducible polymersomes have been extensively studied for the triggered release of anticancer drugs in an intracellular reductive environment. These polymersomes usually possess a bioreducible disulfide bond in the middle or side chain of an amphiphilic polymer. Although the dissociation energy of a disulfide bond (–S–S–) is lower than that of a carbon–carbon bond (–C–C–), it is stable in an oxidizing extracellular environment.21,22 Disulfide bonds are longer (ls–s ∼ 2.03 Å) than carbon–carbon bonds (ls–s ∼ 1.54 Å), making them susceptible to various physical and chemical stimuli. Disulfide bonds are stable under physiological conditions; however, they are readily reduced in an intracellular environment due to the thiol–disulfide exchange reaction with glutathione (GSH), a tripeptide.23 Owing to the fact that several intracellular compartments, including the cytosol, mitochondria and cell nucleus, contain a higher concentration of GSH than the blood, polymersomes containing disulfide bridges are expected to be readily disintegrated and consequently release drugs more readily inside the cell than in circulation. Specifically, the concentration of GSH in the blood plasma is in the micromolar range (2–20 μM), whereas the concentration of GSH is approximately 1–10 mM in the intracellular compartments.24,25 Most GSH (85%–90%) is present in the cytosol, whereas much lower concentrations of GSH (15%–10%) are found in other organelles, including the mitochondria, nuclear matrix, and peroxisomes.26 The high GSH concentration provides an essential reducing environment inside the cell. This change in the concentration gradient has encouraged the development of disulfide-containing polymersomes for the site-specific triggering of drug release.27–29 Such polymersomes with therapeutic payloads may significantly improve the therapeutic efficacy of the drug, because various therapeutic targets are located intracellularly.Hubbell et al. were the first to develop disulfide-containing polymersomes, which consisted of PEG-b-poly(propylene sulfide) bearing an intervening disulfide bond.30 Polymersomes prepared from this block copolymer were demonstrated to destabilize in the presence of GSH or cysteine. The destabilization of polymersomes was examined via the encapsulation of calcein, a hydrophilic fluorescent probe, whose fluorescence in the polymersomes is autoquenched at a specific concentration. However, after the exposure of the polymersome to reducing agents, the released calcein generated a fluorescence signal and the intensity of this signal was measured to estimate the amount of released calcein. This release test indicated that the polymersome ruptured within 10 min of the addition of reducing agents. The cell uptake of polymersomes by J774A-1 cells was monitored; the fluorescence signal of calcein increased over time in the intraendosome and subsequently in the cytoplasm. These polymersomes may deliver therapeutic agents at the endosome and improve the efficacy of the drug by avoiding exposure to the harsh conditions encountered after lysosome fusion.
In another study, Jia et al. developed a novel type of disulfide-containing robust liquid crystal polymersome in which the hydrophobic block is a cholesterol-containing liquid crystal polymer.31 Since the polymersomes were prepared using cholesterol pendant groups, they were robust owing to the physical cross-linking of the cholesterol in the membrane and remained stable for a longer period of time. The destruction of polymersomes was examined based on the encapsulation of calcein, whose release was triggered by the addition of GSH. Although the term polymersome was coined in early 2000, the anticancer properties of drug-loaded polymersomes remain poorly understood. The combination of multiple drugs with different molecular targets can maximize therapeutic efficacy and is more likely to overcome drug resistance.32 Combination drug therapy has been adopted in the clinic as a primary regimen for cancer treatment. To observe the fate of drug-loaded polymersomes, we prepared bioreducible polymersomes for the controlled delivery of dual-drugs (Fig. 3).33 In this study, a triblock copolymer bearing disulfide bonds, PEG-b-p(lysine)-SS-p(caprolactone), that is cleavable in an intracellular environment was synthesized. In an aqueous solution, the copolymer self-assembled into polymersomes and could simultaneously encapsulate the hydrophobic drug camptothecin (CPT) and the hydrophilic drug doxorubicin·hydrochloride (DOX·HCl). The encapsulated drugs were rapidly released in response to 10 mM GSH, which is similar to the concentration encountered in an intracellular environment. An in vitro cytotoxicity test showed that dual drug-loaded polymersomes enhanced the cytotoxicity of drugs to SCC7 cells. Confocal microscopy images showed enhanced fluorescence intensity for the DOX-loaded bioreducible polymersomes in the cytoplasm of SCC7 cells, whereas weaker fluorescence was detected for the disulfide-insensitive control polymersomes. The significantly higher fluorescence of the bioreducible polymersome was due to the high GSH concentration in the cytoplasm, which resulted in the cleavage of the disulfide bond in the polymersomes and enhanced drug release.
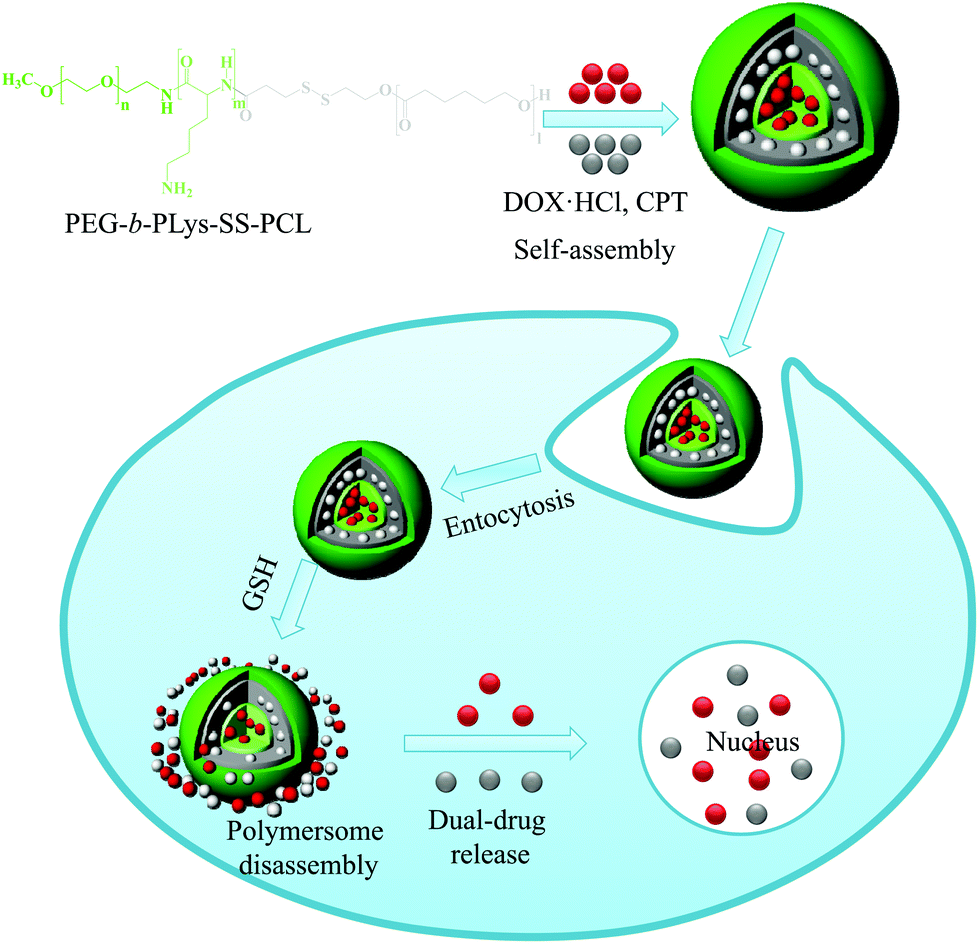 | ||
| Fig. 3 Schematic illustration of bioreducible dual-drug polymersomes for intracellular drug delivery.33 Reprinted with permission from ref. 33. | ||
Although the majority of polymersomes reach their target by the EPR effect, the preparation of functionalized polymersomes with targeting ligands is a topic of interest.34 Such polymersomes can specifically target cancer cells using an active targeting mechanism, in which polymersomes can bind to the cancer cells via overexpressed receptors. Polymersomes prepared using ligands behave like a missile and specifically bind to certain receptors that are overexpressed on cancer cells but not expressed on normal cells. Koul et al. prepared dual-targeting ligand-containing, redox-responsive polymersomes for the targeted delivery of DOX to breast cancer cells.35 Redox-responsive polymersomes, composed of PEG-poly(lactic acid) (PEG-PLA)-based tetrablock copolymers with multiple disulfide bonds, were initially prepared, and their surface was functionalized using folate and trastuzumab ligands. The presence of a higher hydrophobic balance enhanced the DOX loading, whereas the drug release was triggered by exposure to GSH. The folic acid and trastuzumab ligand in the polymersomes enhanced the cellular uptake of polymersomes in breast cancer cells. Owing to these properties, the systemic administration of DOX-loaded polymersomes resulted in complete tumor regression in Ehrlich ascites tumor-bearing Swiss albino mice.
Polymersomes prepared using amphiphilic polymers tend to dissociate after systemic administration, which might be due to their significant dilution in the body condition upon administration.36,37 As a consequence, the polymer concentration falls below the critical aggregation level, resulting in rapid drug release at unwanted sites.38,39 To surmount this issue, polymersomes have been cross-linked to preserve their nanostructure. In particular, cross-linking of polymersomes with a bioreducible cross-linker elegantly resolves extracellular instability and enhances intracellular drug release. We prepared a novel biocompatible and biodegradable polymersome nanotemplate using a PEG-b-PLys-b-PCL triblock copolymer for chemical cross-linking (Fig. 4).40 The primary amine in the PLys block was reacted with a disulfide-containing cross-linker to obtain reversibly cross-linked biostable polymersomes, and the cross-linking took place on the surface of a PCL membrane. The cross-linked polymersomes were remarkably resistant to dissociation by sodium dodecyl sulfate, a nanoparticle disturbing agent. Doxorubicin (DOX), which was selected as a model anti-cancer drug, was effectively encapsulated in the cross-linked polymersomes with a high loading efficiency. The in vitro release test indicated that DOX-loaded cross-linked polymersomes greatly retarded the release of DOX under physiological conditions (pH 7.4), whereas the DOX release rate was markedly increased in the presence of 10 mM GSH, which mimics an intracellular environment. These results suggest that cross-linking the polymersomes reduces the initial burst release and enhances the drug release at the target sites.
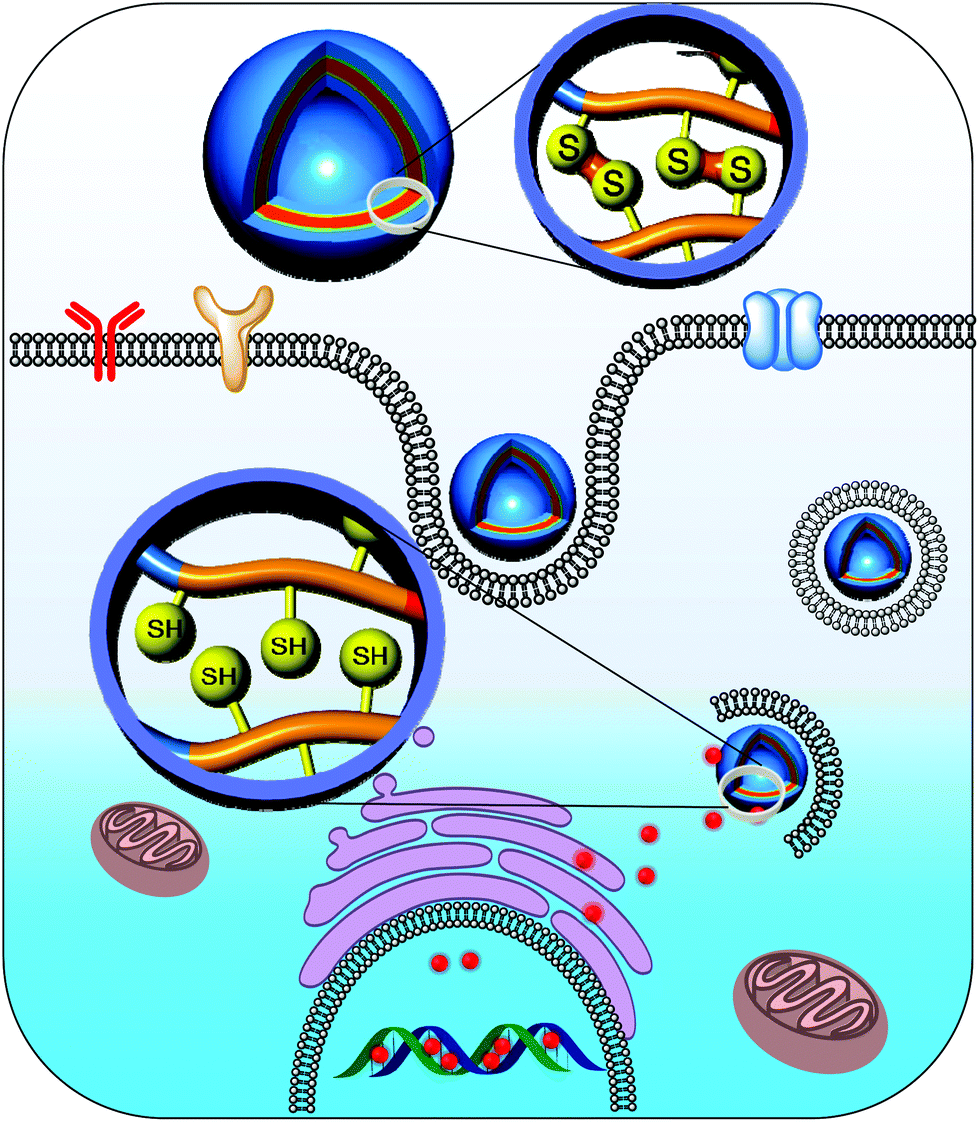 | ||
| Fig. 4 Controlled delivery of drugs into the nucleus of cancer cells from cross-linked polymersomes.40 Reprinted with permission from ref. 40. | ||
Controlled release of proteins at the target site is of great interest. Zhong et al. prepared dual-responsive reversibly cross-linked polymersomes for the site-specific delivery of proteins.41,42 In this study, a water soluble triblock copolymer, PEG-b-poly(acrylic acid)-b-poly(N-isopropylacrylamide) (PEG-b-PAA-b-PNIPAM), was initially prepared using a one-pot sequential reversible addition–fragmentation chain-transfer (RAFT) polymerization reaction. The block copolymers were freely soluble in water at room temperature, whereas they quickly transformed to nano-sized polymersomes when the temperature was increased to 37 °C. This effect may be due to the lower critical solution temperature of the PNIPAM. The carboxylic acid group in the PAA was cross-linked with cystamine, a bioreducible cross-linker, and the cross-linking took place at the interface of the hydrophobic PNIPAM membrane. These cross-linked polymersomes exhibited remarkable stability against high dilution, salt concentration, and temperature. Various proteins, including bovine serum albumin (BSA), lysozyme, cytochrome C, ovalbumin, and FITC-dextran, were effectively loaded into the cross-linked polymersomes with high loading efficiency. The release rate of protein was minimal in a physiological buffer, whereas protein release was enhanced in a reductive environment. The confocal laser scanning microscopy (CLSM) observations demonstrated that the cross-linked polymersomes efficiently delivered proteins into the cytosol of MCF-7 cells. As a result, therapeutic protein-loaded polymersomes enhanced the apoptosis of MCF-7 cells compared with the free proteins.
Although PEG-b-PAA-b-PNIPAM-based polymersomes exhibited appreciable in vitro efficacy, the neutral nature of PNIPAM in polymersomes restricts their application in vivo. To make PEG-b-PAA-b-PNIPAM-based polymersomes suitable for practical applications, Zhong et al. elegantly replaced the PNIPAM with a poly(2-(diethylamino)ethyl methacrylate) (PDEA) block and obtained PEG-b-PAA-b-PDEA (Fig. 5).43 The pH-sensitivity, cell permeability and proton sponge effect of the PDEA copolymer are known, and these materials have been applied for various biomedical uses. To obtain self-cross-linked polymersomes, cystamine was conjugated to PAA and reduced to obtain thiolated PEG-b-PAA-b-PDEA. The copolymer was freely soluble in acidic environments but formed self-assembled polymersomes by increasing the pH to 7.8. These polymersomes could efficiently encapsulate BSA and cytochrome C without loss of its biological activity. The intracellular localization of protein was measured by exposing the protein-loaded polymersomes to MCF-7 cells. Microscopy images indicated that protein-loaded polymersomes were taken up by the MCF-7 cells via endocytosis and subsequently escaped from endosomes due to the proton sponge effect. The released polymersomes in the cytosol were de-cross-linked, which enhanced protein release. As a result, cytochrome C-loaded polymersomes were more cytotoxic to MCF-7 and HeLa cancer cells than 293 T cells.
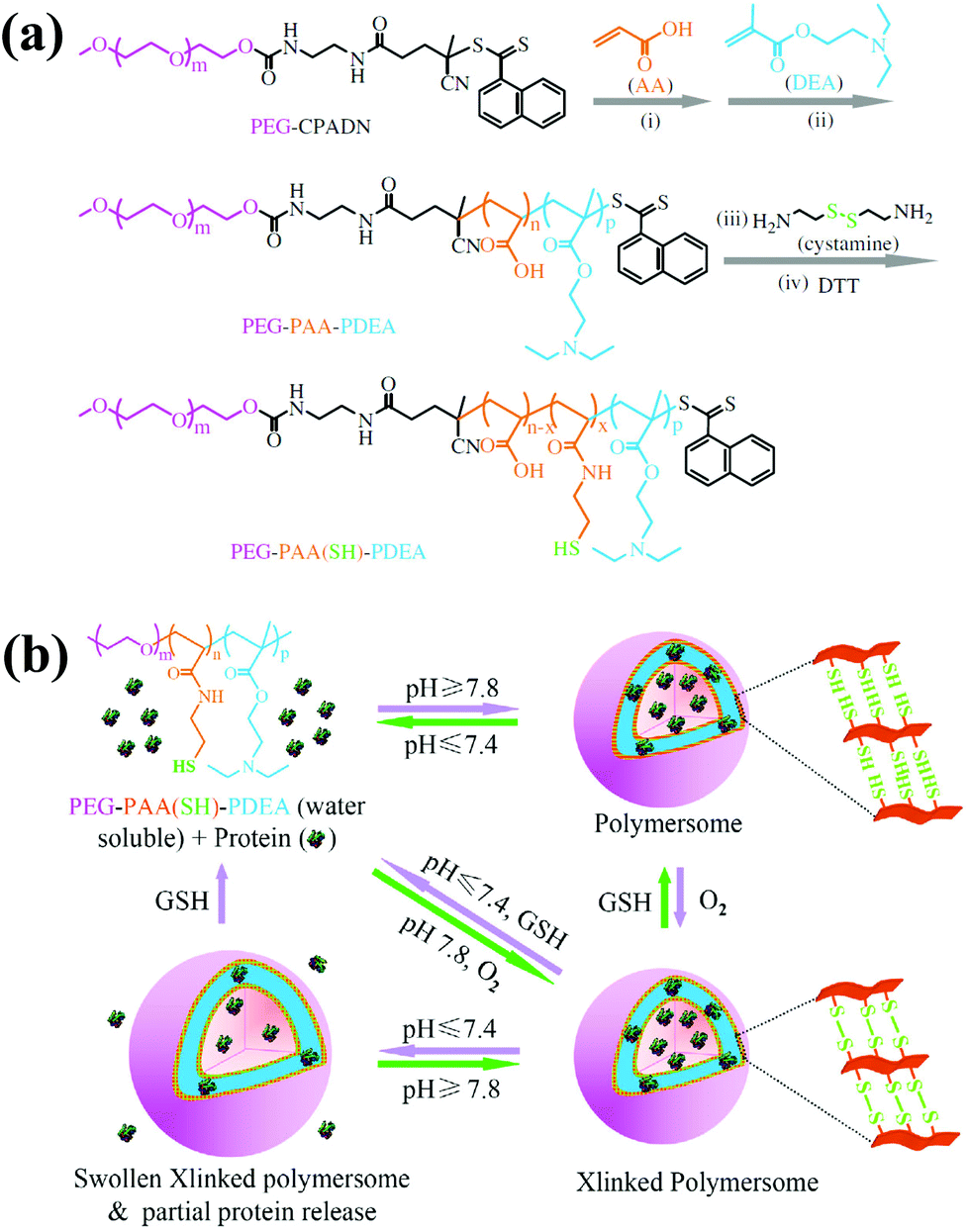 | ||
| Fig. 5 (a) Synthesis scheme for the preparation of the thiolated PEG–PAA–PDEA triblock copolymer. (b) Schematic illustration of pH and reduction dual-responsive cross-linked polymersomes for the triggered release of encapsulated protein.43 Reprinted with permission from ref. 43. | ||
Polymersomes prepared using amphiphilic copolymers are typically inert. These inert carriers are the major component of drug carriers, whereas drugs are the minor component. The major limitation of these nanomedicines is their low drug content. Therefore, a large amount of nanocarriers must be systemically administered to maximize the therapeutic effect of the drug. For practical applications, repeated administration of high doses of these excipients may cause systemic toxicity and impose an additional burden on the patient to excrete the carriers. To overcome these limitations, CPT was conjugated to a very short oligomer of ethylene glycol that could form polymersomes and be used for drug encapsulation.32 SN38 is an active metabolite of irinotecan, a potent anticancer drug, which has been widely used to treat colon cancer. Recently, Gu et al. prepared prodrug (SN38)-anchored polymersomes (OEG-2S-SN38).44 The drug was released from polymersomes in the presence of reactive oxygen species (ROS) and GSH. As a consequence, prodrug polymersomes were highly effective for treatment in a tumor model. These results suggested that drugs can be directly used as part of the carrier to fabricate other highly efficient nanocarriers and substantially reduce the use of other inert carrier materials while increasing the content of the loaded drug and avoiding premature burst release.
4. pH-Responsive polymersomes
The extracellular pH of tumor tissues (∼6.5–7.2) is slightly lower than that of normal tissues and other biological fluids (∼7.4); it is further decreased in intracellular endosomes (pH 5.5–5.0) and lysosomes (pH 4.0–4.5).45–48 Owing to the wide range of pH gradients in physiological systems, the application of pH-responsive polymersomes in cancer therapy is of great interest. In fact, the fabrication of pH-tunable polymersomes has been proposed as the main strategy to control the encapsulation and release pattern of nanoconstructs in vivo. Numerous acid-responsive linkers have been investigated to prepare polymeric nanoconstructs, including hydrazone, imine, orthoester, and acetal.17,49–54 Furthermore, anionic polymers, including poly(acrylic acid), poly(methacrylic acid), and cationic polymers, such as poly(β-amino ester), poly(lysine), and poly(histidine), are sensitive to the pH.47,53,55–58 Such pH-responsive polymersomes can be designed to carry, deliver, and control the release of therapeutic agents to the tumor tissue by relying on the low pH in the vicinity of tumor tissues.pH-Responsive polymersomes, composed of biocompatible and biodegradable polymers, were initially developed by Feijen and Zhong's group.13,59 They prepared PEG- and polyester-based biodegradable polymersomes as a basis for artificial cells.5 Although these polymers can encapsulate and release drugs well, the fine-tuning of the drug release by these polymersomes has been limited by their insensitivity to stimuli. Thus, pH-sensitive polymersomes based on PEG and poly(2,4,6-trimethoxybenzylidene-pentaerythritol carbonate) (PTMBPEC) have been prepared.60 Varying the hydrophobic ratio of the block copolymer resulted in both micelles and polymersomes. Specifically, the PEG(1.9k)-PTMBPEC(6k) and PEG(5k)-PTMBPEC(5.8k) could form polymersomes and micelles, respectively. The acetal linker present in the PTMBPEC is highly sensitive to acidic environments, and its degradation byproduct exhibits low/no cytotoxicity to normal cells. The PTMBPEC-based polymer degrades at mild acidic pH values with a half-life of 0.5 day, and the degraded polymersomes resulted in a significant increase in size (over 1000 nm). Polymersomes could encapsulate hydrophobic (paclitaxel (PTX)) and hydrophilic (DOX·HCl) drugs in their hydrophobic membrane and aqueous core, respectively. Conversely, micelles could encapsulate only PTX. Regardless of the morphology, the drug-loaded nanostructure exhibited pH-dependent drug release, i.e., at mildly acidic pH values, complete drug release was observed.
Liu et al. developed biomimetic pH-responsive biodegradable polymersomes for the simultaneous encapsulation and release of two drugs.61 The copolymer PLA-b-poly(2-methacryloyloxyethyl phosphorylcholine) (PLA-b-PMPC) was prepared by atom-transfer radical-polymerization. The natural PMPC present in the copolymer effectively reduces the protein opsonization due to its hydrophilic nature and may allow the prolonged circulation of polymersomes in the bloodstream. Polymersomes encapsulated hydrophilic DOX·HCl and hydrophobic DOX in their aqueous lumen and hydrophobic membrane, respectively. The drug-loaded polymersomes rapidly released the drug under acidic conditions but not at physiological pH values. The drug-loaded polymersomes rapidly entered HepG2 cells, escaped from endosomes and released the drug in the cytosol.
The presence of a large aqueous core in polymersomes may serve as an ideal candidate for the encapsulation and release of water-soluble proteins and peptides. In particular, polymersomes with an ionic membrane may efficiently encapsulate proteins via electrostatic interactions. Unlike other formulations that require the use of organic solvents for protein loading, which results in the denaturation of the protein, polymersomes with ionizable membranes may allow for the efficient encapsulation of proteins due to their tunable pH. For instance, biodegradable polymersomes with pH-responsive ionizable membranes have been developed for the encapsulation and intracellular release of therapeutic proteins.62 In this study, PEG-poly(carbonate)-based block copolymers containing acrylate, carboxylic acid, and amine were prepared. The simple dispersion of block copolymers in PBS (pH 7.4) yielded nano-sized polymersomes that could encapsulate therapeutic proteins by simple mixing. Rapid protein release was observed when the polymersomes were exposed to endosomal pH, whereas protein release was minimized at physiological pH values. To accelerate the release of protein in the cytosol, the same group introduced disulfide bonds to ionizable polymersomes in another study, and these polymersomes exhibited dual-stimuli sensitivity. The pH and reduction sensitivity of polymersomes allows the endosomal escape of polymersomes via the proton sponge effect, and disulfides in polymersomes are reduced in the cytosol to enhance cytochrome C release for cancer cell apoptosis.
Chimeric polymersomes, a novel type of polymersome prepared from asymmetric ABC triblock copolymers, have recently been developed for the simultaneous encapsulation of drugs and proteins.63,64 Zhong et al. developed biodegradable chimeric polymersomes based on asymmetric PEG–PCL–PDEA triblock copolymers for the site-specific delivery of therapeutic agents.64 The film hydration of triblock copolymers allows the formation of polymersomes with a size of 130–175 nm. These polymersomes could load various proteins, including BSA, cytochrome C, lysozyme, ovalbumin and IgG, and the encapsulation of proteins did not alter their morphology. In addition, chimeric polymersomes simultaneously loaded proteins and DOX, which were then transported to the cytoplasm and nucleus, respectively. Although these chimeric polymersomes exhibited remarkable properties, their practical application has been limited because of the lack of suitable targetability. To improve the efficiency of chimeric polymersomes, anisamide-decorated chimeric polymersomes have been developed for the targeted delivery of an apoptotic protein, granzyme B (GrB).65 Anisamide-decorated chimeric polymersomes, composed of PEG–poly(2,4,6-trimethoxybenzylidene-1,1,1-tris(hydroxymethyl)ethane methacrylate)–PAA (PEG–PTTMA–PAA), were readily synthesized by RAFT polymerization. The GrB-loaded chimeric polymersomes exhibited rapid protein release under mildly acidic conditions, perhaps due to the degradation of acetal bonds in PTTMA. An in vitro cytotoxicity test suggested that anisamide-decorated chimeric polymersomes effectively targeted the overexpressed sigma receptor on the surfaces of H460 and PC-3 cells. As a result, GrB-loaded anisamide-decorated chimeric polymersomes were cytotoxic to cancer cells.
Polypeptide-based chimeric polymersomes, also known as pepsomes, have been developed for the efficient encapsulation and transportation of DOX·HCl into the nuclei of drug-resistant MCF-7 cells.66 Pepsomes, composed of PEG–poly(L-leucine)–poly(L-glutamic acid) (PEG–PLeu–PGA), were synthesized via the sequential one-pot ring-opening polymerization of Leu-NCA and benzyl glutamate NCA in the presence of a PEG macroinitiator. The PEG–PLeu–PGA triblock copolymer formed pepsomes in aqueous solutions and encapsulated DOX·HCl in their aqueous core. Interestingly, pepsomes could disassemble at pH 5.0 due to the ionization of PGA groups. As a result, DOX·HCl-loaded pepsomes exhibited a higher antitumor activity than free DOX·HCl in both RAW 264.7 cells and drug-resistant MCF-7 cells (Fig. 6).
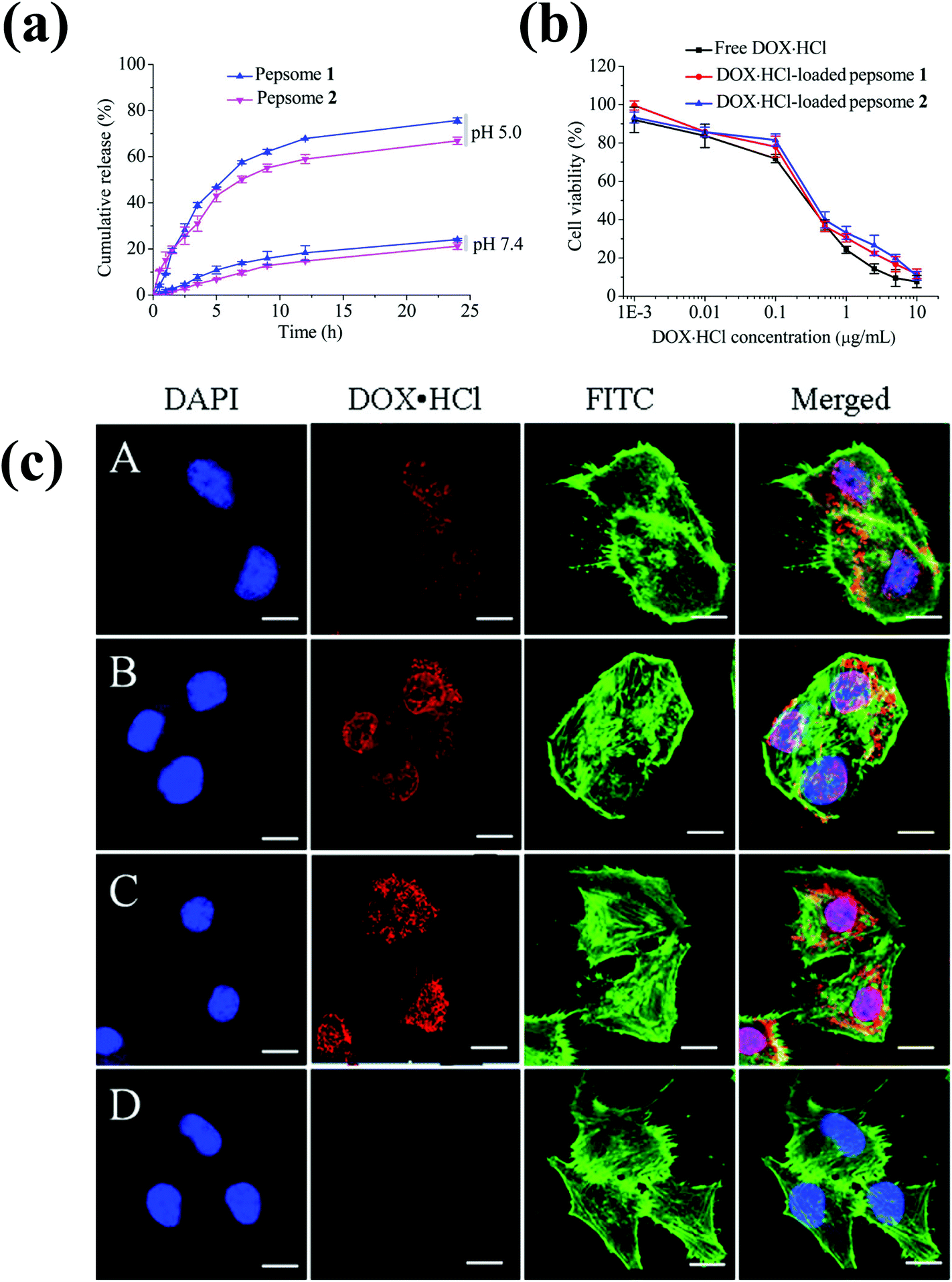 | ||
| Fig. 6 (a) pH-Dependent drug release behavior of pepsomes at 37 °C. (b) Cytotoxicity of DOX·HCl-loaded pepsomes on RAW 264.7 cells. (c) CLSM image of multidrug-resistance MCF-7 cells incubated with DOX·HCl-loaded pepsomes and free DOX·HCl. DOX·HCl-loaded pepsomes (A) 1 h; (B) 2 h; (C) 4 h; and (D) free DOX·HCl, 4 h incubation.66 Reprinted with permission from ref. 66. | ||
5. Enzyme-responsive polymersomes
The development of enzyme-responsive polymersomes is a relatively new area of research.20 In general, enzymes play critical roles in various biological and metabolic processes, serving as the prime protagonists in the chemistry of living organisms.20,67 The upregulation of enzymes often occurs in various diseases including cancer, thrombosis, inflammation, and infectious pathogens.68 Such an enzyme upregulation has been used as a pathological indicator in diagnosis and therapy. In this approach, enzyme-responsive substrates are covalently linked to the copolymer to form polymersomes. These polymersomes undergo physicochemical changes when exposed to certain enzymes found in inflammatory tissues.Cathepsin B (Cath B), a known lysosomal enzyme, is more abundant in tumor tissues than in normal tissues. This enzyme can be used to degrade certain tetrapeptides, such as Gly-Phe-Leu-Gly (GFLG). Owing to this unique property, polymersomes prepared using GFLG peptides may be a good candidate for intracellular delivery. Lee et al. prepared lysosomally cleavable polymersomes, in which a GFLG degradable linker was introduced to the middle of a hydrophilic PEG and a hydrophobic PLA block copolymer.69 The copolymer self-assembled into polymersomes in aqueous solutions and disassembled in the presence of Cath B. Anti-epidermal growth factor receptor-antibody immobilized polymersomes enhanced the cell uptake of polymersomes. Fluorescein-labeled dextran-containing polymersomes demonstrated that peptide linkers were cleaved in the lysosomal compartments, which led to membrane disruption. IgG-immobilized enzyme-responsive polymersomes could specifically bind to the primary antibody-treated SKBR3 cells. These findings identify enzyme-responsive polymersomes as promising tumor-targeting delivery vehicles. The ability of enzyme-responsive polymersomes containing chemotherapeutic drugs or proteins to treat tumors is promising and remains to be tested.
To overcome tumor drug-resistance, the delivery of multiple drugs by a single nanocarrier is promising. In particular, delivering multiple drugs with different chemical properties may improve therapeutic efficacy. For instance, the combination of inhibitors of topoisomerases I and II exhibits synergistic activity when administered together.70 Pavillard et al. demonstrated that the combination of CPT and DOX exhibits the synergistic effect to the C6 glioma cell line.71 CPT is a hydrophobic anti-cancer drug that exhibits anticancer activity by inhibiting DNA topoisomerase 1, an enzyme required for the replication and transcription of DNA.72 On the other hand, DOX·HCl is a hydrophilic compound that exhibits anticancer activity by intercalating DNA strands and subsequently inhibiting macromolecular biosynthesis.17 Since both drugs exhibit anticancer activity at the intracellular level, delivery of these two drugs into the cell could synergistically enhance the anticancer activity. Pramod et al. prepared a new enzyme-responsive polymersome based on a hydrophobic renewable source and hydrophilic dextran.73,74 The renewable source used in this study was pentadecylphenol, which is isolated from cashew nut shells. The dextran polymersomes effectively encapsulated hydrophilic DOX·HCl and hydrophobic CPT. Drug-loaded polymersomes released the drugs upon exposure to esterase, whereas they were stable in PBS buffer. The polymersomes were internalized by the cancer cells via caveolae-mediated endocytosis. As a consequence, enzyme-responsive polysaccharide polymersomes act synergistically to kill cancer cells. The same group also prepared pH- and enzyme-responsive polymersomes utilizing the same approach to deliver DOX into breast cancer cells.75
6. Glucose-responsive polymersomes
In recent years, glucose-responsive polymeric materials have received enormous attention due to their potential application in the treatment of glucose-related human diseases including diabetes.76 In this regard, phenylboronic acid (PBA)-containing polymers have been developed because of their rapid physicochemical changes in response to glucose.77 Generally, at high pKa (8.2–8.86) PBA exists in an equilibrium between the uncharged form and the charged form. The uncharged form is hydrophobic, whereas the charged form is hydrophilic.77,78 The PBA could react reversibly with 1,2-cis-diols of glucose, resulting in the conversion of hydrophobic PBA into the hydrophilic form which might induce the release of entrapped agents. Kim et al. demonstrated glucose-responsive disassembly of the amphiphilic copolymer for the triggered delivery of insulin.79 A glucose-responsive amphiphilic copolymer, composed of PEG and poly(styreneboroxole-alt-N-functionalized maleimide), was synthesized using RAFT polymerization. The amphiphilic polymer could form polymersomes and loaded insulin effectively; the insulin release was triggered by the addition of glucose into the release medium. In another study, Shi et al. prepared glucose-responsive polymersomes based on the complexation between the glucosamine (GA) conjugated block copolymer PEG45-b-P(Asp-co-AspGA) and the PBA conjugated block copolymer PEG113-b-P(Asp-co-AspPBA).80 Vancomycin, chosen as a model drug, was encapsulated into the polymersomes effectively. From the release experiment, it was found that vancomycin was barely released in physiological buffer (pH 7.4). Interestingly, when glucose was added into the release medium, the vancomycin release was triggered.7. Miscellaneous polymersomes
7.1. Photo-responsive polymersomes
Light, an external source, has been used for the design of intelligent delivery systems because the release of entrapped therapeutic agents can be rapidly induced at specific time points and locations by exposure to light.16 Although various stimuli-responsive systems respond quickly, they require the chemical modification of the environment via the addition of acids/bases or other reagents. However, light is a remote stimulus that does not require the addition of external substances. To prepare light-responsive polymersomes, the photolabile chromophores azobenzene, triphenylmethane, 2-nitrophenylalanine, and stilbene have been incorporated into the amphiphilic polymer systems to make the systems susceptible to light.81–84For instance, Burdick et al. presented a modular approach for synthesizing photolabile polymersomes, which allows for the incorporation of various functional groups at the junction of amphiphilic blocks (Fig. 7).85 With the use of this strategy, photo-cleavable 2-nitrobenzyl group or 2-nitrobenzyl group-bearing fluorescein was introduced in the middle of PEG and PCL copolymers. Owing to the amphiphilic nature, the resultant block copolymer formed polymersomes. Exposing these nanoparticles to UV light resulted in the disassembly of polymersomes. The fluorescein-containing polymersomes allowed the labeling of cells. Similarly, Meier et al. synthesized a photocleavable amphiphilic block copolymer, composed of poly(methyl caprolactone)-b-PAA-bearing ortho-nitrobenzyl, which could self-assemble into polymersomes in aqueous solutions (Fig. 8).86 The prepared polymersomes encapsulated a range of payloads, including small molecules (fluorescein) and proteins (eGFP). The polymersomes were completely disintegrated upon UV irradiation and rapidly released payloads. The photocleaving properties of polymersomes were controlled by varying the UV intensity and irradiation time. Zhou et al. prepared block copolymer free hyperbranched supramolecular amphiphiles through the noncovalent coupling of functionalized adamantane or azobenzene (Azo) with hyperbranched polyglycerol (HPG) grafted cyclodextrins (CDs).87,88 Such a host–guest complex self-assembled into vesicles with a narrow size distribution. Interestingly, the vesicles can be disassembled readily by the introduction of competitive hosts. The Janus hyperbranched polyglycerol prepared by Azo/CD complexation between CD-g-HPG and Azo-HBPO could form vesicles in an aqueous solution.88 The Azo group could undergo light-triggered reversible isomerization between the trans and the cis form under UV light irradiation. As a result, the Janus hyperbranched vesicles could be disassembled under irradiation of UV light (365 nm). This kind of system could be useful for pulsatile drug delivery applications.
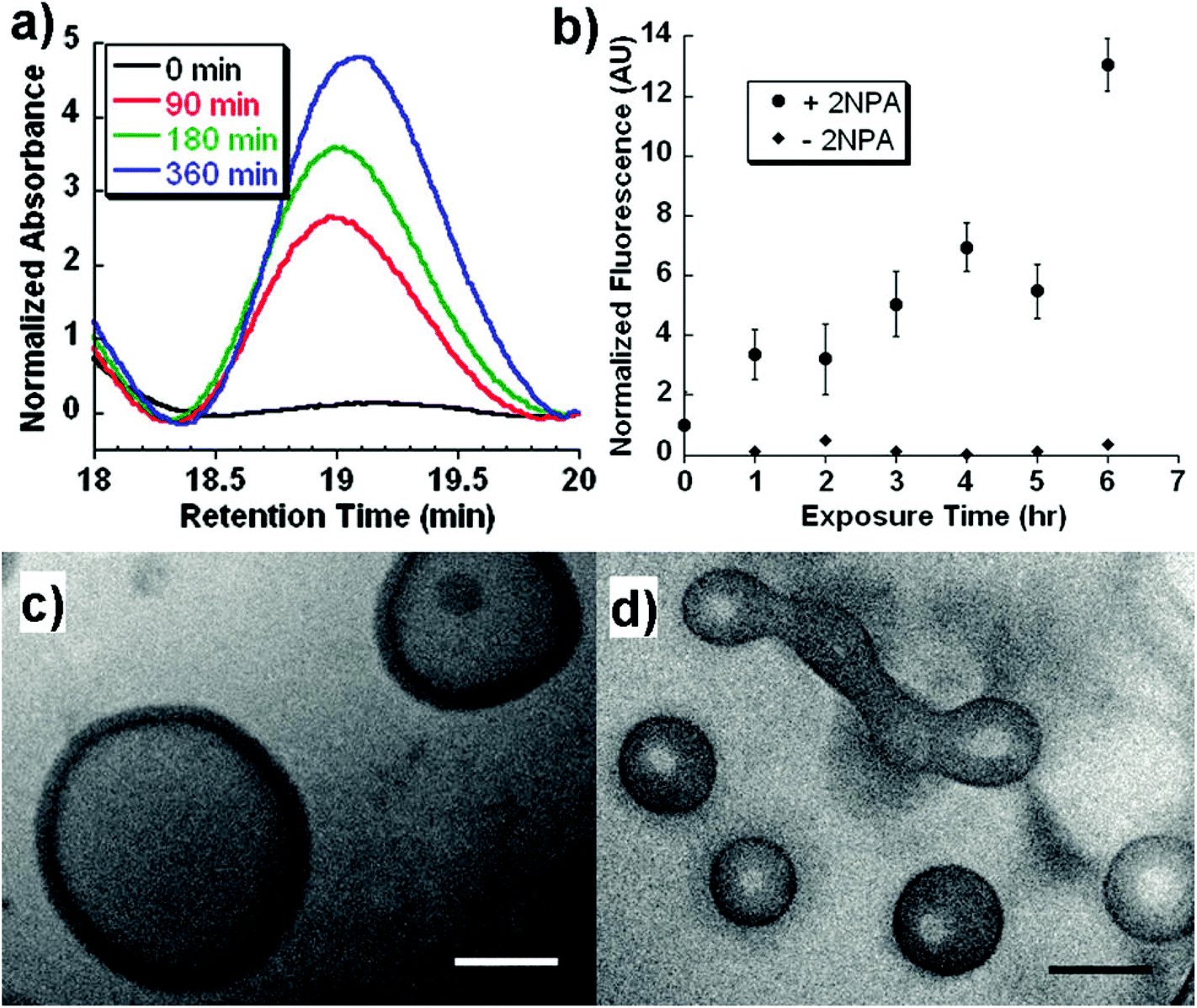 | ||
| Fig. 7 (a) GPC trace of the block copolymers as a function of UV exposure time. (b) In vitro release of biocytin from polymersomes under UV exposure. Cryo-TEM image of polymersomes at (c) 0 h and (d) 6 h after UV exposure.77 Reprinted with permission from ref. 77. | ||
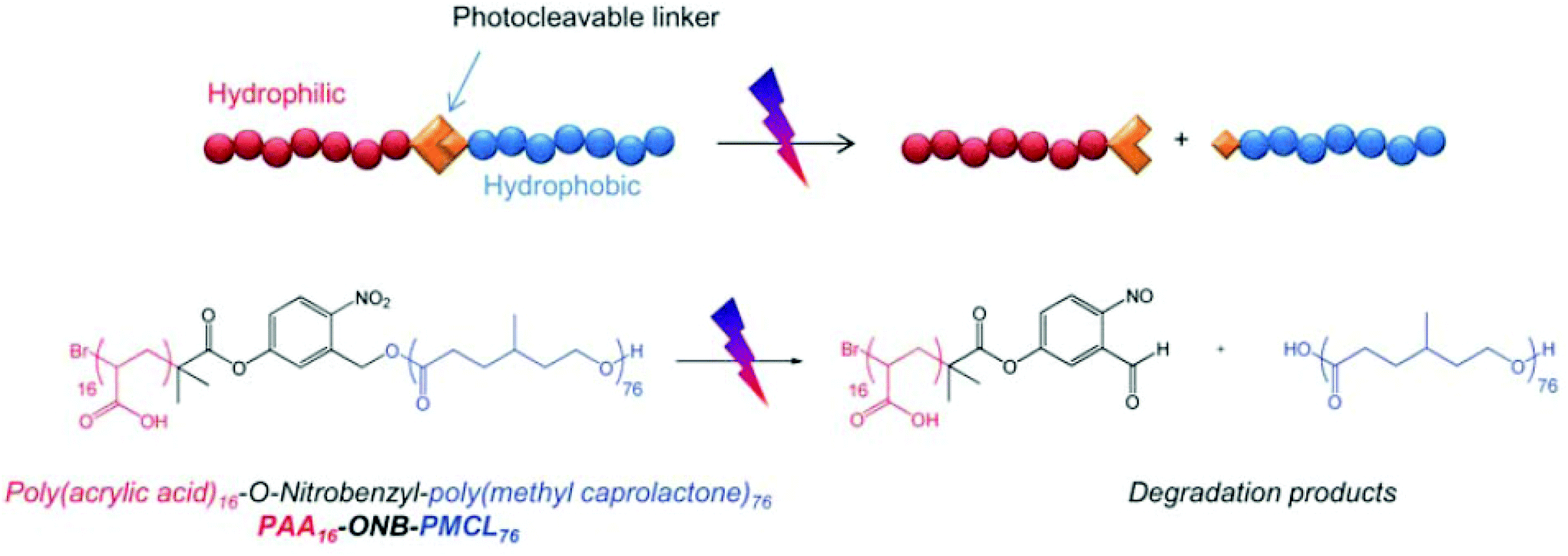 | ||
| Fig. 8 Schematic illustration of photocleavable polymersomes and chemical structure of the O-nitrobenzyl-bearing poly(methyl caprolactone)-b-poly(acrylic acid) copolymer and its degradation reaction after UV irradiation.78 Reprinted with permission from ref. 78. | ||
Although various UV-responsive polymersomes have been shown to possess excellent properties in vitro, this radiation is only applicable for the treatment of certain regions of the human body, such as the eye or the skin. Therefore, the use of UV radiation has been limited by its toxicity and low penetration depth. To circumvent this limitation, photo-sensitive groups that respond to near-infrared (NIR) laser light (∼700–1000 nm) have been utilized. NIR light deeply penetrates tissue and is expected to cause minimal photodamage to the body. Nie et al. fabricated nano-sized gold vesicles from gold nanoparticles containing a PEG-poly(styrene) (PEG-PS) anchoring polymer (Fig. 9).89 The gold-vesicles encapsulated hydrophobic Ce6 with a high loading efficiency (∼18.4 wt%). The vesicles showed strong NIR absorption, as a result of the plasmonic coupling effect of gold nanoparticles. Thus, NIR excitation produced heat and a singlet oxygen from gold nanoparticles and Ce6, respectively, to synergistically kill cancer cells. The heating of gold-vesicles resulted in the disassembly of the nanoconstructs and enhanced the release of Ce6, which allowed NIR/thermal/photo-acoustic trimodal imaging. Also, the combination of PTT/PDT treatment using single continuous wave laser irradiation improved the anti-tumor efficacy.
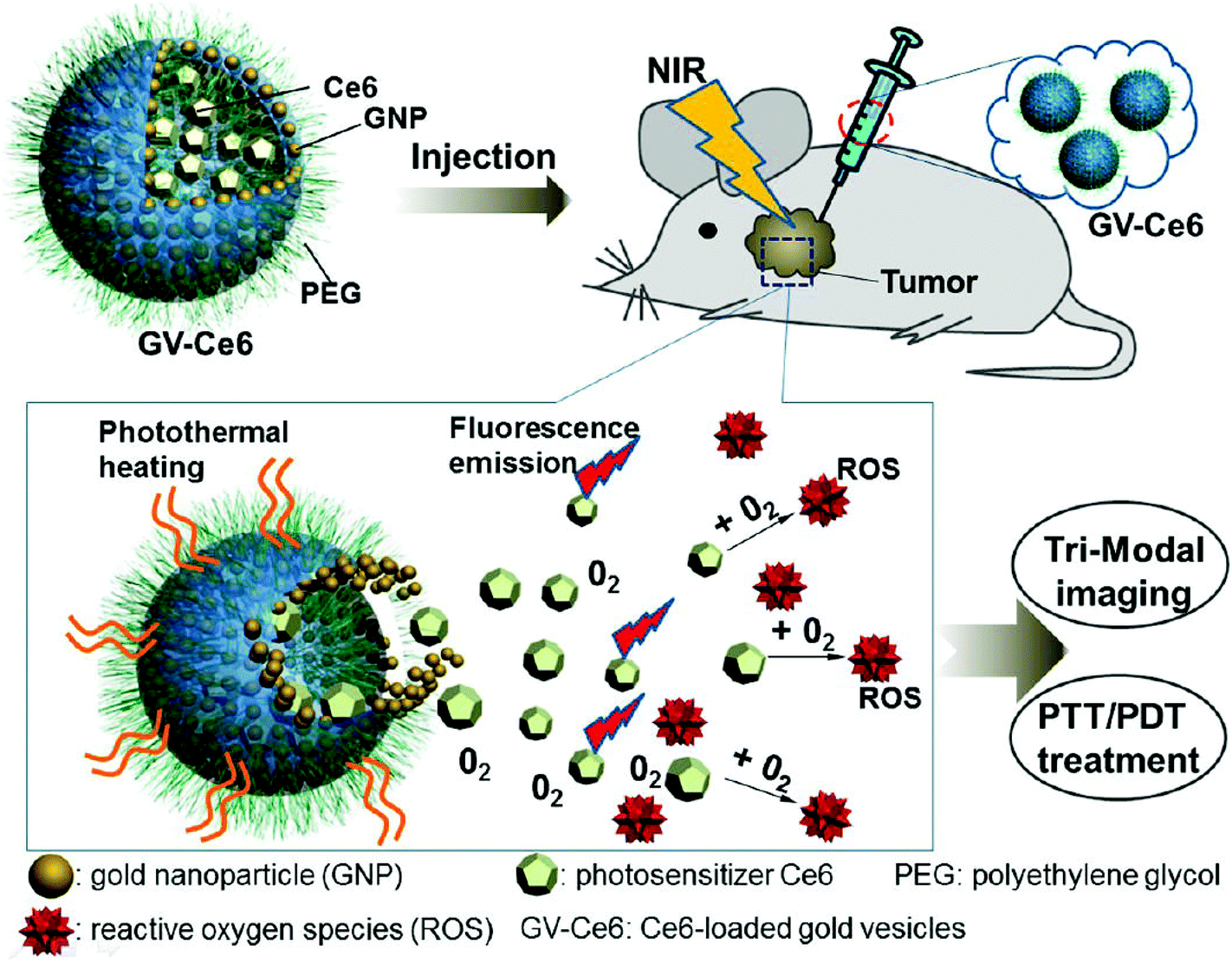 | ||
| Fig. 9 Systemic administration of photosensitizer-loaded gold vesicles for multimodal (fluorescence/thermal/photoacoustic) image-guided photothermal/photodynamic cancer therapy.79 Reprinted with permission from ref. 79. | ||
7.2. Voltage-responsive polymersomes
Numerous studies have attempted to develop stimuli-responsive polymersomes based on block copolymers. Recently, the preparation of block copolymer-free reversible nanoconstructs has received enormous attention because of the easy fabrication and tunable structural properties of these materials. Block copolymer-free polymersomes can be obtained via host–guest interactions between the end group of hydrophilic and hydrophobic homopolymers.90 CDs, composed of cyclic oligosaccharides with high biocompatibility, are known for their ability to form inclusion or host–guest complexes with various hydrophilic and lipophilic molecules via hydrogen bonding or hydrophobic interactions.91–93 Yin et al. prepared voltage-responsive vesicles based on chain end-decorated poly(styrene)-β-CD (PS-β-CD) and PEG-ferrocene (PEG-Fc) homopolymers (Fig. 10).94 These materials could form reversible diblock copolymers (PS-β-CD/PEG-Fc) via the host–guest interaction between CDs and Fc. Under aqueous conditions, the copolymers self-assembled into vesicular structures that were approximately ∼102 nm in size and stable for 3 months. Interestingly, the application of +1.5 V electrochemical stimuli initiated the disassembly of the vesicles, which had completely disrupted within 2 h. The voltage-responsiveness of the polymersomes was further verified by the polymersomes with rhodamine B (RhB). The release rate of RhB was precisely tuned by varying the applied potential. In particular, an abrupt release was observed in response to a high voltage (+4.0 V). The unique features of voltage-responsive polymersomes offer a new approach for stimuli-responsive drug delivery; such systems are well suited for applications that require structural changes without the addition of chemical reagents.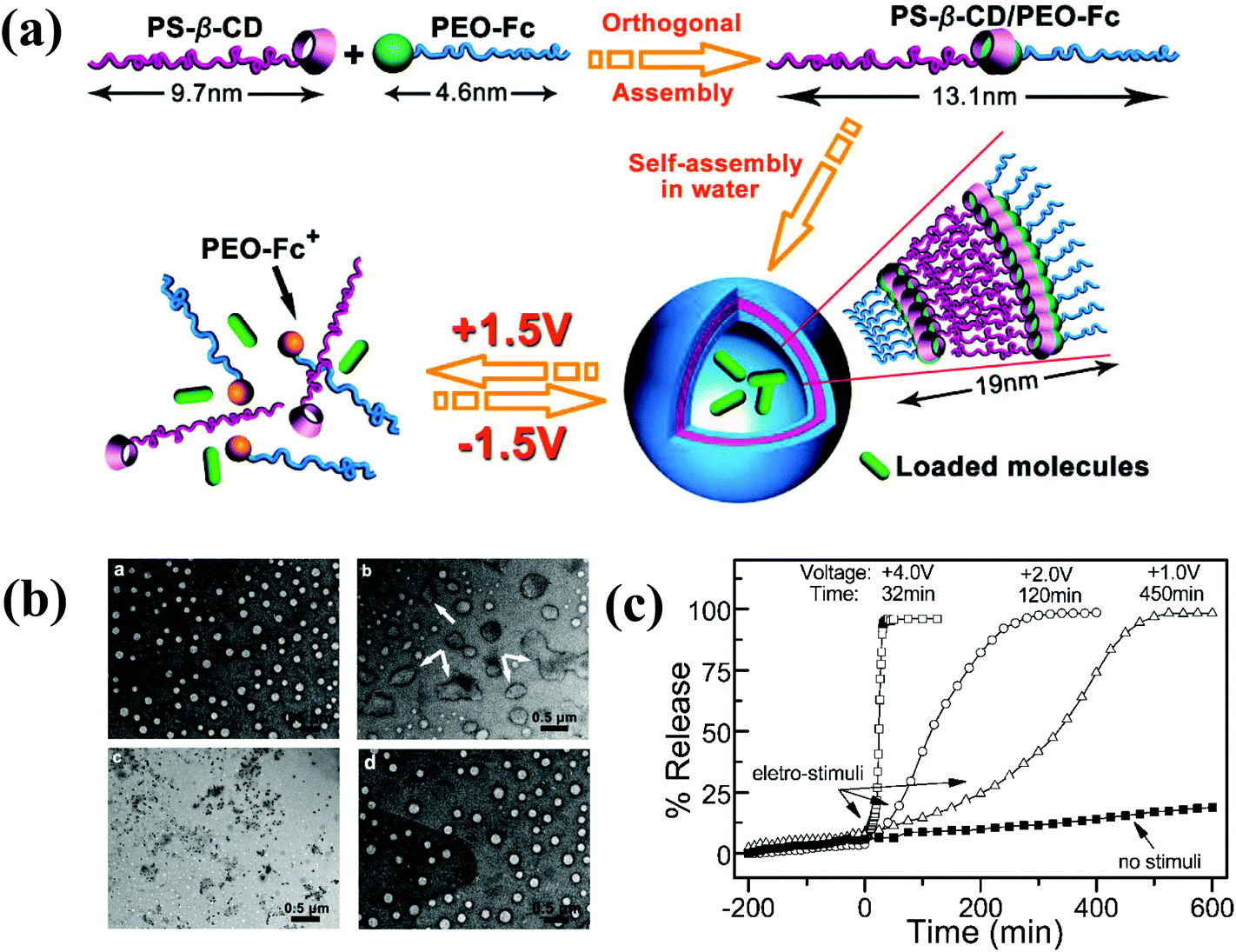 | ||
| Fig. 10 (a) Schematic illustration of the formation of block copolymer free polymersomes from PS-β-CD and PEG-Fc homopolymers and their voltage-responsive assembly and disassembly. (b) TEM image of voltage-responsive polymersomes. The top left figure shows the polymersomes with no external voltage and the top right figure shows the polymersomes 2 h after +1.5 V applied voltage. The bottom left figure shows the polymersomes 2 h after +1.5 V applied voltage and the bottom right figure shows the polymersomes 5 h after −1.5 V applied voltage. (c) In vitro release of RB from polymersomes upon applying varying voltage.84 Reprinted with permission from ref. 84. | ||
Although PS-β-CD/PEG-Fc-based polymersomes showed promising voltage-responsive properties, their disassembly in aqueous solutions resulted in the formation of hydrophobic aggregates due to the hydrophobic PS homopolymer. To surmount this limitation, Park et al. developed electrically switchable polymersomes, in which the polymersome membrane was split by an oxidizing voltage.95 They prepared tetra-aniline-conjugated PEG (TAPEG). In an aqueous solution, TAPEG self-assembled into polymersomes and effectively encapsulated a fluorescein derivative. The membrane was split by the action of applied voltage and could form puck-like micelles. The encapsulation and release pattern of FITC demonstrated the potential of electrically switchable polymersomes.
7.3. Ultrasound and magnetic field-responsive polymersomes
Ultrasound, an external stimulus, is used as an adjuvant of chemotherapy in tumor treatment.96 In chemotherapy, ultrasound can be used as a sensitizer to enhance chemotherapy and overcome drug resistance. Compared with light, which does not have time and target selectivity, ultrasound is effective for deep tissue penetration by tuning the frequency, duty cycles and time of exposure. Recently, Du et al. prepared novel polymersomes that are responsive to both ultrasound and pH (Fig. 11).97 The polymersomes were prepared from a triblock copolymer based on PEG-b-poly[2-(diethylamino)ethyl methacrylate-stat-tetrahydrofuranyloxy)ethylmethacrylate] (PEG-b-p(DEA-stat-TMA)). These polymersomes respond to both ultrasound and pH, and their potential applications were examined based on the entrapment and release of DOX under different conditions. In response to 180 W ultrasound radiation, the vesicles shrank to accelerate DOX release. Upon ultrasound exposure, the PTMA chains hydrolytically reduced and the hydrophobic balance was altered, which resulted in the shrinking of the polymersomes.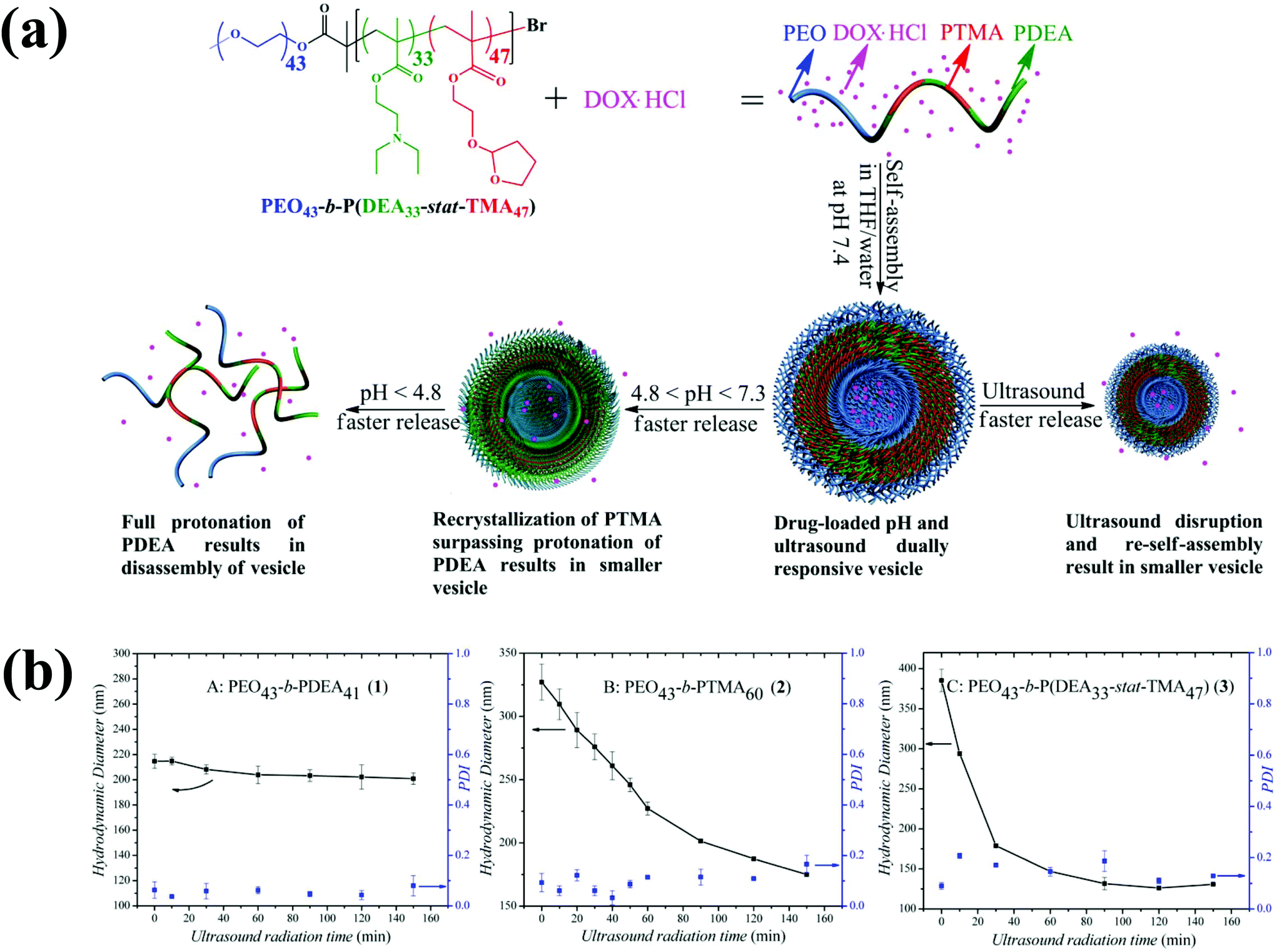 | ||
| Fig. 11 (a) Preparation of ultrasound and pH dual-responsive polymersomes from PEG-b-P(DEA-stat-TMA) copolymers and their drug release triggered by lowering the pH or by ultrasound radiation. (b) Effect of particle size changes of copolymers by ultrasound irradiation as a function of time.87 Reprinted with permission from ref. 87. | ||
Magnetic field-responsive materials are an important class of stimuli-responsive polymeric systems in drug delivery.98 The drug release from these types of polymersomes could be triggered by applying a magnetic field. In general, magnetic field-responsive polymersomes are prepared by encapsulating magnetic nanoparticles into the polymersomes. For instance, Lecommandoux et al. simultaneously encapsulated magnetic nanoparticles and DOX into the poly(trimethylene carbonate)-b-poly(GA)-based polymersomes.99 The controlled release of DOX was studied under magnetic field. The applied magnetic field could generate local heating by the hypothermia effect, and the produced heat energy altered the crystallinity of the polymersome membrane, which resulted in an abrupt drug release.
8. Conclusion and future perspectives
Harnessing the tumor microenvironment in nanocarrier-based drug delivery has proven to be a powerful strategy to improve chemotherapy. In this review, we highlighted several representative examples of stimuli-responsive polymersomes, which are the most promising nanoconstructs for biomedical applications. Polymersomes are self-assembled amphiphilic polymers in which an aqueous compartment is enclosed by a thick bilayer membrane. Unlike conventional polymersomes, stimuli-responsive polymersomes rapidly release drugs at the target site in response to a specific stimulus. Numerous biological and chemical stimuli, including pH, temperature, enzyme, and reduction, have been employed to trigger the hydrophobic–hydrophilic balance of polymersomes and destabilize their nanostructures. It is noteworthy that physical stimuli, including ultrasound, electrochemical potential, and light, can be applied via external sources and have seldom been used to prepare polymersomes. In fact, physical stimuli-responsive polymersomes constitute a non-invasive cancer therapy, which may open a new paradigm in cancer therapy. Although numerous stimuli-responsive polymersomes have been developed for biomedical applications, their potential application has often been proven in vitro, which does not guarantee their efficacy in vivo. In addition, the safety of polymersomes needs to be considered primarily. In particular, for practical applications, it will be important to develop polymersomes based on well established biocompatible materials like PEG and poly(carbonate)-based materials. Furthermore, novel stimuli-responsive polymersomes with suitable targetability and stability with multi-stimuli-linkers that might be cleavable in extracellular and intracellular space in a stepwise manner might be promising.Acknowledgements
This research was supported by the Basic Science Research Program through a National Research Foundation of Korea grant funded by the Korean Government (MEST) (20100027955).Notes and references
- Z. Cheng, A. Al Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338, 903–910 CrossRef CAS PubMed.
- R. Gref, Y. Minamitake, M. Peracchia, V. Trubetskoy, V. Torchilin and R. Langer, Science, 1994, 263, 1600–1603 CAS.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS.
- J. Yang, Curr. Opin. Colloid Interface Sci., 2002, 7, 276–281 CrossRef CAS.
- F. Meng, G. H. M. Engbers and J. Feijen, J. Controlled Release, 2005, 101, 187–198 CrossRef CAS PubMed.
- H. Yin, S.-W. Kang and Y. H. Bae, Macromolecules, 2009, 42, 7456–7464 CrossRef CAS.
- F. Ahmed, R. I. Pakunlu, A. Brannan, F. Bates, T. Minko and D. E. Discher, J. Controlled Release, 2006, 116, 150–158 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2014, 13, 813–827 CrossRef CAS PubMed.
- S. Egli, M. G. Nussbaumer, V. Balasubramanian, M. Chami, N. Bruns, C. Palivan and W. Meier, J. Am. Chem. Soc., 2011, 133, 4476–4483 CrossRef CAS PubMed.
- D. E. Discher, V. Ortiz, G. Srinivas, M. L. Klein, Y. Kim, D. Christian, S. Cai, P. Photos and F. Ahmed, Prog. Polym. Sci., 2007, 32, 838–857 CrossRef CAS PubMed.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS PubMed.
- F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS PubMed.
- T. Thambi, V. G. Deepagan, H. Y. Yoon, H. S. Han, S.-H. Kim, S. Son, D.-G. Jo, C.-H. Ahn, Y. D. Suh, K. Kim, I. Chan Kwon, D. S. Lee and J. H. Park, Biomaterials, 2014, 35, 1735–1743 CrossRef CAS PubMed.
- W. H. De Jong and P. J. A. Borm, Int. J. Nanomed., 2008, 3, 133–149 CrossRef CAS.
- J. Babin, M. Pelletier, M. Lepage, J.-F. Allard, D. Morris and Y. Zhao, Angew. Chem., Int. Ed., 2009, 48, 3329–3332 CrossRef CAS PubMed.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640–4643 CrossRef CAS PubMed.
- R. L. McCarley, Annu. Rev. Anal. Chem., 2012, 5, 391–411 CrossRef CAS PubMed.
- J. Liu, Y. Huang, A. Kumar, A. Tan, S. Jin, A. Mozhi and X.-J. Liang, Biotechnol. Adv., 2014, 32, 693–710 CrossRef CAS PubMed.
- Y. Ding, Y. Kang and X. Zhang, Chem. Commun., 2015, 51, 996–1003 RSC.
- I. Park, S. S. Sheiko, A. Nese and K. Matyjaszewski, Macromolecules, 2009, 42, 1805–1807 CrossRef CAS.
- H. A. Skinner, Trans. Faraday Soc., 1945, 41, 645–662 RSC.
- T. Thambi and J. H. Park, J. Biomed. Nanotechnol., 2014, 10, 1841–1862 CrossRef CAS PubMed.
- D. P. Jones, J. L. Carlson, P. S. Samiec, P. Sternberg, V. C. Mody, R. L. Reed and L. A. S. Brown, Clin. Chim. Acta, 1998, 275, 175–184 CrossRef CAS.
- G. Wu, Y.-Z. Fang, S. Yang, J. R. Lupton and N. D. Turner, J. Nutr., 2004, 134, 489–492 CAS.
- Y.-M. Go and D. P. Jones, Biochim. Biophys. Acta, 2008, 1780, 1273–1290 CrossRef CAS PubMed.
- T. Thambi, H. Y. Yoon, K. Kim, I. C. Kwon, C. K. Yoo and J. H. Park, Bioconjugate Chem., 2011, 22, 1924–1931 CrossRef CAS PubMed.
- T. Thambi, G. Saravanakumar, J.-U. Chu, R. Heo, H. Ko, V. Deepagan, J.-H. Kim and J. Park, Macromol. Res., 2013, 21, 100–107 CrossRef CAS.
- T. Thambi, D. G. You, H. S. Han, V. G. Deepagan, S. M. Jeon, Y. D. Suh, K. Y. Choi, K. Kim, I. C. Kwon, G.-R. Yi, J. Y. Lee, D. S. Lee and J. H. Park, Adv. Healthcare Mater., 2014, 3, 1829–1838 CrossRef CAS PubMed.
- S. Cerritelli, D. Velluto and J. A. Hubbell, Biomacromolecules, 2007, 8, 1966–1972 CrossRef CAS PubMed.
- L. Jia, D. Cui, J. Bignon, A. Di Cicco, J. Wdzieczak-Bakala, J. Liu and M.-H. Li, Biomacromolecules, 2014, 15, 2206–2217 CrossRef CAS PubMed.
- Y. Shen, E. Jin, B. Zhang, C. J. Murphy, M. Sui, J. Zhao, J. Wang, J. Tang, M. Fan, E. Van Kirk and W. J. Murdoch, J. Am. Chem. Soc., 2010, 132, 4259–4265 CrossRef CAS PubMed.
- T. Thambi, V. G. Deepagan, H. Ko, D. S. Lee and J. H. Park, J. Mater. Chem., 2012, 22, 22028–22036 RSC.
- P. V. Pawar, S. V. Gohil, J. P. Jain and N. Kumar, Polym. Chem., 2013, 4, 3160–3176 RSC.
- S. V. Lale, A. Kumar, S. Prasad, A. C. Bharti and V. Koul, Biomacromolecules, 2015, 16, 1736–1752 CrossRef CAS PubMed.
- Y. H. Bae and H. Yin, J. Controlled Release, 2008, 131, 2–4 CrossRef CAS PubMed.
- H. S. Han, T. Thambi, K. Y. Choi, S. Son, H. Ko, M. C. Lee, D.-G. Jo, Y. S. Chae, Y. M. Kang, J. Y. Lee and J. H. Park, Biomacromolecules, 2015, 16, 447–456 CrossRef CAS PubMed.
- A. N. Koo, H. J. Lee, S. E. Kim, J. H. Chang, C. Park, C. Kim, J. H. Park and S. C. Lee, Chem. Commun., 2008, 6570–6572 RSC.
- E. S. Read and S. P. Armes, Chem. Commun., 2007, 3021–3035 RSC.
- T. Thambi, V. G. Deepagan, H. Ko, Y. D. Suh, G.-R. Yi, J. Y. Lee, D. S. Lee and J. H. Park, Polym. Chem., 2014, 5, 4627–4634 RSC.
- H. Xu, F. Meng and Z. Zhong, J. Mater. Chem., 2009, 19, 4183–4190 RSC.
- R. Cheng, F. Meng, S. Ma, H. Xu, H. Liu, X. Jing and Z. Zhong, J. Mater. Chem., 2011, 21, 19013–19020 RSC.
- H. Sun, F. Meng, R. Cheng, C. Deng and Z. Zhong, Acta Biomater., 2014, 10, 2159–2168 CrossRef CAS PubMed.
- J. Wang, X. Sun, W. Mao, W. Sun, J. Tang, M. Sui, Y. Shen and Z. Gu, Adv. Mater., 2013, 25, 3670–3676 CrossRef CAS PubMed.
- I. F. Tannock and D. Rotin, Cancer Res., 1989, 49, 4373–4384 CAS.
- D. Neri and C. T. Supuran, Nat. Rev. Drug Discovery, 2011, 10, 767–777 CrossRef CAS PubMed.
- G. H. Gao, J. W. Lee, M. K. Nguyen, G. H. Im, J. Yang, H. Heo, P. Jeon, T. G. Park, J. H. Lee and D. S. Lee, Nat. Rev. Drug Discovery, 2011, 155, 11–17 CAS.
- R. A. Gatenby and R. J. Gillies, Nat. Rev. Cancer, 2004, 4, 891–899 CrossRef CAS PubMed.
- K. Dan and S. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 7300–7305 CrossRef CAS PubMed.
- T. Thambi, V. G. Deepagan, C. K. Yoo and J. H. Park, Polymer, 2011, 52, 4753–4759 CrossRef CAS PubMed.
- S. J. Lee, K. H. Min, H. J. Lee, A. N. Koo, H. P. Rim, B. J. Jeon, S. Y. Jeong, J. S. Heo and S. C. Lee, Biomacromolecules, 2011, 12, 1224–1233 CrossRef CAS PubMed.
- C. Ding, J. Gu, X. Qu and Z. Yang, Bioconjugate Chem., 2009, 20, 1163–1170 CrossRef CAS PubMed.
- F. Zhan, W. Chen, Z. Wang, W. Lu, R. Cheng, C. Deng, F. Meng, H. Liu and Z. Zhong, Biomacromolecules, 2011, 12, 3612–3620 CrossRef CAS PubMed.
- J. M. Shin, S.-H. Kim, T. Thambi, D. G. You, J. Jeon, J. O. Lee, B. Y. Chung, D.-G. Jo and J. H. Park, Chem. Commun., 2014, 50, 7632–7635 RSC.
- G. H. Gao, Y. Li and D. S. Lee, J. Controlled Release, 2013, 169, 180–184 CrossRef CAS PubMed.
- K. H. Min, J.-H. Kim, S. M. Bae, H. Shin, M. S. Kim, S. Park, H. Lee, R.-W. Park, I.-S. Kim, K. Kim, I. C. Kwon, S. Y. Jeong and D. S. Lee, J. Controlled Release, 2010, 144, 259–266 CrossRef CAS PubMed.
- D. H. Kim, Y. K. Seo, T. Thambi, G. J. Moon, J. P. Son, G. Li, J. H. Park, J. H. Lee, H. H. Kim, D. S. Lee and O. Y. Bang, Biomaterials, 2015, 61, 115–125 CrossRef CAS PubMed.
- S.-H. Kim, J.-H. Kim, D. G. You, G. Saravanakumar, H. Y. Yoon, K. Y. Choi, T. Thambi, V. G. Deepagan, D.-G. Jo and J. H. Park, Chem. Commun., 2013, 49, 10349–10351 RSC.
- F. Meng, C. Hiemstra, G. H. M. Engbers and J. Feijen, Macromolecules, 2003, 36, 3004–3006 CrossRef CAS.
- W. Chen, F. Meng, R. Cheng and Z. Zhong, J. Controlled Release, 2010, 142, 40–46 CrossRef CAS PubMed.
- G.-Y. Liu, L.-P. Lv, C.-J. Chen, X.-S. Liu, X.-F. Hu and J. Ji, Soft Matter, 2011, 7, 6629–6636 RSC.
- S. Li, F. Meng, Z. Wang, Y. Zhong, M. Zheng, H. Liu and Z. Zhong, Eur. J. Pharm. Biopharm., 2012, 82, 103–111 CrossRef CAS PubMed.
- Y. Du, W. Chen, M. Zheng, F. Meng and Z. Zhong, Biomaterials, 2012, 33, 7291–7299 CrossRef CAS PubMed.
- G. Liu, S. Ma, S. Li, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomaterials, 2010, 31, 7575–7585 CrossRef CAS PubMed.
- L. Lu, Y. Zou, W. Yang, F. Meng, C. Deng, R. Cheng and Z. Zhong, Biomacromolecules, 2015, 16, 1726–1735 CrossRef CAS PubMed.
- P. Chen, M. Qiu, C. Deng, F. Meng, J. Zhang, R. Cheng and Z. Zhong, Biomacromolecules, 2015, 16, 1322–1330 CrossRef CAS PubMed.
- Q. Hu, P. S. Katti and Z. Gu, Nanoscale, 2014, 6, 12273–12286 RSC.
- A. Wells and J. Grandis, Clin. Exp. Metastasis, 2003, 20, 285–290 CrossRef CAS.
- J. S. Lee, T. Groothuis, C. Cusan, D. Mink and J. Feijen, Biomaterials, 2011, 32, 9144–9153 CrossRef CAS PubMed.
- H. JJ, M. JL and R. N, Oncology, 2003, 17, 46–51 Search PubMed.
- V. Pavillard, D. Kherfellah, S. Richard, J. Robert and D. Montaudon, Br. J. Cancer, 2001, 85, 1077–1083 CrossRef CAS.
- X.-Q. Li, H.-Y. Wen, H.-Q. Dong, W.-M. Xue, G. M. Pauletti, X.-J. Cai, W.-J. Xia, D. Shi and Y.-Y. Li, Chem. Commun., 2011, 47, 8647–8649 RSC.
- P. S. Pramod, R. Shah, S. Chaphekar, N. Balasubramanian and M. Jayakannan, Nanoscale, 2014, 6, 11841–11855 RSC.
- P. S. Pramod, K. Takamura, S. Chaphekar, N. Balasubramanian and M. Jayakannan, Biomacromolecules, 2012, 13, 3627–3640 CrossRef CAS PubMed.
- P. S. Pramod, R. Shah and M. Jayakannan, Nanoscale, 2015, 7, 6636–6652 RSC.
- R. Ma and L. Shi, Polym. Chem., 2014, 5, 1503–1518 RSC.
- J. N. Cambre and B. S. Sumerlin, Polymer, 2011, 52, 4631–4643 CrossRef CAS PubMed.
- A. Matsumoto, S. Ikeda, A. Harada and K. Kataoka, Biomacromolecules, 2003, 4, 1410–1416 CrossRef CAS PubMed.
- H. Kim, Y. J. Kang, E. S. Jeong, S. Kang and K. T. Kim, ACS Macro Lett., 2012, 1, 1194–1198 CrossRef CAS.
- H. Yang, C. Zhang, C. Li, Y. Liu, Y. An, R. Ma and L. Shi, Biomacromolecules, 2015, 16, 1372–1381 CrossRef CAS PubMed.
- F. B. Peters, A. Brock, J. Wang and P. G. Schultz, Chem. Biol., 2009, 16, 148–152 CrossRef CAS PubMed.
- M. A. Yassin, D. Appelhans, R. G. Mendes, M. H. Rümmeli and B. Voit, Chem. – Eur. J., 2012, 18, 12227–12231 CrossRef CAS PubMed.
- Y.-L. Lin, H.-Y. Chang, Y.-J. Sheng and H.-K. Tsao, Macromolecules, 2012, 45, 7143–7156 CrossRef CAS.
- E. Mabrouk, D. Cuvelier, F. Brochard-Wyart, P. Nassoy and M.-H. Li, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7294–7298 CrossRef CAS PubMed.
- J. S. Katz, S. Zhong, B. G. Ricart, D. J. Pochan, D. A. Hammer and J. A. Burdick, J. Am. Chem. Soc., 2010, 132, 3654–3655 CrossRef CAS PubMed.
- E. Cabane, V. Malinova, S. Menon, C. G. Palivan and W. Meier, Soft Matter, 2011, 7, 9167–9176 RSC.
- W. Tao, Y. Liu, B. Jiang, S. Yu, W. Huang, Y. Zhou and D. Yan, J. Am. Chem. Soc., 2012, 134, 762–764 CrossRef CAS PubMed.
- Y. Liu, C. Yu, H. Jin, B. Jiang, X. Zhu, Y. Zhou, Z. Lu and D. Yan, J. Am. Chem. Soc., 2013, 135, 4765–4770 CrossRef CAS PubMed.
- J. Lin, S. Wang, P. Huang, Z. Wang, S. Chen, G. Niu, W. Li, J. He, D. Cui, G. Lu, X. Chen and Z. Nie, ACS Nano, 2013, 7, 5320–5329 CrossRef CAS PubMed.
- A. Feng, Q. Yan, H. Zhang, L. Peng and J. Yuan, Chem. Commun., 2014, 50, 4740–4742 RSC.
- M. Sivasubramanian, T. Thambi and J. H. Park, Carbohydr. Polym., 2013, 97, 643–649 CrossRef CAS PubMed.
- M. Sivasubramanian, T. Thambi, V. G. Deepagan, G. Saravanakumar, H. Ko, Y. M. Kang and J. H. Park, J. Nanosci. Nanotechnol., 2013, 13, 7271–7278 CrossRef CAS PubMed.
- H. Jang, T. Thambi, M. Sivasubramanian, J. Byun, J. Ahn, S. Chae, D.-G. Jo, J. Jeong, K. Lee and J. Park, Macromol. Res., 2014, 22, 816–819 CrossRef CAS.
- Q. Yan, J. Yuan, Z. Cai, Y. Xin, Y. Kang and Y. Yin, J. Am. Chem. Soc., 2010, 132, 9268–9270 CrossRef CAS PubMed.
- H. Kim, S.-M. Jeong and J.-W. Park, J. Am. Chem. Soc., 2011, 133, 5206–5209 CrossRef CAS PubMed.
- Y. Li, R. Tong, H. Xia, H. Zhang and J. Xuan, Chem. Commun., 2010, 46, 7739–7741 RSC.
- W. Chen and J. Du, Sci. Rep., 2013, 3, 1–9 Search PubMed.
- S. F. Medeiros, A. M. Santos, H. Fessi and A. Elaissari, Int. J. Pharm., 2011, 403, 139–161 CrossRef CAS PubMed.
- H. Oliveira, E. Pérez-Andrés, J. Thevenot, O. Sandre, E. Berra and S. Lecommandoux, J. Controlled Release, 2013, 169, 165–170 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |