
Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens†
Leighton
Pritchard
 *a,
Rachel H.
Glover
b,
Sonia
Humphris
c,
John G.
Elphinstone
b and
Ian K.
Toth
c
*a,
Rachel H.
Glover
b,
Sonia
Humphris
c,
John G.
Elphinstone
b and
Ian K.
Toth
c
aInformation and Computational Sciences, The James Hutton Institute, Invergowrie, Dundee, Scotland DD2 5DA, UK. E-mail: leighton.pritchard@hutton.ac.uk
bThe Food and Environment Research Agency, Sand Hutton, York, YO41 1LZ, UK
cCell and Molecular Sciences, The James Hutton Institute, Invergowrie, Dundee, DD2 5DA, UK
First published on 16th November 2015
Abstract
Soft rot Enterobacteriaceae (SRE) are bacterial plant pathogens that cause blackleg, wilt and soft rot diseases on a broad range of important crop and ornamental plants worldwide. These organisms (spanning the genera Erwinia, Pectobacterium, Dickeya, and Pantoea) cause significant economic and yield losses in the field, and in storage. They are transmissible through surface water, by trade and other movement of plant material and soil, and in some cases are subject to international legislative and quarantine restrictions. Effective detection and diagnosis in support of food security legislation and epidemiology is dependent on the ability to classify pathogenic isolates precisely. Diagnostics and classification are made more difficult by the influence of horizontal gene transfer on phenotype, and historically complex and sometimes inaccurate nomenclatural and taxonomic assignments that persist in strain collections and online sequence databases. Here, we briefly discuss the relationship between taxonomy, genotype and phenotype in the SRE, and their implications for diagnostic testing and legislation. We present novel whole-genome classifications of the SRE, illustrating inconsistencies between the established taxonomies and evidence from completely sequenced isolates. We conclude with a perspective on the future impact of widespread whole-genome sequencing and classification methods on detection and identification of bacterial plant pathogens in support of legislative and policy efforts in food security.
 Leighton Pritchard | Leighton obtained a B.Sc. (Hons) in Forensic and Analytical Chemistry, and a PhD in computational biology from the University of Strathclyde. Following a postdoc in systems biology at the University of Wales, Aberystwyth, he joined the Scottish Crop Research Institute and obtained a B.A. (Hons) in Mathematics. He is now based at the James Hutton Institute. Leighton's research interests include computational and systems biology of microbial plant pathogens, their interactions with host plants, and with each other. He has published more than 40 scientific papers spanning genomics, diagnostics, metabolic modeling, and bioinformatics tools. |
 Rachel H. Glover | Rachel Glover has dually trained in both molecular biology and bioinformatics and works as a computational biologist within the Applied Genomics team at Fera. She is responsible for managing the bioinformatics surrounding Fera's next-generation sequencers in addition to advising on bioinformatics experimental design and analyses for Fera research projects i.e. genome assemblies and annotation, metagenomics, data standards and release. Her research interests surround the metagenomic detection of pathogens, biodiversity informatics and the development of improved algorithms for DNA barcode identifications. |
 Sonia Humphris | Sonia Humphris is a graduate of the University of Abertay, where she obtained a PhD in Microbiology. She is currently a post-doctoral molecular bacteriologist in Ian Toth's lab at the James Hutton Institute in Dundee, Scotland. Sonia investigates diseases caused by enterobacterial plant pathogens, focusing on the potato pathogens Pectobacterium atrosepticum, and Dickeya species. Her research covers pathogenicity and disease epidemiology of these pathogens, and the interactions between them and their hosts. Sonia's research interests also include development of accurate and sensitive diagnostic assays for classification of Pectobacterium and Dickeya species. |
 John G. Elphinstone | Principal phytobacteriologist in the Fera Plant Protection Programme, Sand Hutton, York, UK. Specialist in molecular detection and identification, taxonomy, epidemiology and control of plant pathogenic bacteria of quarantine and other statutory importance. Consultant on risk assessment and plant health policy to Defra, the EC, USDA and to potato and horticultural industries worldwide. Consultant to EU and Defra Plant Health Authorities on biology and management of plant pathogenic bacteria. Member of the EPPO panel on diagnosis of plant pathogenic bacteria and a founding member of the European Association of Phytobacteriologists. Currently managing research projects for Defra, the Agriculture and Horticulture Development Board (AHDB) and the EU. |
 Ian K. Toth | Ian Toth leads the Weeds, Pests and Diseases Theme at the James Hutton Institute. He obtained a PhD from University of Warwick for work on the soft rot enterobacteria, and remained at the university to work the biology of antibiotic resistance in Streptomyces populations, before working for Novo Nordisk in their enzyme discovery division. Ian joined SCRI in 1995, returning to his main interest of plant pathology: the soft rot enterobacteria. In 2006 he became co-ordinator for Government-funded research on plant pathogens at SCRI. |
Background
Soft-rot Enterobacteriaceae (SR)
The Enterobacteriaceae are a large, widespread group of Gram-negative bacteria that includes food-borne human pathogens such as Escherichia coli, Yersinia pestis and Salmonella spp. The soft-rotting Enterobacteriaceae (SRE) are a plant-pathogenic subgroup of these bacteria, several of which (Erwinia amylovora, Dickeya dadantii, D. solani, Pectobacterium atrosepticum, and P. carotovorum) are considered to be amongst the most significant bacterial plant pathogens in terms of scientific and economic importance. They affect global food security by placing limits on yield and quality through their disease burden on several crops including potato, the world's fourth-largest crop.1 The SRE plant pathogens are aggressive necrotrophs, which typically cause maceration of the host plant's cell walls by production of plant cell wall-degrading enzymes (PCWDEs) that disrupt and kill host tissue to provide nutrition.2,3 PCWDEs are the primary virulence determinant of SREs, and are released alongside other virulence factors under quorum sensing (QS) control initiated by environmental stimuli such as oxygen deprivation.4,5 Productive infection produces symptoms or complex syndromes often characterised as ‘soft rot’, ‘wilt’ or ‘blackleg’, depending on the symptoms and how they are sited on the plant, and whether symptoms occur in the field, or in storage.6Although they are phylogenetically closely related to each other and exhibit similar disease symptoms, the SRE vary widely in geographical range, host range, and virulence.7 For example, within the Pectobacterium genus, P. carotovorum subsp. carotovorum has global distribution and a broad host range, whereas P. atrosepticum is largely restricted to potato grown in temperate climates, and P. betavasculorum almost exclusively causes disease on sugar beet.8Dickeya spp. also cause diseases on a broad range of crop and ornamental plants including grain crops (not a known host for Pectobacterium), but appear to do so from much lower initial inoculum levels, more aggressively, and often at warmer ambient temperatures than Pectobacterium spp.6
SRE are present on all farmed continents, can persist in surface and irrigation water, and present a global threat to a wide range of crops in many agricultural contexts.7,9P. wasabiae, although first identified on horseradish in Japan, has now been isolated from potato in Europe, Africa, Australasia, Iran, and North America.10–14P. carotovorum subsp. brasiliensis, which is highly aggressive and causes severe disease, was first isolated as a novel species in Brazil, but has subsequently been isolated from Korea, South Africa, New Zealand and Europe.10 SRE have been introduced to new territories by trade, and there is evidence of persistence and divergence of these bacteria, including adaptation to new host crops once introduced to a geographical region.15,16 The relative disease burden of different species or isolates of SRE pathogens in a region is observed to vary dynamically over time.6,17
Legislation for SRE
The significant threat from SRE to crop and ornamental plants in Europe is recognised in legislation at European and national levels. The European and Mediterranean Plant Protection Organisation (EPPO) recommends that member states regulate D. dianthicola and E. amylovora as quarantine pests, under the A2 list of pests already present in the EPPO region (EPPO 2015 PQR – EPPO database on quarantine pests, available online at http://www.eppo.int). Similarly, under the Seed Potatoes (Scotland) Amendment Regulations 2010, there is a zero tolerance policy for all Dickeya spp. on potatoes in Scotland, as production of ‘clean’ (disease-free) seed potato production for export is of economic significance for this nation. The economic threat from these pathogens is global, and the need for appropriate legislation not only confined to Scotland (even within the UK), but there is no uniform policy on compulsory sampling of soil or plant material, or on tolerance for infection of seed tubers at an international level.6,7Effective diagnostic tests supporting legislation and control discriminate with a level of certainty between ‘target’ and ‘non-target’ samples. An ideal test will be sensitive and accurate, and reflect the principal causative agent, where this can be defined. The efficacy of any test in support of legislative effort requires clear definition of the organisms to be controlled. These definitions may be based on phenotype, such as the ability of the target organism to produce disease symptoms in test plants or analogues of symptoms on test substrates, to present a particular serotype, exhibit particular antibiotic resistance, or to produce a specific toxin.18,19 Alternatively, the definition may be taxonomic and so based on the evolutionary history of the target organism, rather than direct evidence of phenotype, such as pathogenicity. In the legislative examples for SRE above, the target organism to be controlled is specified by taxonomic classification, which codifies an assumption that this is a reliable proxy for phenotype and threat from disease.
Taxonomy and phenotype
Diagnostic tests that assume a taxonomic classification implies a pathogenic phenotype (or vice versa) are potentially vulnerable to misclassification if one characteristic is incorrectly assumed to be a proxy for the other. In general, however, the mapping between genotype (and the taxonomic classification inferred from genotype) and phenotype is complex and not one-to-one.20 It is not guaranteed that a pathogenic bacterium will be capable of producing disease equally well in all environmental or climatic conditions, nor is it guaranteed that a bacterial phenotype, such as the ability to cause disease on a given host under set environmental conditions, is associated only with a single taxonomic class.The mapping between taxonomy and phenotype in bacteria can be particularly difficult to disentangle. In bacterial evolution the majority of functional gene transmission is vertical, but prokaryotic genomes are also subject to extensive exchange of functional genomic DNA by horizontal gene transfer (HGT) mechanisms. These do not correspond well to assumptions of bifurcating evolutionary history, distorting the linkage between taxonomy and phenotype into a ‘phylogenetic net’.21–23 Horizontally-transferred complex traits involving suites of genes (such as methanogenesis) tend to be found highly-conserved in taxonomic clusters but ‘simpler’ traits, such as the ability to exploit specific carbon sources, are often highly dispersed phylogenetically.24
Whole-genome diversity analysis of Lactobacillus lactis, for example, indicates that clustering of isolates based on chromosomal genes corresponds to taxonomic classification, whereas clustering on the basis of plasmid-borne (more likely to be discarded or transferred by HGT) genes reflects niche-adaptation phenotype.25 Similar observations prevail for pathogenic (including plant-pathogenic) bacteria through the effect of movement of pathogenicity islands (PAIs) and metabolic islands (MAIs) in response to stresses such as plant defence responses, so that phenotypes relevant to control and detection of pathogens may not be exactly congruent with taxonomy.22,26–28 In particular, virulence factors and environmental adaptations relevant to disease in planta including protein secretion systems, phytotoxins, nitrogen-fixation, and genes of eukaryotic origin, are known to be associated with acquisition through horizontal gene transfer in SRE.5,29
Some implications for practical pathogen detection in support of legislation follow from the adoption of a taxonomic definition: diagnostic efforts may focus exclusively on taxonomy-based identification of known pathogens; taxonomically distinct organisms posing a similar pathogenic threat may not be identified by these tests; and that members of a controlled taxonomic grouping may already be present undetected in the territory, if previous sampling efforts that focused on suspect samples are incomplete for the group as a whole. These are practical concerns: confusion between phenotype and genotype/taxonomy caused delay in recognition of novel pathogenic strains of E. coli in a recent serious outbreak of disease;19 and Dickeya spp. were identified as having been present unidentified in waterways of South-East England, possibly for several decades.6,9
Correct taxonomic classification is important for diagnostics
In order to implement effectively quarantine and other pathogen control legislation, accurate identification of a controlled organism must be possible to establish its presence, either in the field or the sample of material being stored or transported. This requires the ability to detect accurately the target pathogen in question, and to distinguish it reliably from non-target organisms.Incorrect identification of a controlled plant pathogen incurs societal costs. False positives (Type I error), where a clean sample is wrongly determined to be infected and destroyed or rejected, may result in economically damaging incorrect downgrade or condemnation of uncontaminated produce. It may be ruinous to a farming family's livelihood to quarantine their farm or fields wrongly. Type II errors, or false negatives, where a pathogen is present but undetected, may result in contaminated material being introduced to a previously unexposed farm, or wider geographical region. The import of a novel pathogen to a previously unexposed region may cause widespread economically significant damage, or irreversibly introduce a new pathogen to the regional ecology. Introduced pathogens may jump to novel and unanticipated hosts, as is thought to have been the case for Dickeya spp. in Europe.16
Additional to quarantine and legislative control, precise determination of the causative agent of disease is of considerable value to epidemiology and tracking of disease outbreaks. Forecasting and modelling of SRE ecology and future patterns and spread of disease in affected regions rely on accurate identification of existing and historical pathogen prevalence, their corresponding hosts and environmental preferences and mechanisms of geographical spread. Precise diagnostics are therefore also important for prediction and prevention of pathogen spread, in addition to evaluation of the effectiveness of legislative and other control measures.
Diagnostic methods for SRE
Where it is assumed that all members of a bacterial taxonomic group have the same potential for pathogenicity, the diagnostic question is whether unknown bacteria isolated from a sample can be correctly classified as belonging, or not, to that group. These classifications are typically determined using phenotypic, morphological, or sequence-based techniques.A comprehensive overview of the wide range of techniques for differentiating between Pectobacterium and Dickeya SRE is given in Czajkowski et al. (2015),7 covering selective growth media, biochemical and physiological assays, and DNA sequencing-based methods. For SRE, detection and identification has historically been by a polyphasic approach, involving isolation on semi-selective medium, followed by serological and other biochemical analyses. Historically, classification attempts were limited to identification as E. carotovora subsp. carotovora, E. c. subsp. atroseptica or E. chrysanthemi, and the legacy of these imprecise assignments continues to cause difficulties for linking contemporary taxonomy to historical phenotypic, geographical and host range observations, as the original samples are often not available.8 Biochemical and phenotypic tests may also take considerable time to complete, delaying transit of uncontaminated perishable material, or preventing rapid deployment of countermeasures against a pathogen.18 DNA sequencing-based techniques are now routinely applied that offer advantages of reproducibility, speed, and specificity over traditional biochemical tests. DNA sequencing-based diagnostics can also be used in multiplexed analyses to detect several SRE simultaneously, and are potentially semi-quantitative, even in complex environments.7,30 Routine whole-genome sequencing offers future potential for even more precise classification and identification of virulence determinants in novel pathogens.
Contemporary sequencing-based diagnostic methods rely on the ability to target and amplify by PCR diagnostic sections of DNA sequence that are specific only to a single class of organism, and present in all members of that class. Techniques include quantitative PCR (qPCR), variable number tandem repeat analysis (VNTR), loop-mediated isothermal amplification (LAMP), and multilocus sequence typing (MLST).8,16,31,32 In general, these approaches depend on the ability to design PCR primers or associated reporter probes that are themselves specific only to the diagnostic section of interest, and may in principle be designed at any arbitrary level of taxonomic organisation (genus, species, subspecies, and so on). PCR-based methods have practical advantages, but in diagnostic use assumptions are made that the presence of target-specific DNA implies presence of the organism itself, and that the organism is viable. This may not always be the case, as DNA may persist in the environment in the absence of its originating organism, and is also present in non-viable bacteria. Additionally, it is possible that primers believed to be specific may give false-positive interactions with off-target DNA that has not been previously encountered, or was not used in the evaluation of primer specificity. In both cases, the theoretical bias is towards false-positive overdiagnosis of the presence of a viable pathogen. It is also possible that the nominally specific region of DNA to be amplified is not in fact universal to all members of the class, but perhaps absent in some as yet untested isolate, which may also lead to false negative results.
Two complementary approaches have been used in diagnostic PCR primer design for SRE. The first, and most common, is to identify a region of DNA that is often a target gene associated with virulence, or a phylogenetically-divergent region such as 16S rDNA or intergenic transcribed region (ITS), which has been demonstrated or is expected to be common only to the desired group of organisms and/or divergent or absent in non-target organisms. This has provided widely-used ‘gold-standard’ primers for SRE including those based on pectate lyase genes33,34 and 16S RNA/ITS regions.35 The increasing availability of whole genome sequences for the SRE have recently made it possible to adopt an alternative approach by which a bulk search for thermodynamically viable PCR amplification products is performed across complete genome sequences for the target organisms of interest, and also for related non-target control organisms. The ability of the predicted primers to discriminate between classes of organisms is then tested in silico, before in vitro validation of promising diagnostic test candidates. This latter technique has been successful in producing PCR primers that are able to discriminate between very closely-related Dickeya spp., and between outbreak isolates of E. coli at sub-serotype level.31,36
The tangled taxonomy of SRE
An assumption runs through all the diagnostic techniques based on taxonomic classification that existing classifications of isolates from previous outbreaks and in strain collections, and of genomes in public sequence repositories, are correct.The phylogeny of Gammaproteobacteria, and Enterobacteria has historically been difficult to resolve cleanly, even using complex telescoping multiprotein approaches.37 The taxonomic history of SRE is particularly involved, and has recently undergone several significant revisions including reassignment of genus and species-level classifications, and the introduction of several new species (e.g. Dickeya solani38). The SRE were formerly classified as a single group of pectinolytic Erwinia spp., but are now recognised to comprise at least three taxonomically distinct genera: Erwinia, Pectobacterium, and Dickeya.7 Briefly, the current Pectobacterium classifications were obtained by reclassification of the single species Erwinia carotovora, and simultaneous elevation of several subspecies to species level, as the new genus Pectobacterium.39 The genus Dickeya was formed by initial reclassification of the species E. chrysanthemi to P. chrysanthemi, followed by elevation of P. chrysanthemi to the genus Dickeya, with simultaneous elevation of subspecies.40
For diagnostic tests, it is essential to establish performance statistics such as false positive and false negative rates. Without these measures, the likelihood of, for example, accepting contaminated material or condemning clean material cannot be calculated, and the relative performance of novel tests cannot be compared.41 In order to train classification methods, and to calculate these performance statistics, accurately-determined members of the classes to be tested must be available as ‘gold standards’. In addition to the potential for confusion in tracing historical sample identities through the corresponding nomenclature changes, the taxonomic revision of SRE has demonstrably led to misclassification of sequence data in the public record, and it is possible that similar misclassifications exist in reference collections.31 If these taxonomic classification metadata are used uncritically, evaluation of diagnostic methods against reference collections could compound the potential for classification error in diagnostics.
Polyphasic approaches to species classification, as historically used for SRE, have potential for internal contradiction due to the breakdown of one-to-one mapping between taxonomy and phenotype, and the sometimes subjective choice of which combination of techniques constitutes a diagnosis. In contrast to this, the complete genome sequence of a bacterium provides its complete genetic signature and a full account of its metabolic and biochemical potential, in terms of translated/transcribed elements such as virulence factors. However, the availability of a genome sequence does not imply the ability to reconstruct completely phenotypic or disease-causing potential as we cannot yet completely define the function of all genes, extrapolate comprehensively from genome to phenome, or take into account dynamic effects such as the influence of environmental context (including host response). Nevertheless, a complete genome sequence greatly increases the amount of information available for classification and diagnosis on the basis of both genomic distance and gene complement, and provides the most complete, unbiased measure possible for taxonomic organisation. Whole-genome sequencing technology is increasingly fast, accurate, and economical (though, for now, impractical in the field), and public repositories of sequence information such as NCBI provide a ready resource of information and potential in silico validation.
Whole-genome comparison-based classification methods such as Average Nucleotide Identity (ANI) have been demonstrated to correlate well with the previous ‘gold-standard’ DNA–DNA hybridisation (DDH) approaches to evaluation of genomic relationship.42,43 In particular, an established threshold of 95% sequence identity by ANI was found to correspond to the traditional ‘gold standard’ prokaryotic species threshold of 70% identity by DDH. Recently, the related MiSI (Microbial Species Identifier) method has been proposed as an approach for correction of inconsistent prokaryotic species classification, and for taxonomic assignment of newly-sequenced organisms. For MiSI, a common gene content of at least 60% of the complement, with at least 96.5% nucleotide sequence identity between reciprocally best-matching genes, is found to correspond well to species boundaries. The MiSI method was systematically applied to over 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 sequenced prokaryotes in the NCBI public database. Both ANI and MiSI techniques identify misclassified genomes in the public repositories; up to 18% of all annotated species in public sequence databases were indicated to include potentially misclassified isolates, by MiSI.44
000 sequenced prokaryotes in the NCBI public database. Both ANI and MiSI techniques identify misclassified genomes in the public repositories; up to 18% of all annotated species in public sequence databases were indicated to include potentially misclassified isolates, by MiSI.44
The methods used to classify sequenced prokaryotes are similar to those used to classify isolates in large, unsequenced, public strain collections. It may be reasonable to expect a similar rate of misclassification to obtain in these resources also, and this is a significant possible detriment to the gold-standard training/test datasets when designing and evaluating diagnostic tools.
Average Nucleotide Identity (ANI) analysis
Average Nucleotide Identity (ANI) analysis was applied to the 257 Enterobacteriaceae and Vibrio genomes described in ESI Table 1† (ESI Fig. 1; Table 2†). To generate this genome set, fourteen Pectobacterium genome sequences were obtained from the NCPPB collection, and sequenced at Fera (these sequences have previously been submitted to NCBI, with accessions indicated in ESI Table 1†). Twenty-five Dickeya genomes were also obtained as described in.45,46 The remaining 201 completely sequenced enterobacterial isolates were downloaded from the NCBI/GenBank repository. To generate input data for ANIm, all sequences (chromosome, organelle, draft or complete) for a given isolate were combined into a single multi-FASTA file, in a staging directory, prior to analysis. The isolates, and the accession numbers for all sequences combined in this way, are described in ESI Table 1.†Similar analysis was also performed for the 66 SRE genomes (plus Erwinia and Pantoea spp.) as a group (ESI Fig. 2; ESI Table 4†), and also separately for the 19 Pectobacterium and 34 Dickeya spp. genomes, to clarify relationships within those genera (Fig. 1; ESI Table 8;†Fig. 2; ESI Table 6†). This analysis was performed using the ANIm method described in Richter et al. (2009),43 and implemented in the Python module PYANI (v0.1.2) (https://github.com/widdowquinn/pyani/releases/tag/v0.1.2). A Makefile, datafiles, and associated scripts reproducing the analyses presented in this manuscript are publicly available at GitHub: https://github.com/widdowquinn/SI_Pritchard_etal_2015.
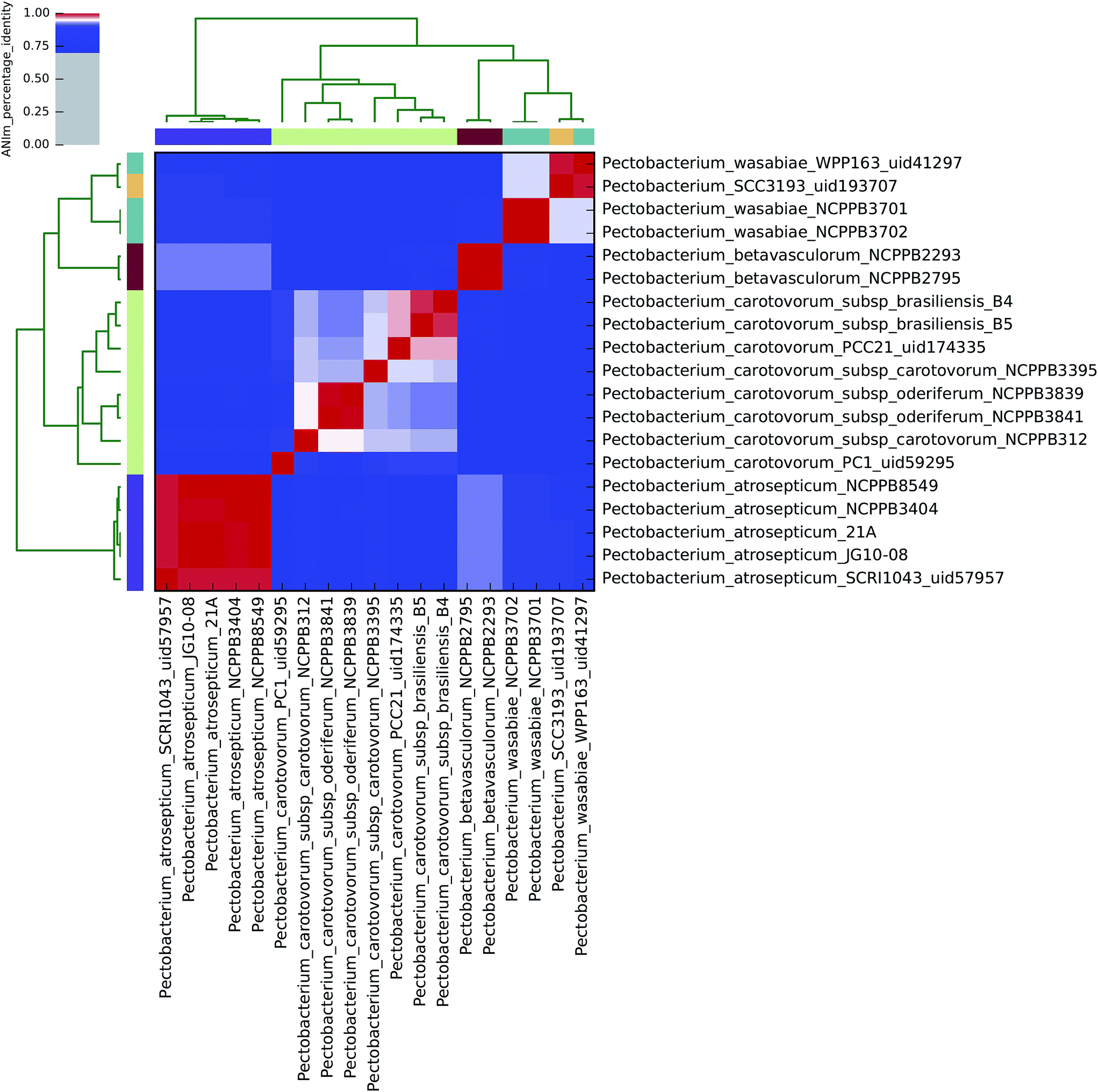 | ||
| Fig. 1 Heatmap of ANIm percentage identity for 19 isolates of Pectobacterium spp. as described in ESI Tables S1 and S8.† Species-level assignments and isolate identifiers as indicated at source are given as row and column labels. Cells in the heatmap corresponding to 95% ANIm sequence identity (and therefore the same species42,43) are coloured red. Blue cells correspond to ANIm comparisons indicating that the corresponding organisms do not belong to the same species. Colour intensity fades as the comparisons approach 95% ANIm sequence identity. Colour bars above and to the left of the heatmap correspond to source species-level assignments for each isolate in the analysis. Hierarchical clustering of the analysis results in two dimensions is represented by dendrograms, constructed by simple linkage of ANIm percentage identities. The analysis indicates up to eight species-level clades along the heatmap diagonal, in contrast to the six currently recognised Pectobacterium spp., suggesting that current taxonomic classifications require revision. | ||
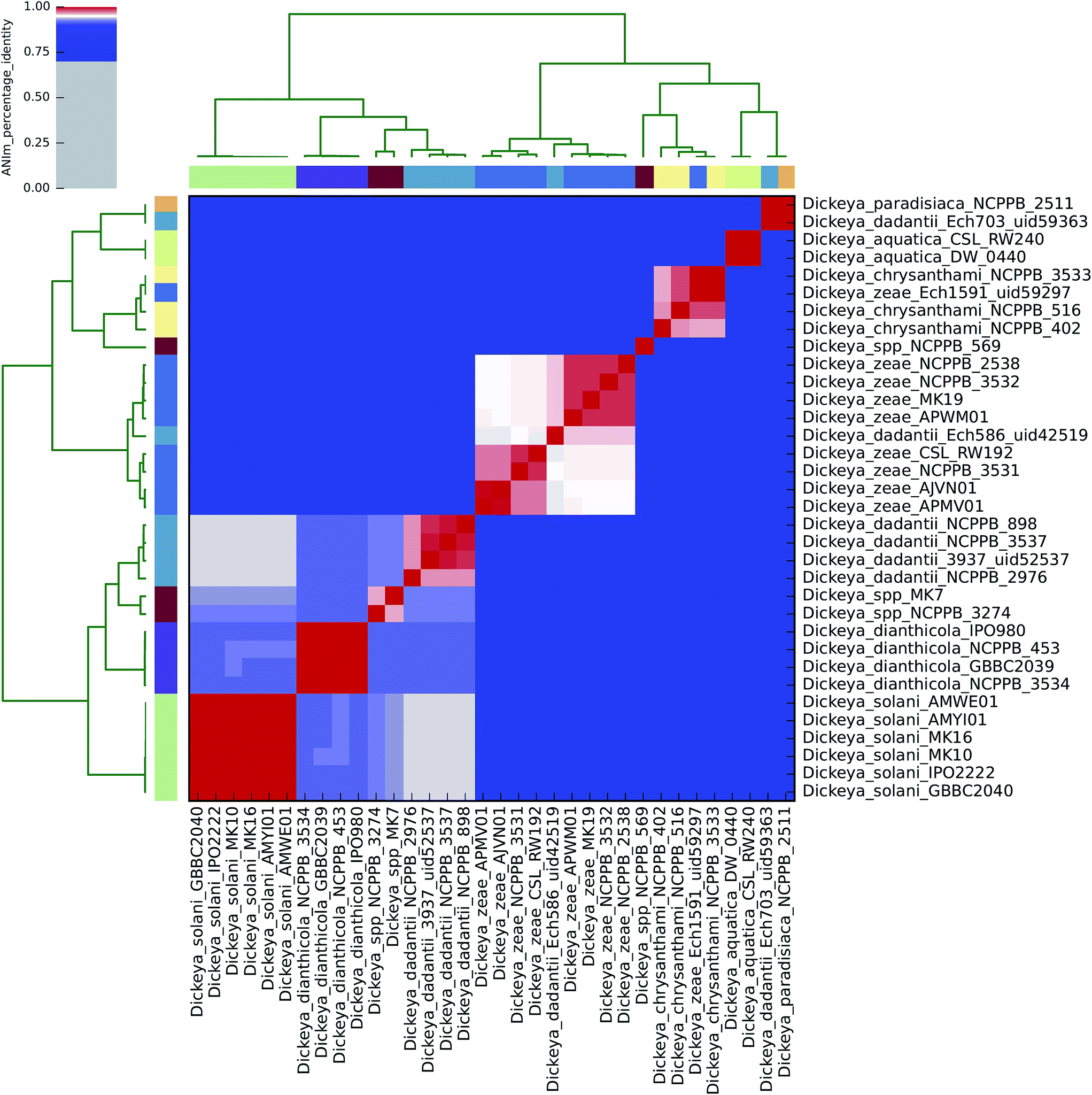 | ||
| Fig. 2 Heatmap of ANIm percentage identity for 34 isolates of Dickeya spp. as described in ESI Tables S1 and S6.† Species-level assignments and isolate identifiers as indicated at source are given as row and column labels. Cells in the heatmap corresponding to 95% ANIm sequence identity (and therefore the same species42,43) are coloured red. Blue cells correspond to ANIm comparisons indicating that the corresponding organisms do not belong to the same species. Colour intensity fades as the comparisons approach 95% ANIm sequence identity. Colour bars above and to the left of the heatmap correspond to source species-level assignments for each isolate in the analysis. Hierarchical clustering of the analysis results in two dimensions is represented by dendrograms, constructed by simple linkage of ANIm percentage identities. The analysis indicates nine species-level clades, in contrast to the seven currently recognised Dickeya spp. Three of the four completely sequenced Dickeya genomes in NCBI/GenBank (Ech703, Ech1591, Ech586) are shown to be incorrectly classified at source, in this analysis. | ||
Using the recommended 95% ANIm identity for species membership,42,43 these analyses exemplify several issues concerning bacterial taxonomic classification with potential impact on sequence-based diagnostics. In particular, they highlight that classification in public databases or strain collections, and in some cases existing species demarcations, may not be consistent with evidence derived from whole genome data.
ANIm analysis of Dickeya spp.
The current taxonomic classification of the Dickeya genus comprises seven species: aquatica, chrysanthemi, dadantii, dianthicola, solani, paradisiaca, and zeae.16,38 ANIm analysis of 34 sequenced Dickeya isolates indicates support for these divisions at the 95% identity threshold, and suggests that further species level assignments may be necessary with the Dickeya genus for two as yet unclassified clades (Fig. 2 and 4).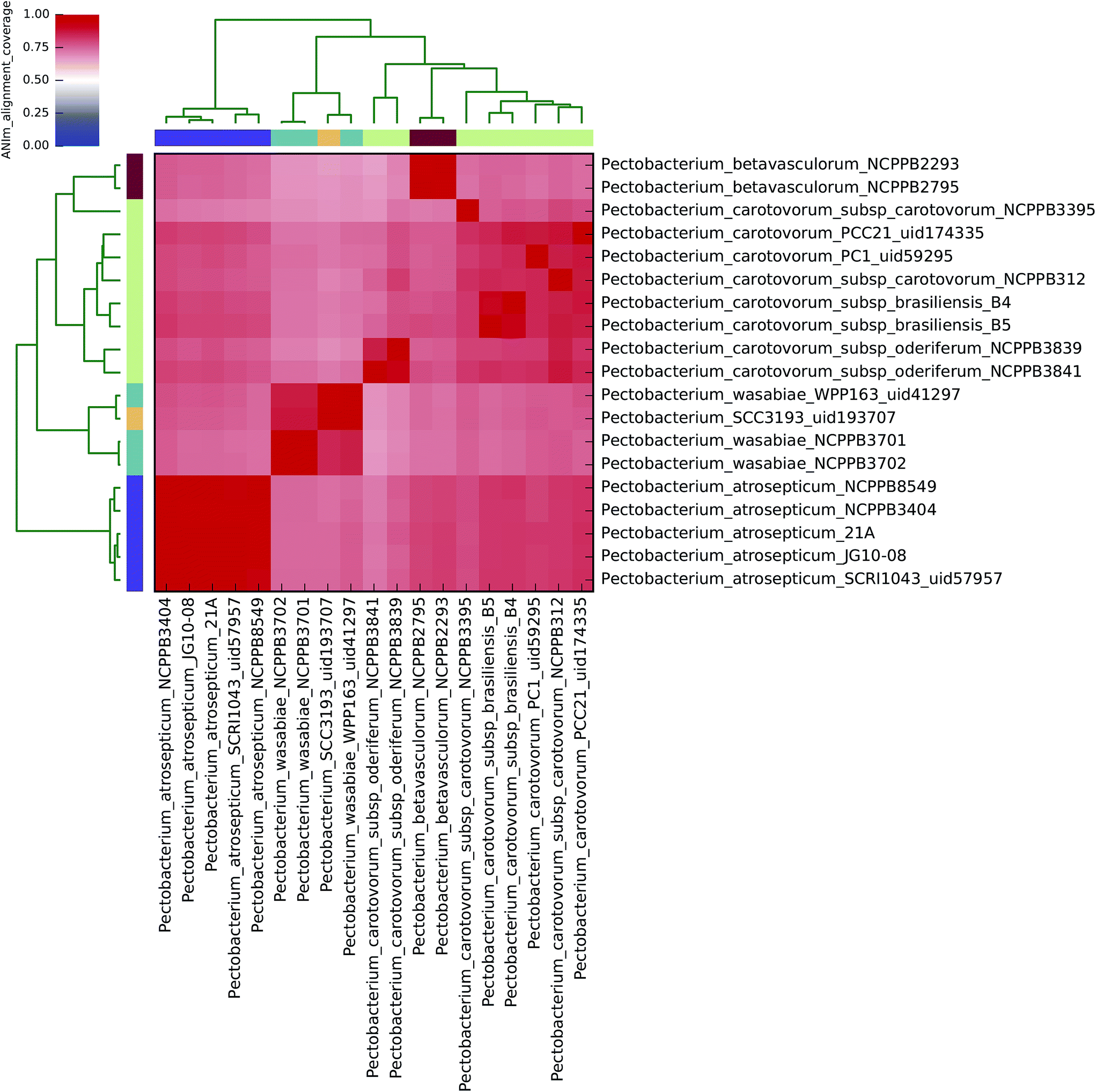 | ||
| Fig. 3 Heatmap of ANIm coverage for 19 bacterial isolates of Pectobacterium as described in ESI Tables S1 and S9.† The isolates and species assignments as indicated at source are given as row and column labels. Cells in the heatmap corresponding to 50% coverage or greater are coloured red. Blue cells correspond to coverage of 50% or less. Colour intensity fades as the comparisons approach 50% coverage. Colour bars above and to the left of the heatmap correspond to genus assignments for each isolate in the analysis. Hierarchical clustering of the data in two dimensions is represented by dendrograms, constructed by simple linkage of ANIm percentage identities. Although there is no standard, accepted interpretation of species or genus boundary by alignment coverage, this analysis identifies a single clade corresponding exactly to Pectobacterium, with minimum aligned genome length above 50%. Internal structure is also seen, corresponding to species divisions as observed in Fig. 1. | ||
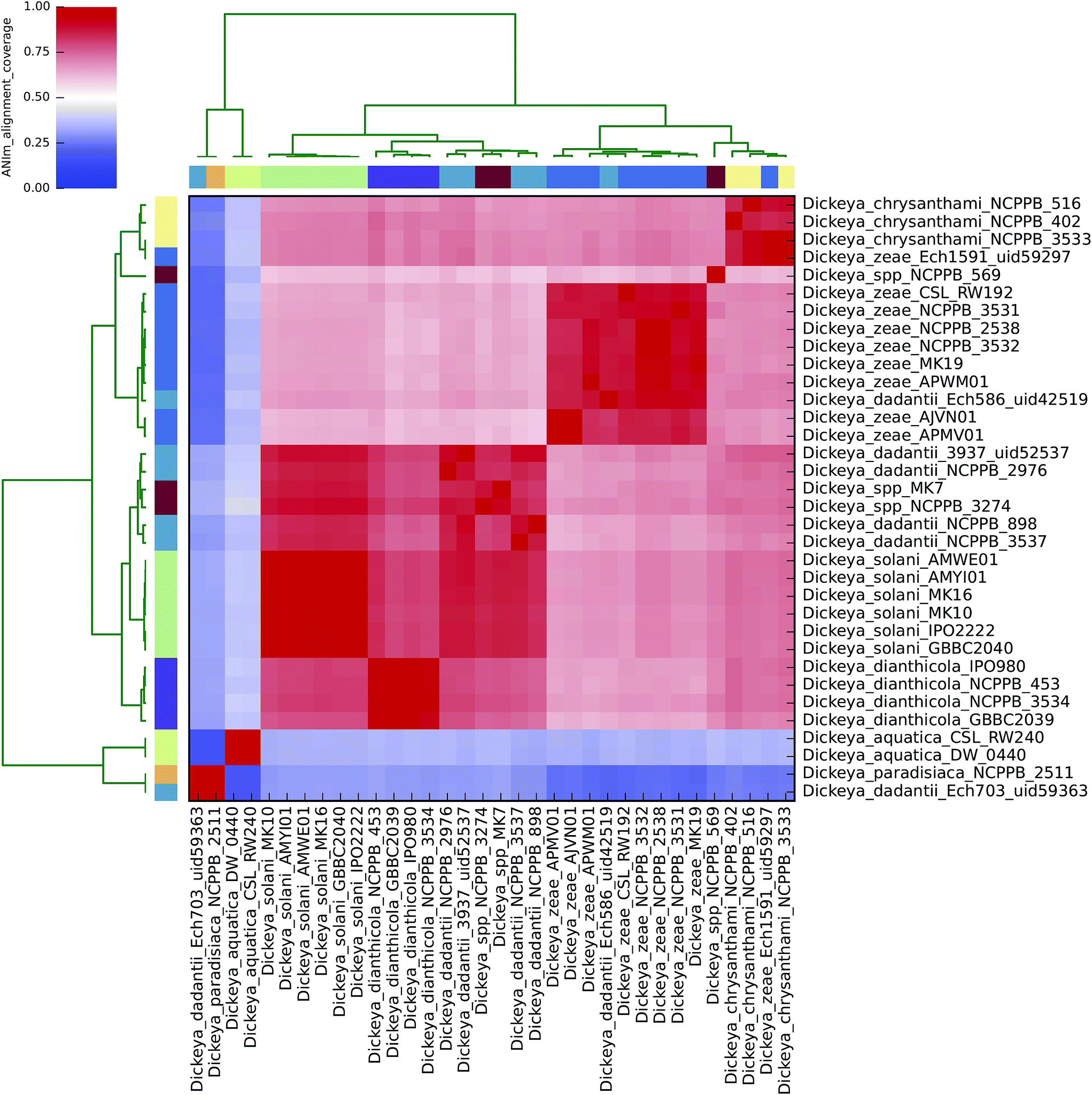 | ||
| Fig. 4 Heatmap of ANIm coverage for 34 bacterial isolates of Dickeya as described in ESI Tables S1 and S7.† The isolates and species assignments as indicated at source are given as row and column labels. Cells in the heatmap corresponding to 50% coverage or greater are coloured red. Blue cells correspond to coverage of 50% or less. Colour intensity fades as the comparisons approach 50% coverage. Colour bars above and to the left of the heatmap correspond to genus assignments for each isolate in the analysis. Hierarchical clustering of the data in two dimensions is represented by dendrograms, constructed by simple linkage of ANIm percentage identities. Although there is no standard, accepted interpretation of species or genus boundary by alignment coverage, this analysis identifies a single clade corresponding to most Dickeya spp., with minimum aligned genome length above 50%. Internal structure is also seen, corresponding to species divisions as observed in Fig. 2. The clades corresponding to D. aquatica and D. paradisiaca align to the remaining Dickeya genomes, and each other, with less than 50% of their genome lengths. This is consistent with the observations in Fig. 2 that these isolates belong to two distinct species groups, and may be productively reclassified in a genus other than Dickeya. | ||
The alignment coverage plot for Dickeya spp. shows three clades, the members of each of which align to each other over more than 60% of their total genome length (Fig. 4). D. aquatica and D. paradisiaca are significantly dissimilar to the other Dickeya spp. and to each other, such that no more than around 40% of these genomes align to any of the main clade genomes. This is a similar level of dissimilarity to that which separates Salmonella spp. from E. coli spp. (ESI Fig. 3, ESI Table 3†), hence reclassification of these two species to genera other than Dickeya might be considered consistent. This result demonstrates that prevailing taxonomic assignments based on polyphasic approaches can be inconsistent with evidence from genome-scale data.
To date, four completely sequenced and closed Dickeya genomes have been deposited in the GenBank public database (“D. dadantii” Ech703 uid59363; “D. zeae” Ech1591 uid59297; “D. dadantii” Ech586 uid42519; and D. dadantii 3937 uid52537), along with several scaffolded genomes. The ANIm analysis conducted here supports earlier suggestions that three of these sequences should be reclassified: Ech703 to D. paradisiaca; Ech1591 to D. chrysanthemi; and Ech586 to D. zeae.31,47,48 This is an instance in which taxonomic classification metadata in public databases is not consistent with evidence from whole genome sequences.
ANIm analysis of Pectobacterium spp.
All 19 sequenced Pectobacterium spp. align to each other with at least 60% coverage of the total genome length (Fig. 3), consistent with their assignment to a single genus. At the 95% identity threshold, ANIm analysis of these isolates, which nominally represent four of the accepted six Pectobacterium spp. (aroidearum, atrosepticum, betavasculorum, cacticida, carotovorum, and wasabiae), subdivides sequences into eight distinct species-level groupings (Fig. 1). A more flexible threshold of sequence identity for species boundaries might reduce this number to six, but even with this revision there would remain inconsistency between existing species classifications in the Pectobacterium genus, and evidence from whole-genome comparison.The five P. atrosepticum isolates were classified by ANIm as belonging to a single species, consistent with current classification, and this was also the case for the two P. betavasculorum isolates (Clades V and IV, respectively8). However, and in agreement with previous analyses,8 novel species-level groupings would better describe the outcome of whole-genome clustering for the other two Pectobacterium species classifications, as detailed below. These results demonstrate inconsistencies between existing species classifications, and classification metadata associated with isolates from strain collections, with evidence gained through whole genome sequencing.
P. wasabiae
The four sequenced P. wasabiae (Clade III8) isolates were divided into two distinct species clusters at the 95% identity threshold; all four isolates align to at least 80% genome coverage. The first cluster contains isolates Pwa_NCPPB3701 and Pwa_NCPPB3702 (99.9% identity); the second cluster contains Pwa_WPP163 and Pwa_SCC193 isolates (98.8% identity). The two clusters share approximately 94% sequence identity in common, which falls just below the species boundary threshold identity,42,43 and suggests that the current P. wasabiae classification could usefully be divided into two species-level groups.P. carotovorum
By ANIm analysis, the eight sequenced P. carotovorum isolates form a complex of four species at the 95% identity threshold; the existing species-level classification was also found to comprise two distinct clades in an earlier multilocus sequence analysis.8 The PC1 isolate is unequivocally classified as a singleton species-level isolate, distinct from the remaining seven P. carotovorum genomes, sharing no more than 91% identity with any other P. carotovorum sequence. This is comparable to the observed sequence identity between isolates of P. atrosepticum and P. wasabiae.At the ANIm 95% identity threshold, the remaining seven P. carotovorum isolates potentially form three distinct species groupings. The two P. c. subsp. oderiferum isolates (sharing 99% identity) are classified together with P. c. subsp. carotovorum NCPPB312 as a single species (95.1% identity). This species group shares no more than 93% identity with the remaining P. carotovorum isolates, which are divided into a singleton: P. c. subsp. carotovorum NCPPB3395; and a distinct species group comprising the two P. c. subsp. brasiliensis isolates and P. c subsp. carotovorum Pcc21, that share at least 96% sequence identity. Hence, at least two species-level clades (approximately P. carotovorum subsp. oderiferum and P. carotovorum subsp. brasiliensis) are suggested by the analysis, consistent with earlier classifications.8
Thus, whole-genome comparisons suggest that reclassification of Pectobacterium from four to at least six, and possibly eight distinct species, accompanied by reclassification of specific isolates, may be justified.
The proposed changes to taxonomic classification for Dickeya and Pectobacterium spp. arising from this ANIm analysis are summarised in ESI Table 10.†
Future perspective
Lessening the effect of taxonomic misidentification
The complications of prokaryotic evolution and corresponding difficulties of polyphasic bacterial taxonomic classification have, as illustrated here, produced culture collection and sequence database resources whose legacy taxonomic assignments may at times be inconsistent with evidence now arising through whole-genome sequencing and comparison. Continued use of existing classifications may cause difficulties for and inaccuracies in development, assessment, and application of sequence-based diagnostic tests, with consequent implications for the effectiveness of policy, legislation and quarantine.Historical inconsistencies in strain collections and sequence databases are, however, correctable with widespread retrospective application of modern sequencing technologies and whole-genome classification techniques to existing collections for re-identification. The SRE have for decades provided a rich basis for collaborative genomic work that has positioned them as a model for bacterial plant pathology studies, and now makes them excellent candidates for this kind of corrective effort.17 With increasing adoption of routine bacterial whole-genome sequencing prior to strain submission, and new approaches to bacterial sequence database organisation, the impact of inconsistent classification on diagnostic testing may diminish greatly over the foreseeable future.
Improving understanding of the phenotype–genotype relationship
It is likely for practical reasons that legislation and other administrative tools of control will continue to prefer precise, quantifiable and bounded classification of controlled organisms, such as taxonomic identity, over potentially qualitative, flexible and context-dependent practical assessments of pathogenic potential. This will require researchers in the field to improve our collective understanding of the relationship between phenotype and genotype and, in particular, the risk of pathogenicity conditioned on taxonomic classification obtained by sequence-based methods. For example, given a single taxonomic classification or a complex multiplexed diagnostic result for the bacterial community associated with a sample, statistical methods similar to those used to predict functional composition (i.e. the likelihood of a function being present) in metagenomic samples may be adaptable to estimation of the likelihood of a sample being contaminated by a viable bacterial pathogen.49The collective gene complements of bacterial groupings can be considered to constitute a pangenome, comprised of sets of ‘core’ genes: those shared by all members of the grouping; and ‘dispensible’ genes: those that are differentially present amongst members of the group.50,51 The ‘dispensible’ (or ‘accessory’) genes may correspond to those that are susceptible to gene loss, or acquisition by horizontal gene transfer, or that are implicated in environmental and niche adaptation, and pathogenicity.5,52,53 The tangled prokaryote ‘phylogenetic net’ may be better represented by defining classifications in terms of genomic subsets of a pangenome, rather than a bifurcating evolutionary tree.
Pangenome analyses of E. coli have provided descriptive insights into the relationships between genotype and phenotype, and also predictive whole-genome models of metabolism, including lineage-specific metabolic traits that when applied to SRE may be informative regarding relationships between the genome, host specificity and pathogenicity, as well as control methods.54–56 Large-scale sequencing and phenomic efforts also have the potential to inform the relationship between pangenome and phenotype.57
In concert with a pangenome-based view of prokaryotic evolution and classification, the adoption of non-binary phylogenetic analyses, such as phylogenetic network, phylogeographic, and forest approaches have potential for clarification of the relationship between phenotype and genotype.58–60 Moving from a traditional to pangenome paradigm of prokaryotic classification in the context of such large amounts of data presents specific challenges to data handling, storage, and bioinformatic tool provision that are a focus of current research.61–63
A future for bacterial plant pathogen diagnostics
Whole-genome sequencing (WGS) has rapidly become a commodity for bacterial sequencing. The complete genome of a prokaryote provides an account of its phenotypic potential, meaning that there is no need for preliminary taxonomic classification followed by inference of pathogenic likelihood, if the genome sequence and therefore the presence of virulence and pathogenicity factors can be determined directly. WGS is an extremely high resolution assay, potentially able to discriminate between two isolates at single-base level, and so presents an unparalleled opportunity for precise tracking of outbreaks and transmission.64 The data obtained by WGS is also standardisable and digital, permitting immediate global sharing for worldwide epidemiological analysis and modelling, as exemplified by the Global Microbial Identifier initiative (http://www.globalmicrobialidentifier.org/).Despite this great potential, service- and benchtop-level sequencing capabilities have not yet become completely routine in diagnostic screening for a number of reasons, including the total cost of sequencing, amount of material required, throughput capacity, turnaround time and, perhaps most pertinently, the inability to place a sequencer in-the-field. In part because of these limitations, PCR methods remain the most widely-used DNA sequence-based technology for diagnostics in the field, and are likely to do so for the foreseeable future.
The situation may begin to change more rapidly, however, with availability of the Oxford Nanopore MinION sequencing instrument. This is a small, portable sequencer that directly senses native, individual DNA fragments without prior amplification, and can produce extremely long read fragments on the order of kilobases in length at high quality, suitable for assembly of a complete bacterial genome.65 At less than the size of a smartphone, it is a candidate technology for widespread deployment of sequencing in the field, but its feasibility in this application has not yet been tested, and its robustness in an agricultural context is not established, though recent work indicates the technology is suitable for metagenomic analyses.66 Nevertheless, a near future in which in-the-field whole genome sequencing of bacteria from agricultural samples, with immediate integration into global epidemiological and sequence databases, and sequence-based-diagnostics design services, is now conceivable. This, in combination with a greater functional understanding of genomic contributions to pathogenicity obtained through sequencing and phenomic studies,20 has the potential to revolutionise bacterial pathogen diagnostics, making it a truly global enterprise for the improvement of agriculture, protection of crops, and the development of policy and legislation in support of food security.
Concluding remarks
In this short review and analysis we have explored how the correct taxonomic and diagnostic identification of plant pathogens plays an essential role in global legislation and control efforts in support of continued food security. The current trajectory from polyphasic, to DNA sequence-based, to whole-genome diagnostics promises increasing precision and accuracy for tests that support disease control and pathogen identification. These techniques also shed new light on historical definitions of pathogenic bacteria, sometimes revealing them to be wanting, with implications for development of new policies and control measures. It is our hope that the increasingly close to point of sample availability, and inexpensive nature of modern sequencing technologies will greatly improve the ability of the field as a whole to identify and classify bacterial pathogen threats in support of epidemiological analyses and legal and practical protection efforts, and inform our understanding of the tangled relationships between genotype, phenotype, and taxonomy.References
- J. Mansfield, S. Genin, S. Magori, V. Citovsky, M. Sriariyanum, P. Ronald, M. Dow, V. Verdier, S. V. Beer, M. A. Machado, I. Toth, G. Salmond and G. D. Foster, Mol. Plant Pathol., 2012, 13, 614–629 CrossRef PubMed.
- P. R. Davidsson, T. Kariola, O. Niemi and E. T. Palva, Front. Plant Sci., 2013, 4, 191 Search PubMed.
- N. Hugouvieux-Cotte-Pattat, G. Condemine and V. E. Shevchik, Environ. Microbiol. Rep., 2014, 6, 427–440 CrossRef CAS PubMed.
- H. Liu, S. J. Coulthurst, L. Pritchard, P. E. Hedley, M. Ravensdale, S. Humphris, T. Burr, G. Takle, M. B. Brurberg, P. R. Birch, G. P. Salmond and I. K. Toth, PLoS Pathog., 2008, 4, e1000093 Search PubMed.
- I. K. Toth, L. Pritchard and P. R. Birch, Annu. Rev. Phytopathol., 2006, 44, 305–336 CrossRef CAS PubMed.
- I. K. Toth, J. M. van der Wolf, G. Saddler, E. Lojkowska, V. Hélias, M. Pirhonen, L. Tsror Lahkim and J. G. Elphinstone, Plant Pathol., 2011, 60, 385–399 CrossRef.
- R. Czajkowski, M. Perombelon, S. Jafra, E. Lojkowska, M. Potrykus, J. van der Wolf and W. Sledz, Ann. Appl. Biol., 2015, 166, 18–38 CrossRef PubMed.
- B. Ma, M. E. Hibbing, H. S. Kim, R. M. Reedy, I. Yedidia, J. Breuer, J. Breuer, J. D. Glasner, N. T. Perna, A. Kelman and A. O. Charkowski, Phytopathology, 2007, 97, 1150–1163 CrossRef PubMed.
- J. R. Lamichhane and C. Bartoli, Plant Pathol., 2015, 64, 757–766 CrossRef.
- P. Panda, M. A. W. J. Fiers, K. Armstrong and A. R. Pitman, New Dis. Rep., 2012, 26, 15 CrossRef.
- A. R. Pitman, P. J. Wright, M. D. Galbraith and S. A. Harrow, Australas. Plant Pathol., 2008, 37, 559 CrossRef CAS.
- S. Baghaee-Ravari, H. Rahimian, M. Shams-Bakhsh, E. Lopez-Solanilla, M. Antúnez-Lamas and P. Rodríguez-Palenzuela, Eur. J. Plant Pathol., 2010, 129, 413–425 CrossRef.
- L. N. Moleleki, E. M. Onkendi, A. Mongae and G. C. Kubheka, Eur. J. Plant Pathol., 2012, 135, 279–288 CrossRef.
- M. L. Pasanen, J. Brader, G. Palva, E. T. Ahola, V. van der Wolf, J. Hannukkala and A. M. Pirhonen, Ann. Appl. Biol., 2013, 163, 403–419 CrossRef.
- D. H. Lee, J. B. Kim, J. A. Lim, S. W. Han and S. Heu, Plant Pathol. J., 2014, 30, 117–124 CrossRef CAS PubMed.
- N. Parkinson, L. Pritchard, R. Bryant, I. Toth and J. Elphinstone, Eur. J. Plant Pathol., 2014, 141, 63–70 CrossRef.
- A. O. Charkowski, J. Lind and I. Rubio-Salazar, in Genomics of Plant-Associated Bacteria, ed. D. C. Gross, A. Lichens-Park and C. Kole, Springer, Berlin, 2014, pp. 37–58 Search PubMed.
- A. Lupo, K. M. Papp-Wallace, P. Sendi, R. A. Bonomo and A. Endimiani, Diagn. Microbiol. Infect. Dis., 2013, 77, 179–194 CrossRef CAS PubMed.
- H. Rohde, J. Qin, Y. Cui, D. Li, N. J. Loman, M. Hentschke, W. Chen, F. Pu, Y. Peng, J. Li, F. Xi, S. Li, Y. Li, Z. Zhang, X. Yang, M. Zhao, P. Wang, Y. Guan, Z. Cen, X. Zhao, M. Christner, R. Kobbe, S. Loos, J. Oh, L. Yang, A. Danchin, G. F. Gao, Y. Song, Y. Li, H. Yang, J. Wang, J. Xu, M. J. Pallen, J. Wang, M. Aepfelbacher and R. Yang, N. Engl. J. Med., 2011, 365, 718–724 CrossRef CAS PubMed.
- A. R. Deans, S. E. Lewis, E. Huala, S. S. Anzaldo, M. Ashburner, J. P. Balhoff, D. C. Blackburn, J. A. Blake, J. G. Burleigh, B. Chanet, L. D. Cooper, M. Courtot, S. Csosz, H. Cui, W. Dahdul, S. Das, T. A. Dececchi, A. Dettai, R. Diogo, R. E. Druzinsky, M. Dumontier, N. M. Franz, F. Friedrich, G. V. Gkoutos, M. Haendel, L. J. Harmon, T. F. Hayamizu, Y. He, H. M. Hines, N. Ibrahim, L. M. Jackson, P. Jaiswal, C. James-Zorn, S. Kohler, G. Lecointre, H. Lapp, C. J. Lawrence, N. Le Novere, J. G. Lundberg, J. Macklin, A. R. Mast, P. E. Midford, I. Miko, C. J. Mungall, A. Oellrich, D. Osumi-Sutherland, H. Parkinson, M. J. Ramirez, S. Richter, P. N. Robinson, A. Ruttenberg, K. S. Schulz, E. Segerdell, K. C. Seltmann, M. J. Sharkey, A. D. Smith, B. Smith, C. D. Specht, R. B. Squires, R. W. Thacker, A. Thessen, J. Fernandez-Triana, M. Vihinen, P. D. Vize, L. Vogt, C. E. Wall, R. L. Walls, M. Westerfeld, R. A. Wharton, C. S. Wirkner, J. B. Woolley, M. J. Yoder, A. M. Zorn and P. Mabee, PLoS Biol., 2015, 13, e1002033 Search PubMed.
- V. Kunin, L. Goldovsky, N. Darzentas and C. A. Ouzounis, Genome Res., 2005, 15, 954–959 CrossRef CAS PubMed.
- M. J. Pallen and B. W. Wren, Nature, 2007, 449, 835–842 CrossRef CAS PubMed.
- P. Puigbo, Y. I. Wolf and E. V. Koonin, Genome Biol. Evol., 2010, 2, 745–756 CrossRef PubMed.
- A. C. Martiny, K. Treseder and G. Pusch, ISME J., 2013, 7, 830–838 CrossRef CAS PubMed.
- R. J. Siezen, J. R. Bayjanov, G. E. Felis, M. R. van der Sijde, M. Starrenburg, D. Molenaar, M. Wels, S. A. van Hijum and J. E. van Hylckama Vlieg, Microb. Biotechnol., 2011, 4, 383–402 CrossRef CAS PubMed.
- C. I. Kado, Mol. Plant Pathol., 2009, 10, 143–150 CrossRef CAS PubMed.
- H. C. Lovell, J. W. Mansfield, S. A. Godfrey, R. W. Jackson, J. T. Hancock and D. L. Arnold, Curr. Biol., 2009, 19, 1586–1590 CrossRef CAS PubMed.
- M. W. Silby, C. Winstanley, S. A. Godfrey, S. B. Levy and R. W. Jackson, FEMS Microbiol. Rev., 2011, 35, 652–680 CrossRef CAS PubMed.
- J. Nykyri, O. Niemi, P. Koskinen, J. Nokso-Koivisto, M. Pasanen, M. Broberg, I. Plyusnin, P. Toronen, L. Holm, M. Pirhonen and E. T. Palva, PLoS Pathog., 2012, 8, e1003013 CAS.
- M. Potrykus, W. Sledz, M. Golanowska, M. Slawiak, A. Binek, A. Motyka, S. Zoledowska, R. Czajkowski and E. Lojkowska, Ann. Appl. Biol., 2014, 165, 474–487 CrossRef CAS PubMed.
- L. Pritchard, S. Humphris, G. S. Saddler, N. M. Parkinson, V. Bertrand, J. G. Elphinstone and I. K. Toth, Plant Pathol., 2013, 62, 587–596 CrossRef CAS.
- H. Uematsu, Y. Inoue and Y. Ohto, J. Gen. Plant Pathol., 2015, 81, 173–179 CrossRef CAS.
- D. Fréchon, P. Exbrayat, V. Helias, L. J. Hyman, B. Jouan, P. Llop, M. M. Lopez, N. Payet, M. C. M. Perombélon, I. K. Toth, J. R. C. M. van Beckhoven, J. M. van der Wolf and Y. Bertheau, Potato Res., 1998, 41, 163–173 CrossRef.
- A. Nassar, A. Darrasse, M. Lemattre, A. Kotoujansky, C. Dervin, R. Vedel and Y. Bertheau, Appl. Environ. Microbiol., 1996, 62, 2228–2235 CAS.
- V. Duarte, S. H. De Boer, L. J. Ward and A. M. R. Oliveira, J. Appl. Microbiol., 2004, 96, 535–545 CrossRef CAS PubMed.
- L. Pritchard, N. J. Holden, M. Bielaszewska, H. Karch and I. K. Toth, PLoS One, 2012, 7, e34498 CAS.
- K. P. Williams, J. J. Gillespie, B. W. Sobral, E. K. Nordberg, E. E. Snyder, J. M. Shallom and A. W. Dickerman, J. Bacteriol., 2010, 192, 2305–2314 CrossRef CAS PubMed.
- J. M. van der Wolf, E. H. Nijhuis, M. J. Kowalewska, G. S. Saddler, N. Parkinson, J. G. Elphinstone, L. Pritchard, I. K. Toth, E. Lojkowska, M. Potrykus, M. Waleron, P. de Vos, I. Cleenwerck, M. Pirhonen, L. Garlant, V. Helias, J. F. Pothier, V. Pfluger, B. Duffy, L. Tsror and S. Manulis, Int. J. Syst. Evol. Microbiol., 2014, 64, 768–774 CrossRef PubMed.
- L. Gardan, C. Gouy, R. Christen and R. Samson, Int. J. Syst. Evol. Microbiol., 2003, 53, 381–391 CrossRef CAS PubMed.
- R. Samson, J. B. Legendre, R. Christen, M. Fischer-Le Saux, W. Achouak and L. Gardan, Int. J. Syst. Evol. Microbiol., 2005, 55, 1415–1427 CrossRef CAS PubMed.
- D. I. Broadhurst and D. B. Kell, Metabolomics, 2006, 2, 171–196 CrossRef CAS.
- J. Goris, K. T. Konstantinidis, J. A. Klappenbach, T. Coenye, P. Vandamme and J. M. Tiedje, Int. J. Syst. Evol. Microbiol., 2007, 57, 81–91 CrossRef CAS PubMed.
- M. Richter and R. Rossello-Mora, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 19126–19131 CrossRef CAS PubMed.
- N. J. Varghese, S. Mukherjee, N. Ivanova, K. T. Konstantinidis, K. Mavrommatis, N. C. Kyrpides and A. Pati, Nucleic Acids Res., 2015, 43, 6761–6771 CrossRef CAS PubMed.
- L. Pritchard, S. Humphris, S. Baeyen, M. Maes, J. VanVaerenbergh, J. Elphinstone, G. Saddler and I. Toth, Genome Announc., 2013, 1, e00087-12 Search PubMed.
- L. Pritchard, S. Humphris, G. S. Saddler, J. G. Elphinstone, M. Pirhonen and I. K. Toth, Genome Announc., 2013, 1, e00978-13 CrossRef PubMed.
- G. Marrero, K. L. Schneider, D. M. Jenkins and A. M. Alvarez, Int. J. Syst. Evol. Microbiol., 2013, 63, 3524–3539 CrossRef PubMed.
- J. Zhou, Y. Cheng, M. Lv, L. Liao, Y. Chen, Y. Gu, S. Liu, Z. Jiang, Y. Xiong and L. Zhang, BMC Genomics, 2015, 16, 571 CrossRef PubMed.
- M. G. Langille, J. Zaneveld, J. G. Caporaso, D. McDonald, D. Knights, J. A. Reyes, J. C. Clemente, D. E. Burkepile, R. L. Vega Thurber, R. Knight, R. G. Beiko and C. Huttenhower, Nat. Biotechnol., 2013, 31, 814–821 CrossRef CAS PubMed.
- S. Bentley, Nat. Rev. Microbiol., 2009, 7, 258–259 CrossRef CAS PubMed.
- H. Tettelin, D. Riley, C. Cattuto and D. Medini, Curr. Opin. Microbiol., 2008, 11, 472–477 CrossRef CAS PubMed.
- S. Khayi, Y. Raoul des Essarts, A. Quetu-Laurent, M. Moumni, V. Helias and D. Faure, Genetica, 2015, 143, 241–252 CrossRef CAS PubMed.
- T. Lefebure, P. D. Bitar, H. Suzuki and M. J. Stanhope, Genome Biol. Evol., 2010, 2, 646–655 CrossRef PubMed.
- D. J. Baumler, R. G. Peplinski, J. L. Reed, J. D. Glasner and N. T. Perna, BMC Syst. Biol., 2011, 5, 182 CrossRef PubMed.
- D. A. Rasko, M. J. Rosovitz, G. S. Myers, E. F. Mongodin, W. F. Fricke, P. Gajer, J. Crabtree, M. Sebaihia, N. R. Thomson, R. Chaudhuri, I. R. Henderson, V. Sperandio and J. Ravel, J. Bacteriol., 2008, 190, 6881–6893 CrossRef CAS PubMed.
- C. Wang, Z. L. Deng, Z. M. Xie, X. Y. Chu, J. W. Chang, D. X. Kong, B. J. Li, H. Y. Zhang and L. L. Chen, FEBS Lett., 2015, 589, 285–294 CrossRef CAS PubMed.
- J. R. Bayjanov, M. J. Starrenburg, M. R. van der Sijde, R. J. Siezen and S. A. van Hijum, BMC Microbiol., 2013, 13, 68 CrossRef PubMed.
- D. Bryant and V. Moulton, Mol. Biol. Evol., 2004, 21, 255–265 CrossRef CAS PubMed.
- T. Pearson, P. Giffard, S. Beckstrom-Sternberg, R. Auerbach, H. Hornstra, A. Tuanyok, E. P. Price, M. B. Glass, B. Leadem, J. S. Beckstrom-Sternberg, G. J. Allan, J. T. Foster, D. M. Wagner, R. T. Okinaka, S. H. Sim, O. Pearson, Z. Wu, J. Chang, R. Kaul, A. R. Hoffmaster, T. S. Brettin, R. A. Robison, M. Mayo, J. E. Gee, P. Tan, B. J. Currie and P. Keim, BMC Biol., 2009, 7, 78 CrossRef PubMed.
- K. Schliep, P. Lopez, F. J. Lapointe and E. Bapteste, Mol. Biol. Evol., 2011, 28, 1393–1405 CrossRef CAS PubMed.
- K. Huang, A. Brady, A. Mahurkar, O. White, D. Gevers, C. Huttenhower and N. Segata, Nucleic Acids Res., 2014, 42, D617–D624 CrossRef CAS PubMed.
- S. Marcus, H. Lee and M. C. Schatz, Bioinformatics, 2014, 30, 3476–3483 CrossRef CAS PubMed.
- Y. Zhao, J. Wu, J. Yang, S. Sun, J. Xiao and J. Yu, Bioinformatics, 2012, 28, 416–418 CrossRef CAS PubMed.
- J. Quick, N. Cumley, C. M. Wearn, M. Niebel, C. Constantinidou, C. M. Thomas, M. J. Pallen, N. S. Moiemen, A. Bamford, B. Oppenheim and N. J. Loman, BMJ Open, 2014, 4, e006278 CrossRef PubMed.
- N. J. Loman, J. Quick and J. T. Simpson, bioRxiv, 2015, DOI:10.1101/015552.
- D. J. Turner, X. Dai, S. Mayes and S. Juul, Complete assembly of novel environmental bacterial genomes by MinIONTM sequencing, 2015 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ay02550h |
| This journal is © The Royal Society of Chemistry 2016 |