
Trapping chlorine radicals via substituting nitro radicals in the gas phase†
Akira
Seto
a,
Yuki
Ochi
a,
Hiroaki
Gotoh
a,
Kazuhisa
Sakakibara
*a,
Shota
Hatazawa
a,
Kanekazu
Seki
a,
Naoaki
Saito
b and
Yuji
Mishima
c
aDepartment of Advanced Materials Chemistry, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan. E-mail: mozart@ynu.ac.jp
bNational Institute of Advanced Industrial Science and Technology, Tsukuba, Japan
cKobe Material Testing Laboratory Co., Ltd, Japan
First published on 8th December 2015
Abstract
Although chlorine radicals are strong atmospheric oxidants, their direct spectroscopic detection in the environment (gas phase) using conventional analytical techniques has not been reported. Herein, chlorine radicals (Cl˙), generated by the YAG laser photolysis (λ = 355 nm) of Cl2 in the gas phase, were captured by using 2,2-diphenyl-1-picrylhydrazyl (DPPH˙). The product was identified as DPPH, wherein NO2 was substituted by Cl˙ at the ortho-carbon on the picryl aromatic ring, and was characterized using ion attachment ionization-quadrupole mass spectrometry. This reaction mechanism is a rare example of the detection and characterization of radical species, where radical spin densities on trapping reagents and frontier orbital interactions play important roles.
Introduction
Reactive organic radicals play important roles in the field of organic chemistry and environmental chemistry as key intermediates or transition-state species. If these reactive radicals can be trapped and characterized efficiently, essential clues and novel insights into reaction mechanisms may be obtained. For example, short-lived reactive radicals such as ˙OH, ˙OOH, and Cl˙ are said to be toxic to humans and can induce chain reactions of volatile components in the atmosphere, leading to environmental concerns such as ozone depletion1 or methane degradation caused by Cl˙. However, there are few reports that show concrete experimental results about radical trapping and the identification of reactive hetero radicals using conventional analytical equipment. We have used the results of computational studies2 to design experiments to capture these radicals by chemical methods using functional organic compounds.3 Radical scavenging4 and spin trapping5 constitute examples of conventional techniques (Scheme 1).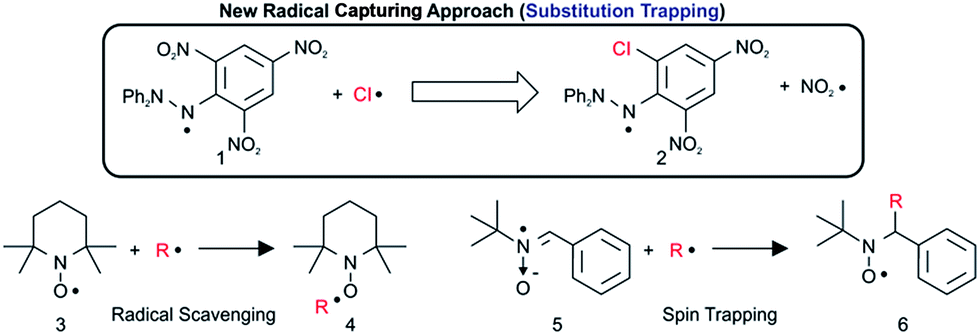 | ||
| Scheme 1 Experiments for capturing short-lived reactive radicals. | ||
However, these radical scavenging and spin trapping methods are only suitable for carbon-centred radicals, and will not be suitable for hetero radicals such as ˙OH, ˙OOH, and Cl˙ because the radical-trapped products are often not stable enough6 (the mean bond enthalpy of the O–Cl bond is 203 kJ mol−1) to be identified. In addition, although methods to detect HO2˙ and RO2˙ in the atmosphere are known,7 direct spectroscopic detection of Cl˙ in the environment with standard equipment has rarely been reported, with the exception of tandem mist chamber experiments combined with photochemical model calculations.8 Sophisticated resonance fluorescence methods9 are suitably sensitive to detect Cl˙; however, such techniques cannot be considered generally applicable because of the strict experimental conditions requisite for vacuum ultraviolet spectroscopy. Indirect analyses based only on the reaction products from radical reactions are liable to lead to uncertain conclusions with regard to essential reactive species. Furthermore, the reactivity of radical-capturing reagents is insufficient to capture Cl˙. Although there are a few reports10 on Cl spin trapping, Cl-adducts often cannot be identified. For actual gas- (104 to 105 atom cm−3) and liquid-phase environmental Cl˙, a novel approach with enhanced sensitivity must be developed; moreover, reliable and rapid analytical methods are necessary for characterization. We have found a new method to enable the capture of Cl˙ by using a popular stable radical reagent 2,2-diphenyl-1-picrylhydrazyl (DPPH˙) through a substitution mechanism on the picryl aromatic ring by elimination of NO2˙. In the present paper, we will show the experimental results for capturing Cl˙ by using DPPH˙ through substitution–trapping and the details of characterization of the Cl˙ trapped product using ion attachment ionization-quadrupole mass spectrometry (IA-QMS) combined with general spectroscopic analyses such as NMR of the reduced Cl˙ trapped DPPH˙ product. In addition, we will study the regioselectivity of Cl˙ substitution on the picryl aromatic ring and the reaction mechanism based on the DFT calculations (UB3LYP/6-31G* or B3LYP/6-31G*) of Cl˙ trapped DPPH˙ paramagnetic or diamagnetic compounds.
Results and discussion
How to capture Cl˙ by using DPPH˙ (substitution–trapping)
We have found a new approach to meet the aforementioned analytical demands by taking advantage of functional paramagnetic organic molecules, wherein Cl˙ is captured by substituting a stable radical (NO2˙) on DPPH˙ 1 (substitution–trapping). Consequently, DPPH˙ functions as a “radical-substitution reagent”, as opposed to taking part in simple radical addition reactions, which occur in conventional radical scavenging experiments. The nitro group is suitable for this radical-substitution reaction because NO2˙ is very stable in ambient air, and is easily eliminated from the aromatic ring because of its lower bond enthalpy DH298(Ar–NO2) (303 kJ mol−1).11 Furthermore, because of the small energy difference between the SOMO/HOMO of DPPH˙ and the LUMO of Cl˙, DPPH˙ (1.1 eV energy profit: refer to the section below describing frontier orbital interactions) undergoes facile nucleophilic substitution reactions with Cl˙ radicals to generate NO2˙. Nevertheless, the substitution of the NO2 group in dia-magnetic substrates with Cl˙ at ambient temperature has seldom been reported.12 Specifically, most chlorination experiments were carried out either at higher temperatures (250 °C)13 or in the presence of light at room temperature,14 resulting in a low yield13 or requiring longer reaction times,14 respectively. Notably, DPPH˙ has many sensitive atoms (as shown in Fig. 1) with significant spin densities that can donate an electron to the LUMO of Cl˙.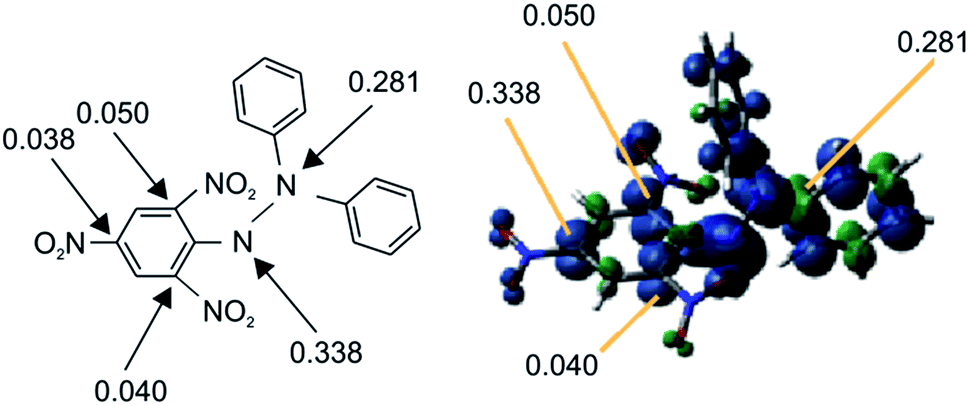 | ||
| Fig. 1 Spin density map of DPPH˙ with UB3LYP/6-31G* calculations. | ||
Although DPPH˙ has been used to assess the radical-scavenging properties of antioxidants in plants,15 it has rarely been used as a radical-capturing reagent.16 Notably, both the substitution and addition reactions of the Cl˙ radical are expected, although substitution is favoured over addition.
Cl˙ capture experiments in the liquid phase
By generating Cl˙ via X-ray irradiation,17 (Scheme 2) we first carried out Cl˙ capture experiments using DPPH˙ in the liquid phase. LCMS (Liquid Chromatography Mass Spectrometry) and ESR (Electron Spin Resonance) of the crude reaction mixture suggested the formation of a paramagnetic product via the reaction between Cl˙ and DPPH˙ through a substitution–trapping mechanism. After irradiating a CHCl3 solution containing NCS (N-chlorosuccinimide) and DPPH˙ for 11 h, compound 2 ([DPPH–NO2 + Cl]˙, where the ortho-NO2 group was substituted by Cl) was isolated and characterized, following the reduction of the crude reaction products with ascorbic acid. Compound 8, obtained from the reduction of 2, was analysed by 1H-NMR (Nuclear Magnetic Resonance), 13C-NMR, HMBC (1H-detected Multi-Bond heteronuclear multiple quantum Coherence), HMQC (1H-detected Multiple Quantum Coherence), IR (InfraRed), and high-resolution MS. The nonisochronous 1H-NMR chemical shifts of the meta-protons and the nonisochronous nature of the two ortho-carbons seen in the 13C-NMR in reduced adduct 8 confirmed the ortho-substitution of the NO2 group (see the ESI†). Notably, NO2˙ substitution by Cl˙ did not proceed at the ortho-position on the picryl ring upon reaction with relevant diamagnetic compounds such as DPPH–H (compound 7) or 2,4,6-trinitrochlorobenzene with Cl˙.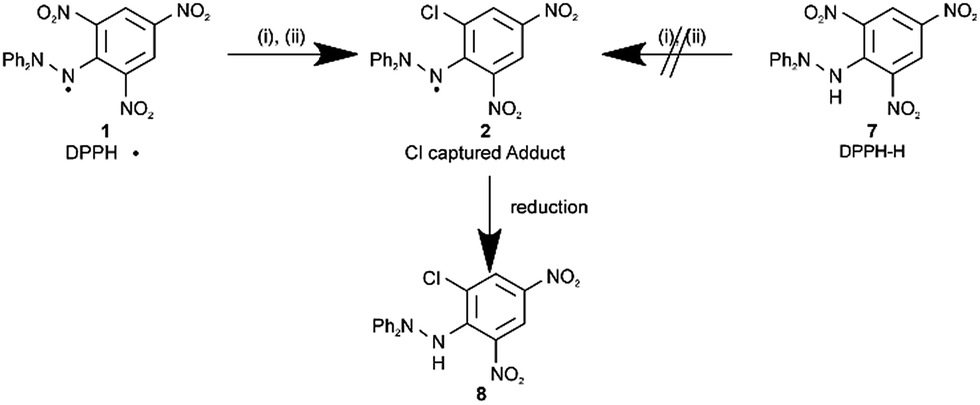 | ||
| Scheme 2 Cl˙ trapping reactions using DPPH˙ and related compounds in the liquid and gas phases. Experimental conditions: (i), NCS, CHCl3, RT 11 h, and X-ray irradiation (liquid); (ii), Cl2 and YAG laser irradiation (λ = 355 nm, 3.2 mJ shot−1) in the gas phase. | ||
The major by-products of this reaction were diamagnetic or paramagnetic compounds with the DPPH˙ framework, produced by the attack of the released NO2˙ on the phenyl ring. Other radical-capturing reagents such as α-phenyl-N-tert-butylnitrone (PBN), tert-butyl(10-phenyl-9-anthryl)nitroxide (BPAN), 2-phenyl-4,4,5,5-tetramethylimidazoline-3-oxide-1-oxyl (PTIO), and 4-oxo-2,2,6,6-tetramethylpiperidine-1-oxyl (4-oxo-TEMPO) were tested under the same conditions as those used to capture Cl˙. Cl˙ captured adducts were not identified via LCMS.
Cl˙ capture experiments in the gas phase and efficient identification of the Cl˙ captured DPPH˙ product by ion attachment ionization-quadrupole mass spectrometry (IA-QMS)
Based on the experimental results obtained using the liquid phase, we attempted to capture Cl˙ using DPPH˙ in the gas phase (Table 1, entry 1). The sample consisted of DPPH˙ and silica filter paper; 2.6 mg cm−2 DPPH˙ in CH2Cl2 was applied on a square of silica filter paper (3 mm edge length). The sample was placed in a reaction vessel with a Cl2 vapour pressure of 1 Torr prior to UV irradiation using a YAG laser (λ = 355 nm, 3.2 mJ shot−1) in 0.1 s intervals. Upon irradiation, the paramagnetic Cl–NO2-exchanged product 2 was confirmed by preparative thin layer chromatography (TLC) and was identified as compound 8 after reduction with ascorbic acid. In addition, chemical species with m/z 46 (NO2) and 30 (NO) were observed by EI-QMS, indicating that stable NO2˙ was eliminated from DPPH˙ upon attack by Cl˙. Without irradiation, the substitution product was not obtained, confirming that the Cl˙ radical was essential. Notably, the Cl˙-substitution reaction was not observed with diamagnetic reagents such as 2,4,6-trinitrochlorobenzene or DPPH–H 7 as shown in Scheme 2. To improve the Cl˙ capture efficiency, the experimental conditions were explored (Table 1, entries 2–5). As the reaction in the gas phase was clean, only the reaction product, starting materials, and a small amount of uncharacterized compounds remained on the silica paper after the reaction. The yields, as judged by NMR and LCMS, were dependent on the reaction time (entries 3–5) and concentration of loaded DPPH˙ (entries 1–3), as shown in Table 1.The yield of Cl–NO2 substituted product 2 is presumed to be controlled by the amount of DPPH˙ existing only in the top surface layer on the filter paper where DPPH˙ can react actually with Cl˙ in the gas phase. Redundant DPPH˙ remaining in the inner layer has no effect on the Cl˙ capturing reaction. This may be partly the reason why counter-intuitive experimental results (Table 1) were observed.
Although the reaction product could be identified by the NMR of the reduced form 8 of 2, mass spectrometry (MS) using a soft-ionization method was adopted to improve the efficiency of the method. Among the available soft-ionization methods for MS analysis, ion attachment ionization-quadrupole mass spectrometry (IA-QMS)18–20 is assumed to be superior owing to fragment-free ionization and high sensitivity. As such, intact samples for reactive radical-capturing experiments can be effectively analysed without separation or derivation prior to characterization and quantitative analysis. In IA-QMS, Li+ ions produced by electrically heating the Li emitter attach to the existing chemical species on the sample silica filter paper prior to ionization (see the ESI†). Therefore, the observed m/z numbers in Fig. 2 denote the sum (M + Li) of the mass numbers of the chemical species (M) and lithium (i.e., 6.94). Because fragmentation and side reactions are negligible in ion attachment ionization, the approximate yield can be determined directly from the ratio of the ion currents of the product and starting material in the mass-chromatogram; thus, quantitative in situ sample analysis is possible. The yields of the Cl-substituted product determined by NMR and IA-QMS were very similar (IA QMS 4.3% vs. NMR 4.5% in Table 1, entry 2).
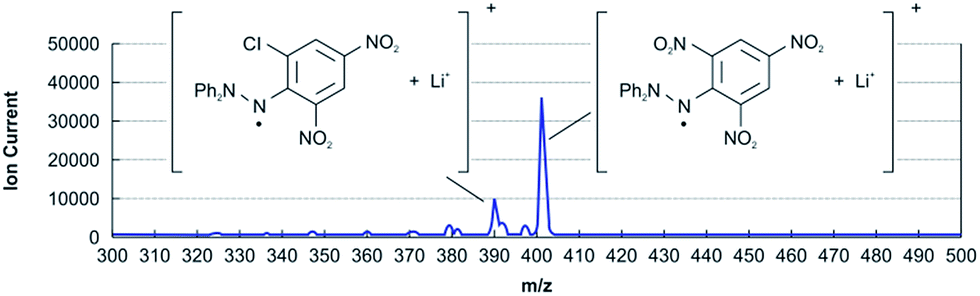 | ||
| Fig. 2 IA-QMS spectrum of reaction products formed between Cl˙ + DPPH˙ in the gas phase. | ||
Theoretical consideration on the substitution–trapping reaction based on the DFT-calculations
In order to clarify why substitution–trapping occurred on DPPH˙ resulting in the formation of the Cl˙ captured product, DFT-calculations were carried out using the Gaussian09 program. Notably, the regioselectivity of Cl˙ substitution at the ortho-carbon can be explained not only by thermodynamics, but also by frontier orbital considerations. Because the hydrazyl N˙ is sterically hindered,21,22 simple Cl˙ attack on the hydrazyl N˙ (spin density: 0.338) is not thermodynamically favoured because the mean bond enthalpy ΔH0 of the newly formed N–Cl bond is rather small (200 kJ mol−1)23 and thus easily dissociative. From the bond dissociation energy data of the pertinent bonds associated with the DPPH˙ capture of Cl˙, the thermodynamically favourable path involves the cleavage of an Ar–NO2 bond (ΔH0(Ar–NO2): 303 kJ mol−1) and formation of an Ar–Cl bond (ΔH0(Ar–Cl): 406 kJ mol−1).11 With regard to the higher reactivity of paramagnetic DPPH˙ toward Cl˙ than that of diamagnetic substrates such as DPPH–H, B3LYP/6-31G* calculations revealed that an energetically favourable frontier orbital interaction between the electron-accepting LUMO of Cl˙ and electron-donating SOMO/HOMO of DPPH˙ was achieved at the ortho-position. As radicals are soft species, their reactions are driven by the coefficient (spin density) at that atom and the energy of the frontier orbitals. A schematic energy diagram illustrating the optimal frontier orbital interaction between Cl˙ and DPPH˙ is shown in Fig. 3. The large frontier orbital spin density value at the ortho-carbon (0.050 in Fig. 1) on the picryl ring supports the selective affinity toward Cl˙; this ortho-attack was calculated to be the most energetically favourable as the net enthalpy gain was −91.9 kJ mol−1 without a significant activation barrier. In contrast, frontier orbital interactions between the HOMO of diamagnetic reagents and the LUMO of Cl˙ are not energetically feasible because additional energy is necessary to prevent the bonding between the pairing electrons in the HOMO.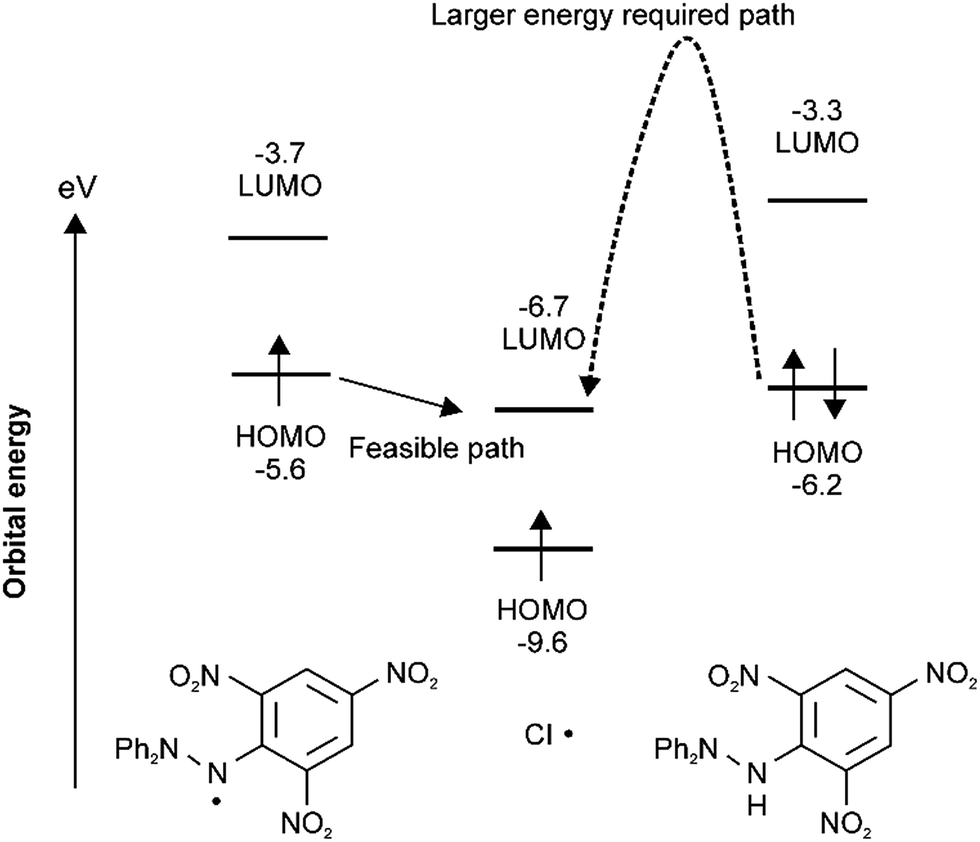 | ||
| Fig. 3 Frontier orbital interactions between the LUMO of Cl˙ and SOMO/HOMO of DPPH˙ by B3LYP* or UB3LYP/6-31G* calculations. | ||
Conclusions
We have developed a novel approach toward capturing Cl˙ using DPPH˙ in the gas and liquid phases at room temperature through substitution–trapping, which is thermodynamically reasonable and is controlled by frontier orbital interactions. This method is a rare example of the detection and characterization of Cl˙ species. In addition, IA-QMS is practical and sensitive enough to identify trapped radicals in situ. Further research is needed to clarify the selectivity of the substitution–trapping radical reaction and mechanism. Applications of this method in actual environmental gas-phase Cl˙ capture and theoretical investigations regarding the transition states are in progress to clarify the reaction mechanism.Acknowledgements
This research was supported by JSPS Grant-in-Aid for Young Scientists (B) Number 2681055.References
- J. Liao, L. G. Huey, Z. Liu, D. J. Tanner, C. A. Cantrell, J. J. Orlando, F. M. Flocke, P. B. Shepson, A. J. Weinheimer, S. R. Hall, K. Ullmann, H. J. Beine, Y. Wang, E. D. Ingall, C. R. Stephens, R. S. Hornbrook, E. C. Apel, D. Riemer, A. Fried, R. L. Mauldin III, J. N. Smith, R. M Staebler, J. A. Neuman and J. B. Nowak, Nat. Geosci., 2014, 7, 91 CrossRef CAS.
- H. Hoshino, K. Sakakibara and K. Watanabe, Chem. Lett., 2008, 37, 774 CrossRef CAS.
- K. Kawasaki, K. Sakakibara, F. Mizukami and T. Ebina, Clay Sci., 2008, 13, 217 CAS.
- P. J. Wright and A. M. English, J. Am. Chem. Soc., 2003, 125, 8655 CrossRef CAS PubMed.
- E. G. Janzen, Acc. Chem. Res., 1971, 4, 31 CrossRef CAS.
- P. Marsal, M. Roche, T. Tordo and P. de Sainte Claire, J. Phys. Chem. A, 1999, 103, 2899 CrossRef CAS.
- K. Miyazaki, A. E. Parker, C. Fittschen, P. S. Monks and Y. Kajii, Atmos. Meas. Tech., 2010, 3, 1547 CrossRef.
- A. A. P. Pszenny, W. C. Keene, D. J. Jacob, S. Fan, J. R. Maben, M. P. Zetwo, M. Springer-Young and J. N. Galloway, Geophys. Res. Lett., 1993, 20, 699 CrossRef CAS.
- A. Notario, G. Le Bras and A. Mellouki, Chem. Phys. Lett., 1997, 281, 421 CrossRef CAS.
- E. G. Janzen, H. J. Stronks, D. E. Nutter Jr, E. R. Davis, H. N. Blount, J. L. Poyer and P. B. McCay, Can. J. Chem., 1980, 58, 1596 CrossRef CAS.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255 CrossRef CAS PubMed.
- J. W. Engelsma and E. C. Kooyman, Recl. Trav. Chim. Pays-Bas, 1961, 80, 537–544 CrossRef CAS.
- K. Tanemura, T. Suzuki, Y. Nishida and T. Horaguchi, Tetrahedron, 2010, 66, 2881 CrossRef CAS.
- C. R. Everly and J. G. Traynham, J. Org. Chem., 1979, 44, 1784 CrossRef CAS.
- Z.-Q. Liu, Chem. Rev., 2010, 110, 5675 CrossRef CAS PubMed.
- P. Ionita, Chem. Pap., 2005, 59, 11 CAS.
- D. Pace, K. Ezell and L. D. Kispert, J. Chem. Phys., 1979, 71, 3971 CrossRef CAS.
- T. Fujii, Mass Spectrom. Rev., 2000, 19, 111 CrossRef CAS PubMed.
- N. Saito, J. Nanjyo, Y. Taneda, Y. Shiokawa and M. Tanimoto, Rapid Commun. Mass Spectrom., 2007, 21, 2654 CrossRef CAS PubMed.
- Y. Kitahara, S. Takahashi, N. Kuramoto, M. Šala, T. Tsugoshi, M. Sablier and T. Fujii, Anal. Chem., 2009, 81, 3155 CrossRef CAS PubMed.
- M. C. Foti, C. Daquino, I. D. Mackie, G. A. DiLabio and K. U. Ingold, J. Org. Chem., 2008, 73, 9270 CrossRef CAS PubMed.
- C. T. Kiers, J. L. De Boer, R. Olthof and A. L. Spek, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 2297 CrossRef.
- K.-S. Song, L. Liu and Q.-X. Guo, J. Org. Chem., 2003, 68, 262 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General remarks in the Experimental section; preparation of a DPPH–Cl paramagnetic adduct ([DPPH + Cl–NO2]˙) and its reduced diamagnetic DPPHH–Cl adduct ([DPPHH + Cl–NO2]˙) via an organic reaction in the liquid phase; spectral characterization data of DPPH adducts (high-resolution mass spectrometry, ESR, IR, 1H-, 13C-NMR, 2D-HMQC, and 2D-HMBC spectra of the reduced DPPHH–Cl); procedure for Cl˙ capture by using DPPH˙ in the gas-phase and characterization of [DPPH + Cl–NO2]˙ using IA-QMS. See DOI: 10.1039/c5ay01118c |
| This journal is © The Royal Society of Chemistry 2016 |