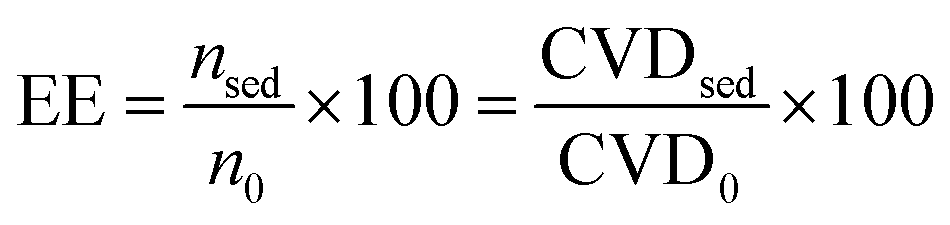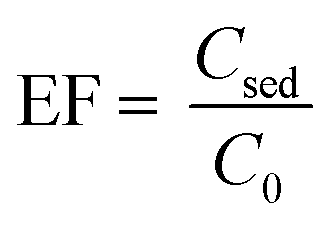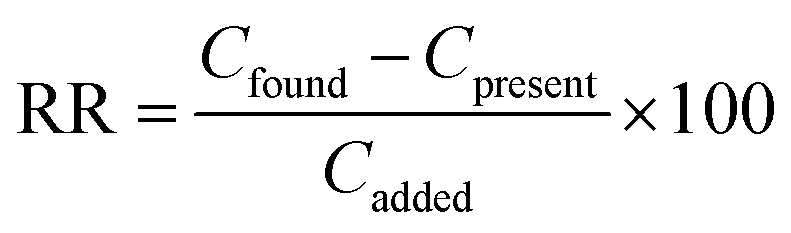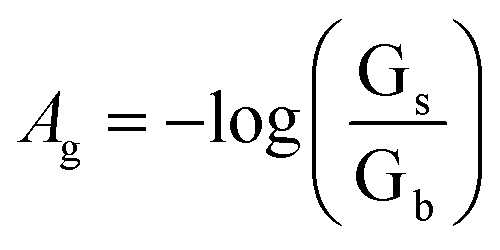DOI:
10.1039/C5AY01981H
(Paper)
Anal. Methods, 2016,
8, 111-118
Reflection scanometry as a new detection technique in temperature-controlled ionic liquid-based dispersive liquid phase microextraction
Received
28th July 2015
, Accepted 1st November 2015
First published on 6th November 2015
Abstract
Coupling of temperature-controlled ionic liquid-based dispersive liquid phase microextraction (TCIL-DLPME) with reflection scanometry for quantitative determination of colored species in solution is introduced for the first time. To evaluate the performance and applicability of the methodology, the ferrous ion which forms a red complex with 2,2′-bipyridyl (bipy) was used as the model analyte. The ionic liquid (IL) 1-hexyl-3-methylimidazolium hexafluorophosphate was used for extracting the complex by TCIL-DLPME after diluting the IL phase with dimethylformamide, which was injected into the detection chamber in a Plexiglas® sheet. It was scanned with a conventional flatbed-scanner. The color of the image was analyzed for its R (red), G (green), and B (blue) components using Adobe Photoshop software. Optimization of the parameters affecting the extraction process as well as the calibration curve was based on the G (green) color value as the analytical signal. Some essential parameters including the IL chemical structure, volume of IL, bipy concentration, pH, temperature, centrifugation time, and aqueous phase volume have been studied in detail. Under optimum experimental conditions the method was successfully applied for the determination of Fe(II) in iron supplement oral drop (IroVit) and boiler water samples.
1. Introduction
Over the last decades, much progress has been made in terms of variety and quality of analytical instruments, but in most analytical procedures sample preparation is still a tedious unavoidable step before instrumental analysis. This is because of the complexity of the sample, the low concentration of the target analytes, the presence of a great number of potential interferents and even the incompatibility with the detection system.1–4 The sample preparation step typically consists of an extraction procedure that results in the isolation and enrichment of the components of interest from a sample matrix. Among different techniques proposed for the sample preparation step, liquid phase microextraction (LPME) has been attracting much more attention in analytical chemistry.5–10 As a useful and popular category of LPME, dispersive liquid phase microextraction (DLPME) has been well established and widely applied in analytical chemistry due to its simplicity of operation, cheap cost, rapidity, good recovery, high enrichment factor achievement, and low consumption of organic solvents.11,12
In recent years, a variety of approaches of DLPME has been well developed and established and there have been major developments in this field, especially in attempts to use ionic liquids (ILs) with their unique physical and chemical properties13 as environmentally friendly extracting solvents instead of traditional organic solvents. Applying a set of dispersant tools, such as temperature-controlling,14 ultrasound,15in situ solvent formation,16 and salt addition,17 to the sample solution instead of the dispersive solvent is also reported in DLPME procedures.
Temperature-controlled ionic liquid-based DLPME (TCIL-DLPME) has been successfully used for the preconcentration and extraction of metal ions, such as vanadium (V4+/V5+),18 chromium (Cr3+/Cr6+),19 silver,20 lead,21,22 aluminium,23 and non-metal compounds, such as poly aromatic hydrocarbons (PAHs),11 pesticides,14 and chlorobenzenes.24
Until now, different analytical instruments such as flame,25,26 graphite furnace,27–30 cold vapor,31 and modified atomic absorption spectrometries,32,33 spectrophotometry,34,35 fiber optic-linear array detection spectrophotometry,36,37 inductively coupled plasma spectrometry,38–42 high performance liquid chromatography,43,44 gas chromatography,45 fluorescence spectrometry,46,47 and some online combinations48–51 have been widely coupled with DLPME for metal detection. However, these techniques are complicated, expensive, energy and time consuming, laborious, and usually need skilled workers. Moreover, in the case of viscose (i.e., ionic liquid) as well as low volume (in μL) extracting phases, some of the above conventional detection techniques are inefficient and have serious limitations for direct coupling to DLPME. In search of a simpler, cheaper, faster, greener, and efficient technique for quantitation of colored species in low volumes, reflection scanometry may be useful as a detection technique in TCIL-DLPME.
In 2009, for improving the paptode methodology,52–54 the sample solution was introduced into a bed of cylindrical cavities created in the body of a Plexiglas® sheet which was then scanned; the method was termed as reflection scanometry.55 In this method, homogeneity, rate of analysis, detection limit and precision of analysis were reported to be significantly improved.
The objective of this work was to combine the advantages of TCIL-DLPME and reflection scanometry, as a detection technique, in order to develop a simple, cheap, fast, green, and efficient methodology for preconcentration and quantification of colored species in solution media. For evaluating the method, the ferrous ion and 2,2′-bipyridyl (bipy) that form a red complex with a structural ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (Fe(II):bipy) were used.56 The ionic liquid 1-hexyl-3-methylimidazolium hexafluorophosphate was used as the extracting solvent. To the best of our knowledge, there has been no reports on the investigation of colored species by using TCIL-DLPME coupled with reflection scanometry.
:3 (Fe(II):bipy) were used.56 The ionic liquid 1-hexyl-3-methylimidazolium hexafluorophosphate was used as the extracting solvent. To the best of our knowledge, there has been no reports on the investigation of colored species by using TCIL-DLPME coupled with reflection scanometry.
2. Experimental
2.1. Preparation of the cell array
The individual cylindrical cavities (0.5 cm in diameter) in a Plexiglas® sheet (thickness of 0.6 cm), which have been fabricated with a laser beam, serve as detection chambers (cells). To construct these cavities, one end of a series of cylindrical holes created in the body of a Plexiglas® sheet was covered by another sheet of Plexiglas® having a thickness of 0.1 cm. In this design, twelve cells were aligned in 4 columns and 3 rows, which are called “detection chambers” in this text (Fig. 1).
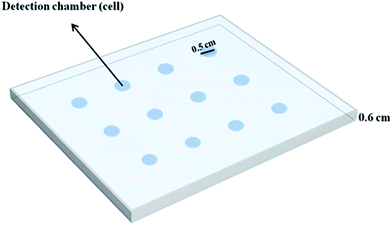 |
| | Fig. 1 A schematic presentation of the cell array created in the body of a Plexiglas® sheet. | |
2.2. Apparatus and software
The acquisition of the images was performed when the detection chamber was illuminated by a cold cathode fluorescent lamp of the scanner (model Canoscan-4200F) for scanning purposes. The reflected light was collected and converted into a series of electronic signals by using a charge coupled device (CCD) as the detection system while the resolution of the scanner was regulated at 600 dpi. A tag image file format (TIFF) was selected to have images with good quality and to maintain all color information for translating them into numerical information (i.e. R (red), G (green), and B (blue) color values) without loss of information. This file format, however, can be intrinsically supported by all types of hardware environments such as personal computers. Calculation of the R, G, and B color values was performed with an optional histogram palette from an Adobe Photoshop element Cs5 ver.12.0 (Adobe System Inc., CA, USA). The dimension of the working “region of interest” (ROI) was 400 pixels (20 × 20 pixels) from the maximum available size of 120 × 120 pixels. An Eppendorf micropipette was used for injecting samples into the conical glass tubes and the detection chamber. The pH measurements were made with a Metrohm (780) pH meter using a combined glass electrode.
2.3. Chemicals
The chemicals 1-bromobutane, 1-bromohexane, 1-bromooctane, 1-methylimidazolium, ammonium iron(II) sulfate hexahydrate, (NH4)2Fe(SO4)2·6H2O, sodium chloride, hydrochloric acid, glycine, and dimethylformamide (DMF) were of the highest available purity purchased from Merck Chemical Company. Ammonium hexafluorophosphate, hydroxylamine hydrochloride (NH2OH·HCl), and 2,2′-bipyridyl (bipy) were purchased from Fluka Chemical Company. Methanol and diethyl ether were purchased from Panreac Chemical Company and were used without further purification. Ionic liquids 1-butyl-3-methylimidazolium hexafluorophosphate, [C4mim][PF6], 1-hexyl-3-methylimidazolium hexafluorophosphate, [C6mim][PF6], and 1-octyl-3-methylimidazolium hexafluorophosphate, [C8mim][PF6], were synthesized as described by Bonhote et al.57 Chemical structures of ionic liquids were determined by using NMR spectroscopy. Distilled deionized water was used throughout this work.
For preparation of standard solutions of Fe(II) ion in the extracting phase (bipy-IL), appropriate volume (in μL) of an aqueous standard solution of Fe(II) was mixed with 50 μL bipy-IL in a 10 mL conical glass tube and the tube was thermostated at 50 °C for 60 min in order to remove its water content. The residual liquid in the tube was the standard solution of Fe(II) ion in the extracting phase.
2.4. Recommend procedure
An aliquot of 5.5 mL of 279.25 μg L−1 Fe(II) ion was brought into contact with 50 μL of 0.15 mol L−1 bipy in [C6mim][PF6] in a 10 mL conical glass tube. The tube was heated in a temperature-controlled water bath at 77 °C for 10 min, followed by shaking for a period of 1 min. The tube was then cooled in ice water for 20 min to obtain a turbid solution. After centrifugation at 1200 rpm for 40 min and resolving the phases, the upper aqueous phase was removed with a μL-syringe. The residue was diluted with 25 μL DMF, followed by shaking in order to have a homogeneous solution before being injected into the chamber. The Plexiglas® sheet was scanned afterward and the resulting image was transferred to the computer for evaluating its R, G, and B color values by using the optional histogram palette from Adobe Photoshop software. The schematic diagram of this procedure is shown in Fig. 2.
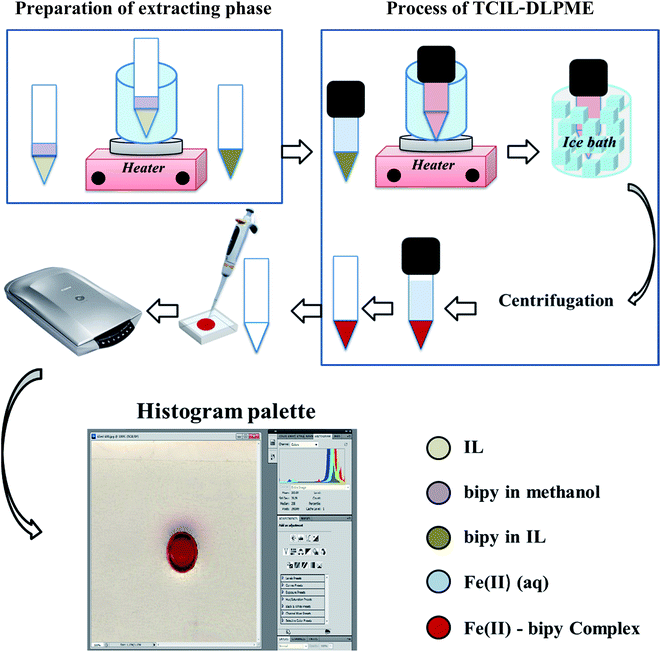 |
| | Fig. 2 Schematic presentation of the reflection scanometry as a new detection technique in temperature-controlled ionic liquid-based dispersive liquid phase microextraction. | |
It should be mentioned that for the preparation of bipy-IL, 100 μL of the standard solution of bipy in methanol was added into 50 μL of IL in a 10 mL conical glass tube. The mixture was placed in a water bath for 5.0 min at 77 °C for removing methanol. The resulting residue was bipy-IL solution that was used as the extracting phase.
2.5. Procedure for real sample analysis
The oral drop sample, without any treatment, was diluted with the buffer solution and then analyzed before and after spiking the standard Fe(II) solution. The boiler water sample was first centrifuged and filtered through a disposable syringe filter (0.22 μm pore-size membrane). Then, a sufficient amount of NH2OH·HCl solution was added to the filtrate and the contents were mixed well. The mixture was diluted with the buffer solution and then analyzed before and after spiking the standard Fe(II) solution.
2.6. Analytical parameters
Some main parameters that were considered for the evaluation of the developed methodology including, extraction efficiency (EE), enrichment factor (EF), and relative recovery (RR) are defined by eqn (1), (2), and (3), respectively,11,58 as follows:| | 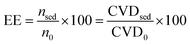 | (1) |
where nsed is the amount of analyte in the sediment (extracting phase) and n0 is the initial amount of analyte in the sample solution. The CVD stands for “color value difference” which is related to the amount of Fe–bipy as the detection form of Fe(II). Since the scanned images revealed that the G color value could provide significant results as the analytical signal to be probed in this investigation; the CVDsed is the difference between the G color values of the sediment and its corresponding blank. The CVD0 is the difference between the G color values of the standard solution and its corresponding blank. The enrichment factor (EF) is defined as follows:| | 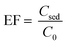 | (2) |
where Csed is the concentration of the extracted analyte in the IL phase and C0 is the initial concentration of the analyte. The relative recovery (RR) is obtained as below:| | 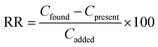 | (3) |
where Cfound, Cpresent, and Cadded are the concentration of the analyte after the addition of a known amount of standard in the real sample, the concentration of the analyte in the real sample obtained by the proposed method, and the concentration of a known amount of standard which was spiked to the real sample, respectively. It should be mentioned that each result reported in this work was the average of four replicate extractions.
3. Results and discussion
3.1. Effect of the IL chemical structure
To determine the appropriate IL for extracting Fe(II), aliquots of 5.0 mL of 279.25 μg L−1 Fe(II) solutions were brought into contact with 50 μL of bipy-IL as the extracting phase. The selected ILs, with different hydrophobicities, that were used for preparing bipy-IL solutions were [C4mim][PF6], [C6mim][PF6], and [C8mim][PF6]. It was revealed that bipy-[C6mim][PF6] was a desirable extracting phase as it was easily dissolved during the heating/shaking period and then was dispersed during the cooling process. After centrifugation, the aggregate showed a volume of 40.0 ± 2.4 μL (n = 4). This ionic liquid was selected as the extracting phase for the rest of the work. It should be mentioned that bipy-[C4mim][PF6] was easily dissolved in the aqueous phase during the heating/shaking process and after centrifugation no aggregation was observed for this IL. Moreover, bipy-[C8mim][PF6] was hardly soluble in the aqueous phase during the heating/shaking process and after centrifugation, the aggregate showed a volume of 43.0 ± 3.7 μL (n = 4). However, characteristics of bipy-[C8mim][PF6] were not desirable for extraction of Fe(II); it could not provide a highly dispersed medium because its hydrophobicity is known to be higher than other ILs in this work.59 This resulted in a poor extraction efficiency for Fe(II).
3.2. Effect of extracting phase volume
The effect of the volume of the extracting phase; i.e. bipy-[C6mim][PF6], on the extraction efficiency of Fe(II) was studied under the experimental conditions of: 5.0 mL of 279.25 μg L−1 Fe(II); 0.10 mol L−1 bipy; pH 2.5; heating time of 10 min; dissolution temperature of 77 °C; extraction time of 20 min; centrifugation time of 20 min; and diluent volume of 40 μL. The Fe(II) solutions were brought into contact with different volumes of the extracting phase. Results, shown in Fig. 3, indicate that the extraction efficiency increased with the extracting phase volume over the range of 30–50 μL and decreased over the range of 55–70 μL. As reported in the literature,14 when the volume of the extracting phase exceeds the “target dispersed amount”, a portion of the extracting phase containing the analyte is adsorbed onto the inner surface of the tube. Practically, this portion of the extracting phase could not be assembled at the bottom of the tube where the bulk of the extracting phase is collected. An aliquot of 50 μL of the extracting phase was found to be the “target dispersed amount” and larger volumes resulted in a decrease in extraction efficiency of Fe(II).
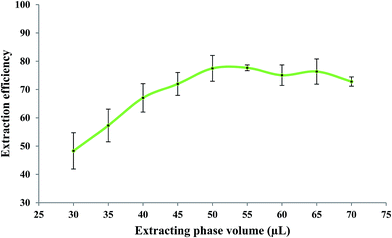 |
| | Fig. 3 Effect of extracting phase volume on the extraction efficiency of Fe(II). Experimental conditions: Fe(II) solution, 5.0 mL of 279.25 μg L−1; bipy concentration, 0.10 mol L−1; pH, 2.5; heating time, 10 min; dissolution temperature, 77 °C; extraction time, 20 min; centrifugation time, 20 min; and diluent volume, 40 μL. | |
3.3. Effect of 2,2′-bipyridyl concentration
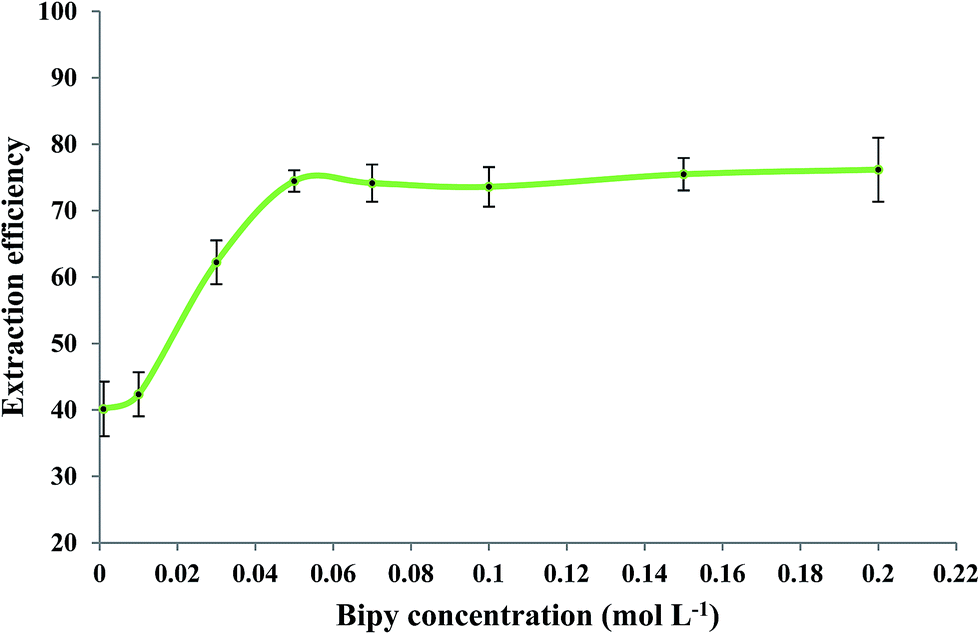
The effect of 2,2′-bipyridyl concentration on the extraction efficiency of Fe(II) was evaluated while other experimental conditions were similar to those mentioned in Section 3.2. Fig. 4 shows that Fe(II) extraction was improved by increasing the bipy concentration up to 0.15 mol L−1. No significant change in extraction efficiency was observed when higher concentrations of bipy were used. A concentration of 0.15 mol L−1 bipy was selected as an optimum experimental value.
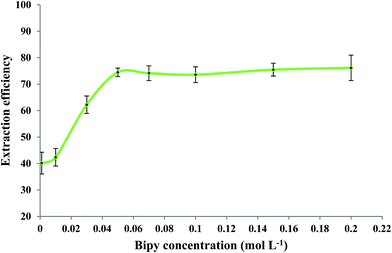 |
| | Fig. 4 Effect of bipy concentration on the extraction efficiency of Fe(II). Experimental conditions: Fe(II) solution, 5.0 mL of 279.25 μg L−1; extracting phase volume, 50 μL; pH, 2.5; heating time, 10 min; dissolution temperature, 77 °C; extraction time, 20 min; centrifugation time, 20 min; and diluent volume, 40 μL. | |
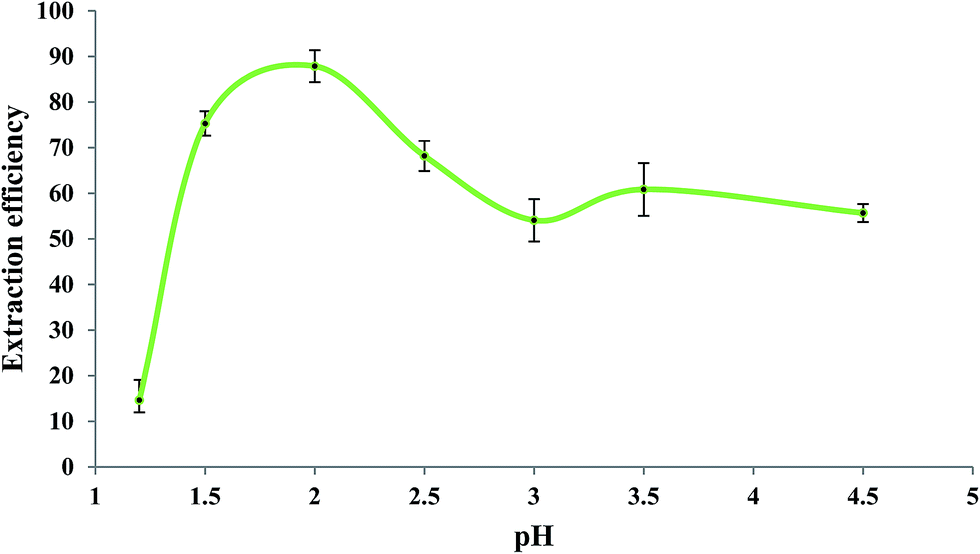
3.4. Effect of pH of the sample solution
Fig. 5 shows the influence of pH of the sample solution on the extraction efficiency of Fe(II) studied in the pH range of 1.2 to 4.5. As reported in the literature,60,61 the rate of oxidation of Fe(II) to Fe(III) is pH-dependent so that it increases by increasing pH. So, in order to prevent oxidation of Fe(II) to Fe(III), the measurements should be performed in acidic solutions. For this experiment, a mixture of glycine, NaCl, and HCl as buffer solution62 was prepared with different pH values. Aliquots of 5.0 mL of 279.25 μg L−1 Fe(II) solutions with different pH values were individually brought into contact with 50 μL of the extracting phase. Other experimental conditions were the same as those mentioned in the previous section. As shown in Fig. 5, the extraction efficiency was high at about pH 2.0; at pH > 2.0, it decreased which could be due to increase in the oxidation rate of Fe(II).62 Lower extraction efficiencies at pH < 2.0 could be due to the competition of H3O+ with Fe(II) for interaction with bipy. Based on these results, pH = 2 was selected as the optimum operating pH. It should be mentioned that the precipitation of colloidal Fe(OH)2 is not expected at pH < 4.5 since the ion product of Fe(II) (5.0 × 10−6 mol L−1) and OH− (3.2 × 10−10 mol L−1) was 5.1 × 10−25 in this experiment which is far less than the Ksp of Fe(OH)2 (8.0 × 10−16).
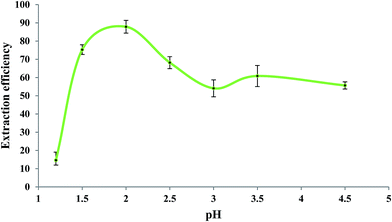 |
| | Fig. 5 Effect of sample pH on the extraction efficiency of Fe(II). Experimental conditions: Fe(II) solution, 5.0 mL of 279.25 μg L−1; extracting phase volume, 50 μL; bipy concentration, 0.15 mol L−1; heating time, 10 min; dissolution temperature, 77 °C; extraction time, 20 min; centrifugation time, 20 min; and diluents' agent volume, 40 μL. | |
3.5. Effect of temperature
To obtain the optimum temperature, the mixture of the extracting phase and aqueous sample solution was first subjected to a high temperature for a period of time followed by shaking. Afterward, the solution was cooled down in order to achieve a dispersed phase in the bulk of the sample solution. To optimize the temperature, a series of microextraction experiments were done in the temperature range of 25–97 °C under other optimized experimental conditions which are already mentioned. The extraction efficiency was higher almost in the temperature range of 40–60 °C. Lower extraction efficiencies at lower temperatures may be due to poor dispersion of the extracting phase and subsequently the lower migration of Fe(II) into the IL-phase. At higher temperatures, dissociation of the complex may occur leading to lower efficiency results. Based on these results, a temperature of 50 °C was selected as the optimum operating temperature.
3.6. Effect of cooling time
After the heating/shaking step, the mixture of IL and aqueous phases was brought into contact with an ice water bath for different periods of time (cooling/extraction time) in order to produce a dispersed phase for collecting the analyte species. The best result was obtained for a cooling time of 20 min. No significant change in extraction efficiencies was observed when solutions were cooled for longer periods of time.
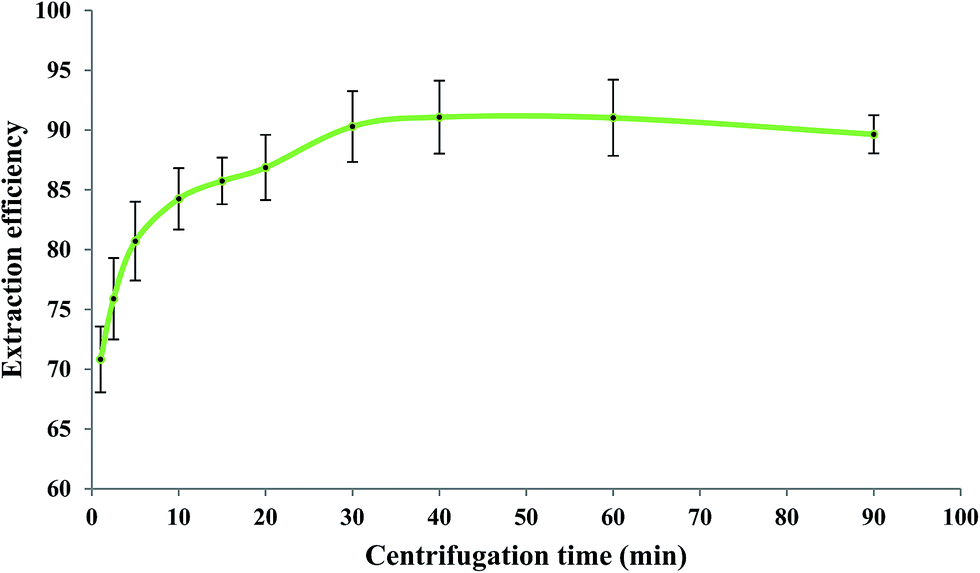
3.7. Effect of centrifugation time
After elapsing the cooling period, the centrifugation process (1200 rpm) was performed for different periods of time at the optimized values of the other experimental conditions as before. Generally, centrifugation provides complete separation of phases through aggregation of the dispersed IL phase. When a short centrifugation time is applied, the dispersed IL drops cannot be well aggregated. However, a long centrifugation time causes dissolving of some portions of the IL phase into the aqueous phase due to local heat production. Consequently, both short and long centrifugation times reduce the extraction efficiency of measurements as it has also been reported in the literature.14 According to Fig. 6, a centrifugation time of 40 min at 1200 rpm was selected as the optimum value.
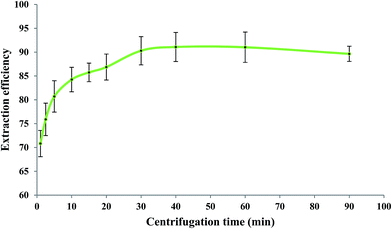 |
| | Fig. 6 Effect of centrifugation time on the extraction efficiency of Fe(II). Experimental conditions: Fe(II) solution, 5.0 mL of 279.25 μg L−1; extracting phase volume, 50 μL; pH, 2.0; bipy concentration, 0.15 mol L−1; heating time, 10 min; dissolution temperature, 50 °C; extraction time, 20 min; and diluent volume, 40 μL. | |
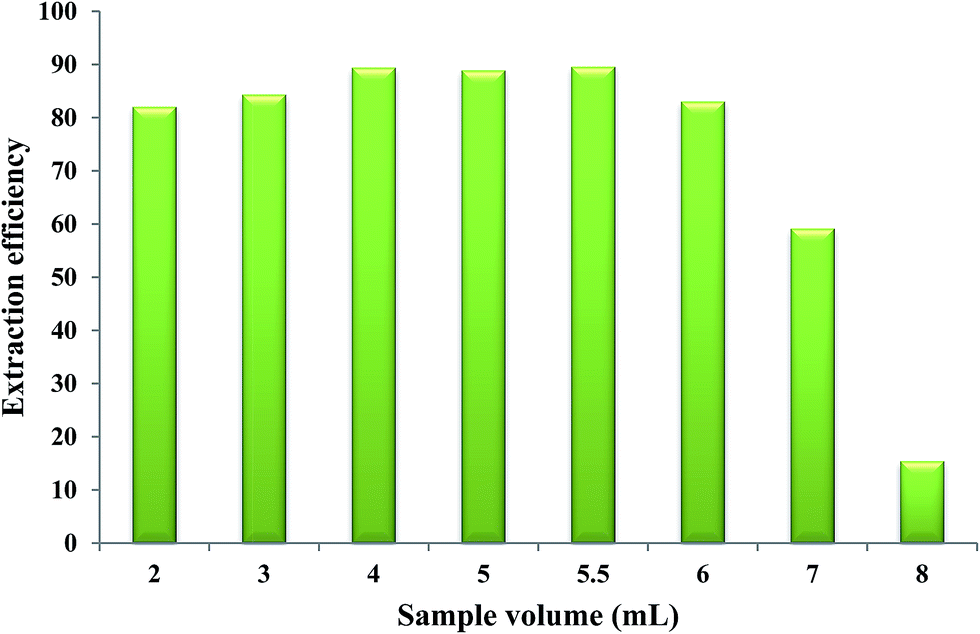
3.8. Effect of aqueous phase volume
In order to determine the breakthrough volume, different volumes of aqueous solutions, each containing 1.40 μg Fe(II), were brought into contact with 50 μL of the extracting phase and the microextraction procedure was performed at the optimized values of the other parameters. The results indicated that by using aqueous solution volume higher than 5.5 mL (“breakthrough volume”), a decrease in extraction efficiency was observed. By increasing the volume of the aqueous sample, the mole fraction of the IL phase in the water/IL mixture decreased causing more dissolution of IL in water which leads to incomplete separation of two phases and consequently results in a lower extraction efficiency of Fe(II). Aqueous solution volumes equal to or less than 5.5 mL could provide higher extraction efficiencies when 50 μL of the extracting phase was used. Therefore, a volume of 5.5 mL was selected as the optimum volume of the aqueous phase (Fig. 7).
3.9. Diluent volume
As mentioned in the procedure, after the TCIL-DLPME process, the ionic liquid phase was diluted with 25 μL DMF before being transferred into the chamber in order to produce a homogeneous phase and to wash out the IL sticking onto the inner surface of the test tube. Among different types of diluents (ethanol, acetonitrile, acetone, diethyl ether, dimethylformamide, and tetrahydrofuran) that were tested, dimethylformamide (DMF) was the best choice for the developed methodology because it does not dissolve the Plexiglas® sheet (detection chamber); it dissolves the IL phase rapidly and completely; and it has good stability and lack of evaporation during the analyzing period. The volume of DMF was optional, however, it must be selected in such a way that not only produces a homogeneous phase but also gives an acceptable value of the enrichment factor.
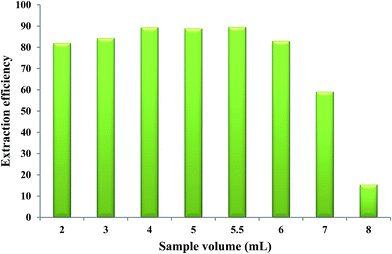 |
| | Fig. 7 Effect of aqueous phase volume on the extraction efficiency of Fe(II). Experimental conditions: Fe(II) solution, different volumes each containing 1.40 μg Fe(II); extracting phase volume, 50 μL; pH, 2.0; bipy concentration, 0.15 mol L−1; heating time, 10 min; dissolution temperature, 50 °C; extraction time, 20 min; centrifugation time, 40 min; and diluent volume, 40 μL. | |
3.10. Analytical figures of merit
Changes in color of the extracting phase (i.e. bipy-IL phase) after the TCIL-DLPME process were analyzed by using the Adobe Photoshop software. The color information of each cell was obtained from the ROI. Then it was decomposed into red (R), green (G), and blue (B) values. The effective intensity of the G color value as the analytical signal, based on ROI information obtained in the 8 bit RGB color system in the computer, was calculated for each cell as follows:
where Ag refers to effective intensity of the G color value; Gs and Gb refer to G color values of the standard and blank solutions, respectively. To obtain the calibration curve, Ag was plotted vs. Fe(II) concentration.
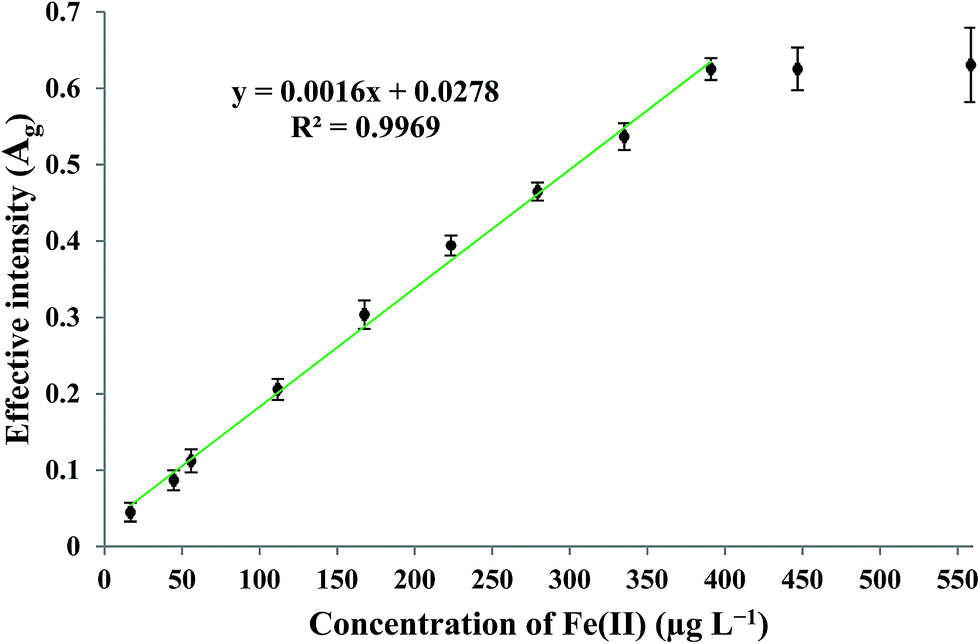
By applying the optimum experimental conditions, the calibration curve was found to be linear in the concentration range of 16–390 μg L−1 Fe(II). The practical detection limit (defined as 3Sb/m; where Sb and m are the standard deviation of the blank measurements (n = 4) and the slope of the calibration curve, respectively) and theoretical63 detection limit were 9.75 and 3.64 μg L−1, respectively. The enrichment factor and precision for the determination of 279.25 μg L−1 Fe(II), measured under the optimum experimental conditions, were found to be 72 and 4.16% (n = 4), respectively. The linear relationship between the effective intensity of the G color value and the concentration of Fe(II) is shown in Fig. 8.
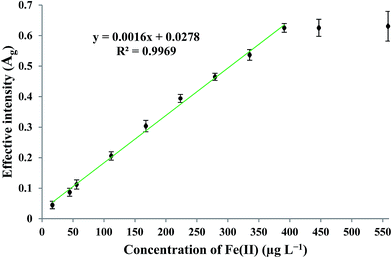 |
| | Fig. 8 Calibration curve under optimum experimental conditions of G (green) color values. | |
3.11. Interference studies
The influence of common coexisting ions in natural water samples on the determination of 279.25 μg L−1 Fe(II) was investigated under optimum experimental conditions. The tolerance limit for a coexisting ion was considered as the largest amount of that ion which caused an error not more than ±5% in measuring the analytical signal. The results of this investigation are summarized in Table 1.
Table 1 Tolerance limits of some common metal ions in natural water for the determination of 279.25 μg L−1 Fe(II) as complexes with bipy
| Coexisting ions |
Tolerance limits (μg L−1) |
| Co(II), Pb(II), Ca(II), Al(III) |
1000 |
| Cu(II) |
500 |
| Ni(II), Cd(II), Zn(II), Mg(II) |
100 |
3.12. Real sample analysis
The applicability of the developed method for the determination of Fe(II) in two different matrices was evaluated by extracting Fe(II) in the chemical form of the Fe–bipy complex into the IL. The samples were an iron supplement oral drop (IroVit components: CFe(II) = 15 g L−1 and strawberry flavor as pleasant) as well as a boiler water sample kindly supplied by Shiraz Serum Company (Shiraz city, Fars Province, Iran). The results are summarized in Table 2 indicating that the precision and accuracy are in acceptable levels for the analysis of Fe(II) in both samples.
Table 2 Determination of the ferrous ion by the developed method in the oral drop sample (containing 15 g L−1 Fe(II) as reported by the manufacturer) and in the boiler water sample (0.0835 g L−1 as measured by ICP-OES) after diluting these samples
| Real samples |
Fe(II) added (μg L−1) |
Fe(II) found (μg L−1) for n = 4 |
Relative error (%) |
RRc (%) |
|
Iron supplement oral drop components (Fe(II) and strawberry flavor as pleasant; Vitane pharmaceuticals company, Germany).
Boiler water sample from Shiraz Serum Company in Shiraz city, Fars province, Iran.
Relative recovery.
|
| Diluted IroVit oral dropa |
0.0 |
175.2 ± 2.1 |
+4.5 |
113.2 |
| 50.0 |
231.8 ± 2.4 |
| Diluted boiler waterb |
0.0 |
173.7 ± 2.0 |
+3.6 |
106.8 |
| 50.0 |
227.1 ± 2.2 |
4. Conclusions
For the first time, reflection scanometry has been used as a new detection technique for TCIL-DLPME. To evaluate the applicability of this setup, the ferrous ion and 2,2′-bipyridyl were used as the model analyte and complexing agent, respectively. The procedure is simple, fast, safe, easy to use, and cost effective. It does not need expensive apparatus in comparison with other TCIL-DLPME methods where sophisticated detection devices have been used. The results showed that the practical and theoretical detection limits for Fe(II) were 9.75 and 3.64 μg L−1, respectively, and the enrichment factor was 72. The precision for determination of Fe(II) in a solution containing 279.25 μg L−1 Fe(II) was found to be 4.16% (n = 4). The linear relationship between the effective intensity of the G color value with the concentration of Fe(II) was in the concentration range of 16–390 μg L−1 Fe(II). The procedure was successfully applied for the determination of Fe(II) in an iron supplement oral drop (IroVit) as well as a boiler water samples.
Acknowledgements
We gratefully acknowledge the Shiraz University Research Council for financially supporting this study.
References
- M. Cruz-Vera, R. Lucena, S. Cárdenas and M. Valcárcel, J. Chromatogr. A, 2009, 1216, 6459–6465 CrossRef CAS PubMed.
- M. Baghdadi and F. Shemirani, Anal. Chim. Acta, 2008, 613, 56–63 CrossRef CAS PubMed.
- L. Xin, F. Rao, L. Min, G. Li-Ping and Y. Li, Chin. J. Anal. Chem., 2013, 41, 1919–1922 CrossRef.
- S. Dong, Q. Hu, Z. Yang, R. Liu, G. Huang and T. Huang, Microchem. J., 2013, 110, 221–226 CrossRef CAS.
- M. A. Jeannot and F. F. Cantwell, Anal. Chem., 1996, 68, 2236–2240 CrossRef CAS PubMed.
- A. L. Theis, A. J. Waldack, S. M. Hansen and M. A. Jeannot, Anal. Chem., 2001, 73, 5651–5654 CrossRef CAS PubMed.
- S. Pedersen-Bjergaard and K. E. Rasmussen, Anal. Chem., 1999, 71, 2650–2656 CrossRef CAS PubMed.
- L. S. de Jager and A. R. J. Andrews, Chromatographia, 1999, 50, 733–738 CAS.
- L. S. de Jager and A. R. J. Andrews, Analyst, 2000, 125, 1943–1948 RSC.
- T. Ligor and B. Buszewski, Chromatographia, 2000, 51, 279–282 Search PubMed.
- M. Rezaee, Y. Assadi, M. R. Milani Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, J. Chromatogr. A, 2006, 1116, 1–9 CrossRef CAS PubMed.
- M. Pirsaheb, N. Fattahi and M. Shamsipur, Food Control, 2013, 34, 378–385 CrossRef CAS.
- V. Vičkačkaitė and A. Padarauskas, Cent. Eur. J. Chem., 2012, 10, 652–674 CrossRef.
- Q. Zhou, H. Bai, G. Xie and J. Xiao, J. Chromatogr. A, 2008, 1177, 43–49 CrossRef CAS PubMed.
- S. Li, S. Cai, W. Hu, H. Chen and H. Liu, Spectrochim. Acta, Part B, 2009, 64, 666–671 CrossRef.
- M. Baghdadi and F. Shemirani, Anal. Chim. Acta, 2009, 634, 186–191 CrossRef CAS PubMed.
- H. Ma, Y. Li, H. Zhang, S. M. Shah and J. Chen, J. Chromatogr. A, 2014, 1358, 14–19 CrossRef CAS PubMed.
- P. Berton, E. M. Martinisa, L. D. Martinezc and R. G. Wuilloud, Anal. Chim. Acta, 2009, 640, 40–46 CrossRef CAS PubMed.
- S. Sadeghi and A. Z. Moghaddam, Talanta, 2012, 99, 758–766 CrossRef CAS PubMed.
- G. Absalan, M. Akhond, L. Sheikhian and D. M. Goltz, Anal. Methods, 2011, 3, 2354–2359 RSC.
- F. Shah, T. G. Kazi, N. Ullah, H. I. Afridi and M. Soylak, Microchem. J., 2012, 101, 5–10 CrossRef CAS.
- H. Bai, Q. Zhou, G. Xie and J. Xiao, Talanta, 2010, 80, 1638–1642 CrossRef CAS PubMed.
- M. S. Arain, S. A. Arain, T. G. Kazi, H. I. Afridi, J. Ali, N. Ullah, S. S. Arain, K. D. Brahman and M. A. Mughal, Spectrochim. Acta, Part A, 2015, 137, 877–885 CrossRef CAS PubMed.
- F. Kamarei, H. Ebrahimzadeh and Y. Yamini, Talanta, 2010, 83, 36–41 CrossRef CAS PubMed.
- T. A. Kokya and K. Farhadi, J. Hazard. Mater., 2009, 169, 726–733 CrossRef CAS PubMed.
- C. X. Wu, Q. H. Wu, C. Wang and Z. Wang, Chin. Chem. Lett., 2011, 22, 473–476 CrossRef CAS.
- E. Z. Jahromi, A. Bidari, Y. Assadi, M. R. Milani Hosseini and M. R. Jamali, Anal. Chim. Acta, 2007, 585, 305–311 CrossRef PubMed.
- M. T. Naseri, M. R. Milani Hosseini, Y. Assadi and A. Kiani, Talanta, 2008, 75, 56–62 CrossRef CAS PubMed.
- M. Mirzaei, M. Behzadi, N. Mahmoud Abadi and A. Beizaei, J. Hazard. Mater., 2011, 186, 1739–1743 CrossRef CAS PubMed.
- P. Liang, L. Zhang and E. Zhao, Talanta, 2010, 82, 993–996 CrossRef CAS PubMed.
- E. Stanisz, J. Werner and H. Matusiewicz, Microchem. J., 2013, 110, 28–35 CrossRef CAS.
- M. T. Naseri, P. Hemmatkhah, M. R. Milani Hosseini and Y. Assadi, Anal. Chim. Acta, 2008, 610, 135–141 CrossRef CAS PubMed.
- S. R. Yousefi and F. Shemirani, Anal. Chim. Acta, 2010, 669, 25–31 CrossRef CAS PubMed.
- X. Wen, L. Kong, M. Chen, Q. Deng, X. Zhao and J. Guo, Spectrochim. Acta, Part A, 2012, 97, 782–787 CrossRef CAS PubMed.
- M. Gharehbaghi, F. Shemirani and M. Baghdadi, Int. J. Environ. Anal. Chem., 2009, 89, 21–33 CrossRef CAS.
- M. Ezoddin, F. Shemirani and M. R. Jamali, J. Anal. Chem., 2010, 65, 153–158 CrossRef CAS.
- N. Shokoufi, F. Shemirani and Y. Assadi, Anal. Chim. Acta, 2007, 597, 349–356 CrossRef CAS PubMed.
- Y. Yamini, M. Rezaee, A. Khanchi, M. Faraji and A. Saleh, J. Chromatogr. A, 2010, 1217, 2358–2364 CrossRef CAS PubMed.
- M. Rezaee, Y. Yamini, A. Khanchi, M. Faraji and A. Saleh, J. Hazard. Mater., 2010, 178, 766–770 CrossRef CAS PubMed.
- H. Sereshti, V. Khojeh and S. Samadi, Talanta, 2011, 83, 885–890 CrossRef CAS PubMed.
- X. Jia, Y. Han, X. Liu, T. Duan and H. Chen, Microchim. Acta, 2010, 171, 49–56 CrossRef CAS.
- I. Razmislevicien, A. Padarauskas, B. Pranaityt and E. Naojalis, Curr. Anal. Chem., 2010, 6, 310–315 CrossRef.
- M. A. Farajzadeh, M. Bahram and M. R. Vardast, Clean: Soil, Air, Water, 2010, 38, 466–477 CrossRef CAS.
- X. Jia, Y. Han, X. Liu, T. Duan and H. Chen, Spectrochim. Acta, Part B, 2011, 66, 88–92 CrossRef.
- A. Ranji, M. Ghorbani Ravandi and M. A. Farajzadeh, Anal. Sci., 2008, 24, 623–626 CrossRef CAS PubMed.
- Q. Zhou, N. Zhao and G. Xie, J. Hazard. Mater., 2011, 189, 48–53 CrossRef CAS PubMed.
- J. Liu, D. Wu, C. Duan and Y. Guan, Talanta, 2013, 105, 87–92 CrossRef CAS PubMed.
- A. N. Anthemidis, C. Mitani, P. Balkatzopoulou and P. D. Tzanavaras, Anal. Chim. Acta, 2012, 733, 34–37 CrossRef CAS PubMed.
- A. N. Anthemidis and K. G. Loannou, Talanta, 2009, 79, 86–91 CrossRef CAS PubMed.
- A. N. Anthemidis and K. G. Ioannou, Anal. Chim. Acta, 2010, 668, 35–40 CrossRef CAS PubMed.
- A. N. Anthemidis and K. G. Ioannou, Talanta, 2011, 84, 1215–1220 CrossRef CAS PubMed.
- A. Abbaspour, M. A. Mehrgardi, A. Noori, M. A. Kamyabi, A. Khalafi-Nezhad and M. N. Soltani Rad, Sens. Actuators, B, 2006, 113, 857–864 CrossRef CAS.
- A. Abbaspour, A. Khajehzadeh and A. Noori, Anal. Sci., 2008, 24, 721–725 CrossRef CAS PubMed.
- A. Abbaspour, E. Mirahmadi and A. Khajehzadeh, Anal. Methods, 2010, 2, 349–353 RSC.
- A. Abbaspour, A. Khajezadeh and A. Ghaffarinejad, Analyst, 2009, 134, 1692–1698 RSC.
- S. Summer, A. Perveen and I. I. Naqvi, Arabian J. Sci. Eng., 2009, 34, 75–85 CAS.
- P. Bonhote, A. P. Dias, N. Papageorgion, K. Kalyanasundaram and M. Gratzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef CAS PubMed.
- M. Rajabi, S. Asemipour, B. Barfi, M. R. Jamali and M. Behzad, J. Mol. Liq., 2014, 194, 166–171 CrossRef CAS.
- J. Ranke, A. Othman, P. Fan and A. Muller, Int. J. Mol. Sci., 2009, 10, 1271–1289 CrossRef CAS PubMed.
- G. Absalan, M. Arabi, R. Khalifeh, H. Sharghi and J. Tashkhourian, IEEE Sens. J., 2014, 14, 349–356 CrossRef CAS.
- B. Morgan and O. Lahav, Chemosphere, 2007, 68, 2080–2084 CrossRef CAS PubMed.
-
J. Lurie, Handbook of Analytical Chemistry, translated by Nicholas Bobrov, Mir Publishers, Moscow, Russia, 1st edn, 1975 Search PubMed.
-
J. N. Miller and J. C. Miller, Statistics and Chemometrics for Analytical Chemistry, Pearson Education Limited, Harlow, UK, 5th edn, 2005, ISBN 0-131-291920 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.