
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A sensitive and versatile method for characterization of protein-mediated transformations of quantum dots†
Magdalena
Matczuk
*a,
Joanna
Legat
a,
Andrei R.
Timerbaev
*b and
Maciej
Jarosz
a
aChair of Analytical Chemistry, Faculty of Chemistry, Warsaw University of Technology, Noakowskiego St. 3, 00-664 Warsaw, Poland. E-mail: mmatczuk@ch.pw.edu.pl
bVernadsky Institute of Geochemistry and Analytical Chemistry, Kosygin St. 19, 119991 Moscow, Russian Federation. E-mail: andrei.timerbaev@univie.ac.at
First published on 15th March 2016
Abstract
We report the development and application of an analytical system consisting of capillary electrophoresis (CE) interfaced with inductively coupled plasma mass spectrometry (ICP-MS) for sensitive and high-resolution characterization of quantum dots (QDs) interacting with serum proteins. Separation resolution between the intact CdSeS/ZnS QDs and their protein conjugates was optimized by varying the type and concentration of background electrolyte, applied voltage, and sample loading. Special attention was paid to the CE system compatibility with physiological conditions, avoiding aggregation effects, and analyte recovery. Optimization trials allowed for acquiring satisfactory stability of migration times (within 6.0% between different days), peak area precision of 5.2–8.0%, capillary recoveries in the range of 90–96%, and a lower limit of detection of 7.5 × 10−9 mol L−1 Cd. With the developed method distinct metal-specific profiles were obtained for the QDs in combination with individual serum proteins, their mixtures, and in human serum. Particularly, it was found that albumin binding to the particle surface is completed after 1 h, without noticeable disruption of the core–shell integrity. The transferrin adsorption is accompanied by the removal of the ZnS shell, resulting in evolving two different metal–protein conjugated forms. On the other hand, proteinization in real-serum environment occurs without binding to major transport proteins, the QDs also lose their the shell (the higher the dose the longer is the time they stay unbroken). The concomitant changes in migration behavior can be attributed to interactions with serum proteins other than albumin and transferrin. Speciation information provided by CE-ICP-MS may shed light on the mechanism of QD delivery to the target regions of the body.
Introduction
Single crystal semiconductor nanocrystals, or quantum dots (QDs), are identified as one of the fastest growing products for fluorescence biosensing and bioimaging applications (see ref. 1–5 to mention a few). However, little is known about the behavior of QDs in vivo. Given that they are systematically introduced intravenously, the interactions with blood components will govern the stability, safety, circulation, delivery, cellular uptake and organ distribution of the QDs.6,7 Perhaps the most critical among these issues is the targeting capability of QDs.8 Because of nonspecific binding and difficulty of intracellular delivery only a few percent of the administered dose can be accumulated in the desired cells. Simply administering a larger dose to offset the targeting inefficiency of QDs would barely improve therapeutic efficacy but rather pose toxic side effects. In order to better understand the factors that affect the delivery mechanism, it is particularly important to study the interactions that exist between QDs and plasma proteins.The analytical methodology used for probing nano-bio interactions and characterization of the protein corona formed on the QD surface is of a wide variety.9–11 For the QD–protein systems, fluorescence measurements find the most frequent application (due to native fluorescence of both QDs and proteins), in different measurement modes or following capillary electrophoresis (CE) separation (see the ESI, Table S-1† for a summary of recent research highlighting the methods in use). Several alternative spectroscopy methods, such as photoluminescence, circular dichroism, Fourier transform infrared, or ultraviolet (UV)-visible absorption spectroscopy, have also been utilized. As can be seen from Table S-1,† the binding information acquired includes stoichiometry, association constants and thermodynamic parameters, types and number of binding sites, protein conformational changes upon association with QDs, etc. However, few of the studies on the interaction between QDs and plasma proteins have been advanced for testing more than a single protein in a mixture with QDs and even less on the real plasma/serum environment. There is no need to emphasize that only such a binding scenario is true to a real-life situation, where the whole proteome as well as low molecular-mass matrix constituents will compete for binding to the QD surface. The pointed-out shortcoming ultimately devaluates the bulk of results obtained. A few exceptions to mention here are: a spectroscopic study by Morgner et al.,12 who could not find a clear trend of specific interactions of plasma components with the QDs, some preliminary data attained by CE with fluorescence detection,13 demonstrating that even after 24 h after introduction into plasma or blood the QD signal is still observable, and a contribution,14 which is biokinetic in character in which the concentration (but not the speciation) of QDs in plasma samples taken from mice was determined using inductively coupled plasma mass spectrometry (ICP-MS).
The objective of this work was, therefore, to develop a hyphenated separation and detection platform for quantitative characterization of the speciation of CdSeS/ZnS QDs in human serum and assessment of the impact of serum proteins on QDs on their way from the point of administration to the cell. To meet this objective, CE was used for the separation of QDs and their protein conjugates and was combined with ICP-MS to specifically and sensitively detect the metal-containing nanosized species of interest. It is worth mentioning that several research groups have employed the CE method in the bioconjugation studies of QDs (see Table S-1† and a recent review15). However, the use of common UV or fluorescence detection appears to be a limitation for performing nano-bio analysis on real-world samples. In the current study, CE-ICP-MS is introduced for the first time with this aim in mind. The developed platform was first tested by using the mixtures of QDs with individual serum proteins and their combinations (at physiological concentration ratio) to demonstrate the robustness with respect to possible analyte aggregation or adsorption. Such a step-by-step approach also enabled the acquisition of additional information on revealing the biotransformations of QDs in serum. The latter task was facilitated by simultaneous recording of 111Cd (core) and 66Zn (shell) isotopes. Finally, the method's feasibility was verified by analyzing QDs exposed to human serum via ex vivo incubation.
Experimental
Materials
The core/shell type CdSeS/ZnS QDs (with the alloyed core) functionalized with 3-mercaptopropionic acid (at 6 nm in nominal diameter, 1 mg L−1 according to the producer) were obtained from Sigma-Aldrich (St Louis, USA) and stored in the dark at 4 °C prior to analysis. All protein standards (lyophilized powders, >97%) and human serum (from human male AB plasma; total protein 40–90 g L−1), as well as three buffer types based on Na2HPO4–NaH2PO4, 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid, or HEPES, and piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) were also from Sigma-Aldrich. Ultrapure Milli-Q water was obtained from a Millipore Elix 3 apparatus (Saint-Quentin, France) and used throughout.Instrumentation
Analyses were performed on a HP3DCE system (Agilent Technologies, Waldbronn, Germany) coupled to a 7500a ICP mass spectrometer (Agilent Technologies, Tokyo, Japan). Polyimide-coated fused-silica capillaries (i.d. 75 μm; o.d. 375 μm; length 70 cm) were purchased from CM Scientific Ltd (Silsden, UK). The liquid-introduction interface was based on a model CEI-100 nebulizer (CETAC, Omaha, USA) equipped with a low-volume spray chamber and a cross-piece to merge the sheath liquid flow. Electrical circuit of the CE was completed via a grounded platinum wire. The electrolyte buffer diluted 10 times and containing 20 μg L−1 Ge was used as the make-up solution. The mass isotopes of 111Cd, 66Zn, and 57Fe were monitored in order to observe the speciation changes upon binding of QDs with serum proteins (holo-transferrin in the case of 57Fe). The signal of 72Ge was recorded to control stability of the CE flow and hyphenation performance, as well as the efficiency of nebulization. Instrumental control and data analysis were performed using Agilent ChemStation software. Operation conditions of the optimized CE-ICP-MS setup are summarized in Table 1.| CE system | |
| Capillary | Fused silica capillary, I.D. 75 μm, O.D. 375 μm, length 70 cm |
| Capillary electrolyte | HEPES 20 mM, pH 7.4 |
| Voltage | 15 kV |
| Temperature | 37 °C |
| Current | 9–11 μA |
| Sample injection | Hydrodynamic, 50 mbar, 6 s |
| ICP-MS system | |
| RF power | 1380 W |
| Sample depth | 6.7 mm |
| Plasma gas | 15.0 L min−1 |
| Nebulizer gas flow | 0.9 L min−1 |
| Monitored isotopes | 111Cd, 66Zn, 57Fe, 72Ge |
The protocols for capillary initialization and pre- and between-run conditioning are described elsewhere.16 The temperature of the capillary cassette was set at 37 °C (physiological temperature). The samples were introduced by applying 20–50 mbar pressure for a specified time. The applied voltage for carrying-out the separation was in the range 5–30 kV. The pH of the background electrolyte was adjusted to 7.4 by adding 1 M NaOH. All solutions to be introduced into the capillary were filtered through 0.2 μm syringe filters (Carl Roth, Karlsruhe, Germany).
The samples were incubated at 37 °C in a WB 22 thermostat (Memmert, Schwabach, Germany). For ultrafiltration experiments, an MPW-350R centrifuge (JW Electronic, Warsaw, Poland), operating at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, and Amicon Ultracel filters (10 kDa cut-off; Merck Millipore, Molsheim, France) were employed.
000 rpm, and Amicon Ultracel filters (10 kDa cut-off; Merck Millipore, Molsheim, France) were employed.
Determination of Cd in QD stock suspension
The stock suspension after 2500 times dilution with 2% nitric acid was subjected to analysis following direct nebulization into the ICP-MS (111Cd) and was quantified against an external calibration curve using Y (89Y) as an internal standard. The concentration of Cd in the stock suspension was determined at 8.77 (±0.03) × 10−4 mol L−1 (n = 10).Sample preparation
Dilution of the QD suspension to the desired concentration (four levels, from 0.585 to 1.17 μmol L−1 Cd) was done with 10 mM phosphate buffer (pH 7.4) containing 100 mM NaCl. Such a concentration range was chosen to approximate a dose of 5 nmol Cd per mouse (∼22 g) used in in vivo studies on the biodistribution of QDs.14 This dose was recalculated taking into account the average mass of the human body, the volume of blood and dilution of samples (10 times) to give 0.32 μmol L−1 Cd. It is important to note that while other type of QDs were used in the study by Chen et al.,14 the CdSeS/ZnS QDs also showed a promising cytotoxicity in in vitro tests on the A549 cancer line.17An aliquot of QD stock solution was added to the individual protein solution in a 10 mM phosphate buffer (pH 7.4) containing 100 mM NaCl (final concentration of albumin and transferrin 4.5 or 0.3 g L−1, respectively, that corresponds to 10-fold diluted serum). The same concentrations of both proteins were maintained in their mixtures used for proteinization of QDs. For mixtures of holo- and apo-transferrin (30/70, w/w), their total concentration was maintained at 0.3 g L−1. All the mixtures were incubated at 37 °C for a specified period of time prior to sample introduction into the CE-ICP-MS system. In the case of affinity CE mode, the capillary was filled with a 0.3 g L−1 transferrin solution in background electrolyte, while the sample contained only QDs (0.877 μmol L−1 Cd).
The samples of QDs mixed with 10 times diluted human serum were incubated and analyzed as before. None of the preparations showed visible signs of particle aggregation regardless of the amount of QDs contained (0.585, 0.675, 0.877, or 1.17 μmol L−1 Cd). Additionally, the serum was ultrafiltered (14000 rpm, 4 °C, 30 min) though a 10 kDa cut-off filter. Both the filtrate and high molecular-mass fraction (isolated by reverse ultrafiltration) were mixed with QDs (final concentration 0.675 μmol L−1 Cd) and then incubated and analyzed similarly to the whole serum samples. As a blank sample 10 times diluted serum was analyzed before each experiment, but no Zn or Cd signals were detected in electropherograms. Furthermore, the sample containing only QDs was analyzed between analyses of QD–serum mixtures to carry out the control experiments.
Results and discussion
Optimization of CE conditions
In order to optimize the separation efficiency and resolution and to attain quantitative elution of QDs and their protein conjugates from the separation capillary, several experimental parameters were evaluated. First, three different electrophoretic buffers were tested. Although previous studies have suggested that background electrolytes with pH 9–11 ensure satisfactory migration behavior of free and conjugated nanoparticles,15 the buffers which feature a marked buffering capacity around the physiological pH, i.e. pH 7.4, were preferred here. This is due to the fact that such buffer conditions will secure the protein conjugates formed in a real biological system from pH-induced changes during electrophoresis. It was determined that among the three buffer systems under investigation (see the Experimental section), tested at identical concentrations (10 mM), the highest peak efficiency, signal intensity, and current stability were obtained with the HEPES buffer solution (see the ESI, Fig. S-1,† for more details). The effect of the HEPES concentration on the peak area was examined in the range of 10–60 mM. The results shown in the ESI, Table S-2† suggest that a concentration of 20 mM represents the optimal conditions for QD detectability. Accordingly, 20 mM HEPES, pH 7.4 was chosen for the following investigations.The next operational parameter optimized was the applied voltage, affecting the migration times and hence the resolution. Higher voltages produced less broadened peaks over a shorter time frame (data not shown), and the separation ability of the CE-ICP-MS system was found to be optimal at 15 kV (generating a current of 10 μA). Sample loading was investigated in order to obtain the highest signals without impairing capillary recovery. It was noticed that the loading is limited by the adsorption of both nanoparticles and excessive proteins in the sample on capillary walls, leading to nonquantitative recovery (<90% QDs) at above 300 mbar s. Separations with loadings lower than this value were not studied as these conditions would have an adverse effect on detectability. Consequently, the experiments described below were performed with a separation voltage of 15 kV and sample loading at 50 mbar for 6 s.
Analytical figures of merit of CE-ICP-MS
Results collected in Table 2 indicate an acceptable precision of the optimized CE-ICP-MS assay, with relative standard deviations (RSDs) of the 111Cd peak area of 5.2% and 8.0% for intraday and interday measurements, respectively. Such precision thresholds are in good agreement with the data of other CE procedures for the quantification of QDs based on UV-vis and florescence detection.18,19 With the ICP-MS detector, a linear response between the peak area and different QD concentrations (0.44–7.04 μmol L−1 Cd) was obtained with a correlation coefficient better than 0.99 (see Fig. S-2†). The detection limit of free QDs was 7.5 × 10−9 mol L−1 Cd which compared favorably to other methods cited in Table S-1.† As shown in the previous section, quantitative recovery from the separation capillary and interface system can be a challenge. Mass loss and sample recoveries were characterized via comparing the ICP-MS signals obtained after electrophoresis and pressure-driven elution (at 50 mbar). This procedure was already successfully implemented and validated in CE-ICP-MS of another class of metal species, metal complexes,20 and showed reasonable results for Au nanoparticles.16 Recoveries on the order of 90.0–96.2% for the nanoparticulate species were deemed satisfactory for systematic studies of QD–protein interaction.Interaction with individual plasma proteins
It stands to reason that albumin, the most abundant serum protein, has received an increased amount of attention in binding studies of QDs (though often tested is the protein obtained from bovine serum; see Table S-1†). The majority of research efforts have been focused on the equilibrium aspects of the interaction between albumin and QDs, whereas the kinetics of the formation of the albumin adsorption layer and possible changes in the composition of nanoparticles upon proteinization remain unexplored. The formation of the albumin corona, having apparently a biphasic kinetics, was clearly seen in our experiments (Fig. 1a; data obtained with the other three concentrations of QDs have a similar character and are not shown for the sake of conciseness). While after 30 min of incubation only about 4% of nanoparticles were converted into the protein-covered form, no peak of free nanoparticles was recorded already after 1 h. Thereafter, the albumin conjugate stayed virtually intact till 20 h of observation, which indicates the formation of a thermodynamically stable nanostructure. A two-step character of QD conjugation was also observed in the case of denatured bovine serum albumin.21 However, conversely, the authors found the first initial stage much faster than the following saturating stage.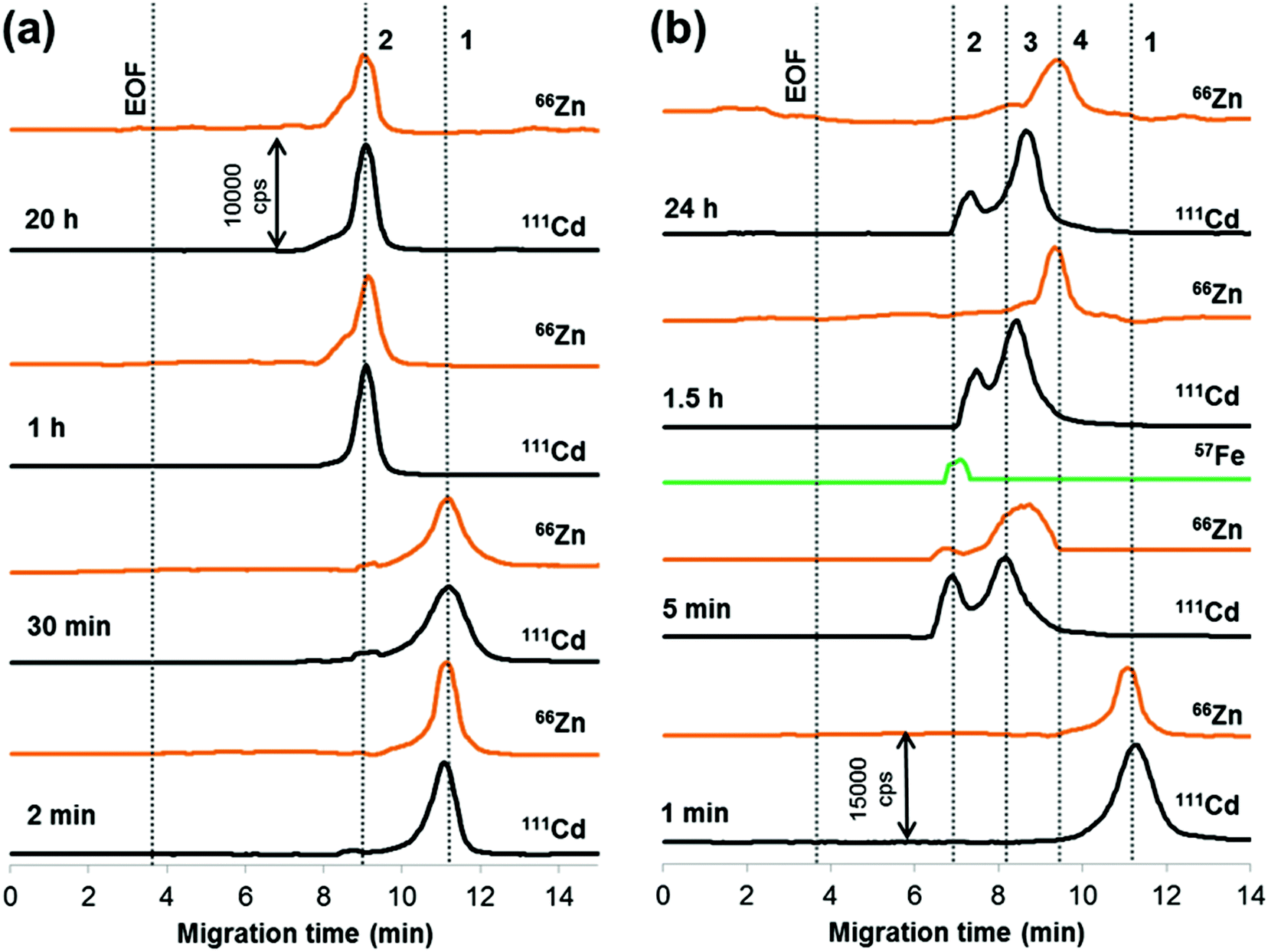 | ||
| Fig. 1 Electropherograms illustrating the progress in conjugation between QDs and (a) albumin and (b) transferrin. Sample: (a) 0.585 μmol L−1 Cd, 4.5 g L−1 albumin; (b) 0.877 μmol L−1 Cd, 0.3 g L−1 transferrin in 10 mM phosphate buffer (pH 7.4), 100 mM NaCl. Peak assignment: (a) 1 – QDs; 2 – QD–albumin conjugate; (b) 1 – QDs; 2 – Cd–holo-transferrin conjugate; 3 – Cd–apo-transferrin conjugate; 4 – zinc conjugate. See Table 1 and Experimental section for CE-ICP-MS conditions. | ||
It is important to mention that association with albumin, as well as with essentially any biomolecule, greatly increases the size of QDs.14 Consequently, the effective electrophoretic mobility decreases and the QDs emerge out of the separation capillary (moving toward the anode placed in our case in the interface) at a notably shorter time (9.1 versus 11.2 min). Another interesting observation is that albumin binding does not impair the integrity of QDs, the ZnS shell remains attached to the core and thus provides a ‘pier’ to the protein molecules. This can be judged from the similar position of 111Cd and 66Zn signals for the conjugate peak (see peak 2 in Fig. 1a).
There is only a single published account14 known to the authors on QD–transferrin binding. This circumstance gave us occasion to investigate this conjugation system in more detail. It was assumed that for transferrin, existing in human blood in two forms: iron-free or apo-transferrin (about 70%) and iron-saturated, holo-transferrin,22 interaction with QDs may give rise to two types of conjugates. This indeed takes place as already after 5 min of incubation of nanoparticles with human serum transferrin (diluted with 10 mM phosphate buffer, pH 7.4, 100 mM NaCl), two peaks are seen in a cadmium specific electropherogram at migration times much smaller than that of the unbound QDs (Fig. 1b). As indicated by the ESI†57Fe-trace, the early migrating peak is due to binding to holo-transferrin. In order to make the peak assignment more unambiguous, a mixture of holo- and apo-transferrin (30/70, total concentration of transferrin 0.3 g L−1) was used for conjugation with QDs (incubation time 1.5 h). The same two peaks at 7.0 and 8.2 min were recorded (data not shown), as in Fig. 1b.
Compared with albumin, two features are distinctive for the binding behavior of transferrin. First, the formation of the transferrin corona proceeds much faster, notably with a similar rate for both protein forms (the apparent rate constants for holo- and apo-transferrin are 2.8 × 10−4 and 2.3 × 10−4 min−1, respectively). This allowed us to explore the QD–transferrin system using affinity CE mode, where the capillary filled with a solution of transferrin in the background electrolyte (see the Experimental section for more details) served as a microreactor.23 Results obtained upon introduction of the QD sample (0.877 μmol L−1 Cd) were essentially the same as for the mixture of QDs and transferrin incubated prior to the CE analysis. This implies that the composition and the structure of the corona are equilibrated during electrophoresis, i.e., on the timescale of a typical CE run.
Another type of distinction is that in contrast to albumin attachment, binding of transferrin tends to release the ZnS shell which becomes completely disintegrated from the core after 1.5 h of interaction (see peak 4 in Fig. 1b). From the peak shape and migration time, it can be assumed that the released ZnS species has a compact, spherical form and is not ‘naked’ but covered with protein. The QD breakdown has a certain influence on the Cd–transferrin conjugates that are subjected to alterations in migration times (enlarging) and the ratio of peak areas (toward a lower proportion of the holo-transferrin conjugate). The latter observation is detailed in Fig. S-3, ESI.† The CE-ICP-MS analysis of samples with varying QD concentration revealed the same conjugate alterations. However, the time taken for the shell removal appears to be a function of the applied dose of QDs, as shown in Fig. 2. At the highest concentration of nanoparticles tested (1.17 μmol L−1), it takes them about 5 h to have the ‘coat’ of ZnS replaced by the two transferrin forms.
 | ||
| Fig. 2 Effect of the concentration of QDs on their disintegration in the course of binding of transferrin. | ||
Concurrent binding to albumin and transferrin
In view of the marked differences in binding behavior of albumin and transferrin that may bear a competitive character in serum and to test the suitability of the method to more complex samples, we have examined a binding scenario where both proteins exist in a mixture with QDs. Concentrations of albumin and transferrin were adjusted to average concentrations in 10-fold diluted human serum. From the results obtained (as summarized in Fig. S-4†), it is most noteworthy that under equilibrium conditions (about 7 h) conjugation takes place with both proteins and, in addition, the ZnS shell remain attached to the core. Therefore, it can be inferred that albumin, being in a 15-molar surplus over transferrin in the reaction mixture and having in general higher affinity to QDs,14 shields the QDs from disintegration. Perhaps this effect also explains why no formation of the QD conjugate with apo-transferrin was observed.Speciation in human serum
Apparently, when the QDs encounter the entire serum proteome, the formation of the protein corona will be a multiplex process influenced by the abundance and binding affinity of individual proteins, as is the general case of metal-based nanoparticles.7,24–26 Typical metal-specific profiles acquired for QDs exposed to human serum for different periods of time are shown in Fig. 3. Obviously, the speciation alterations of QDs occur in a different way than in any of the modelling systems examined above, with albumin and transferrin taking no part in the protein corona. This finding is not considered surprising, because a few tens of plasma proteins, commonly existing in the hard corona,27 rarely correspond to the most abundant proteins in plasma and are not necessarily those with the highest individual affinity to the nanoparticle surface.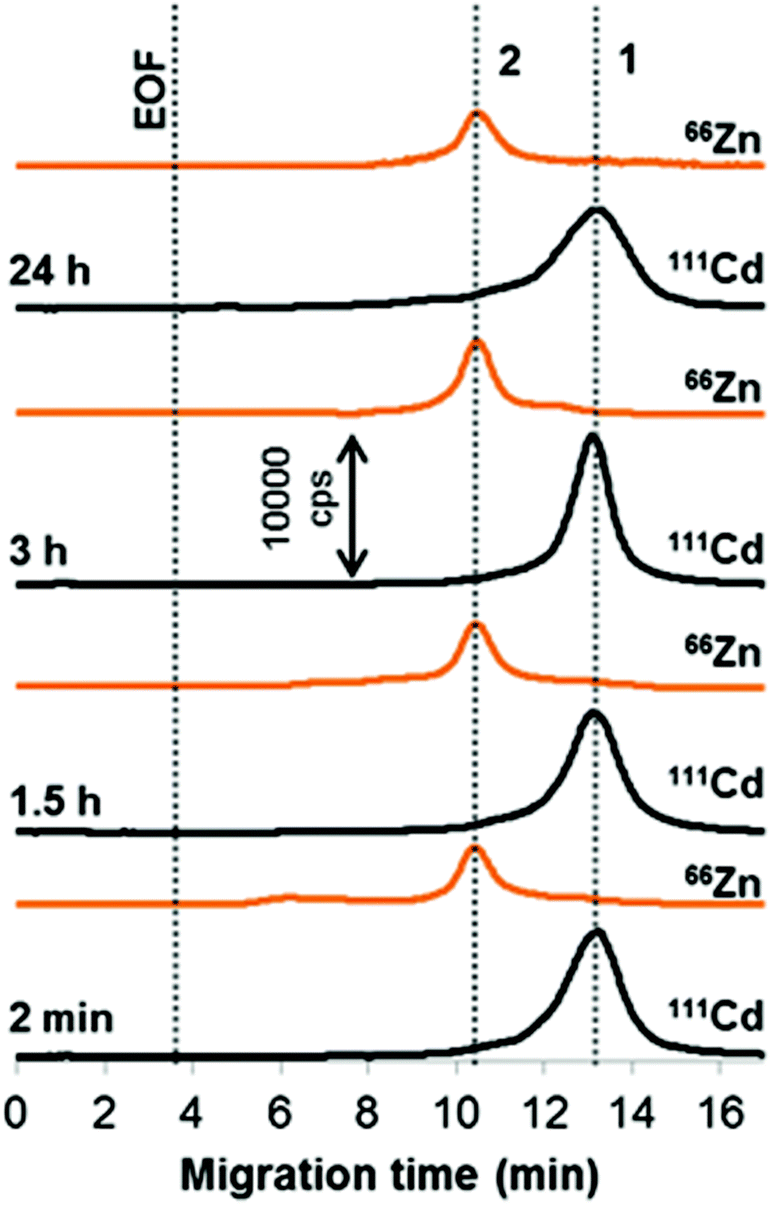 | ||
| Fig. 3 Biotransformation of QDs in human serum. Sample: QDs (0.585 μmol L−1 Cd) in 10-times diluted serum. Peak assignment: core (1) and shell (2) species originating from QDs. See Table 1 and Experimental section for CE-ICP-MS conditions. | ||
Shortly after mixing with serum the QDs lose the shell and become predominately converted into a novel Cd species. The latter migrates at a definitely longer time (13 min) than bare QDs (11.2 ± 0.2 min as determined in the course of the optimization trials), which indicates its greater charge-to-size ratio. The reason for such migration behavior is that the species turn out to be smaller due to the shell release, being bound by some of the serum medium components (but still smaller than the original QDs). In another study,14 the QDs were found to be stable in mice plasma, with an intact core–shell structure, that can be attributable to the different nature of their shell (silica) and functionalization (hydroxyl groups), suggesting that not every QD is alike!
To clarify the role of different (in molecular mass) serum constituents in forming the corona, we carried out the same binding experiments but with two separate fractions obtained after serum ultrafiltration (see the Experimental section). In the case of high molecular-mass fraction (>10 kDa), the speciation changes were very similar to those in the whole serum. On the contrary, when the particles were mixed with a low molecular-mass fraction, only a single peak of intact QDs was recorded (at least till 2 h). All these observations prove that it is a protein (but not albumin or transferrin; see above) that renders the disintegrated QDs (peak 1 in Fig. 3). There exist some indications in the literature that serum histidine-rich glycoprotein, possessing a high complexing ability toward Zn(II),12 or γ-globulin whose attraction to QDs is higher than that of transferrin,14 could be responsible for this (and possibly for the proteinization of the shell). However, the matter is left open, if the same protein(s) constitutes the coronas of the CdSeS core and ZnS shell.
Likewise unanswered and requiring an additional investigation is the question whether losing the shell would evolve the toxicity of a given type of QDs (the ZnS shell is designed to prevent a possible release of toxic cadmium ions to the bloodstream) or if the protein shell structure is able to take over the protecting function. In this context, it is worthwhile to note that for samples with higher QD content (0.675, 0.877, and 1.17 μmol L−1 Cd), the stability of QDs increases, with no observable detachment up to about 2.5 h at the highest dose (Fig. S-5;†cf., Fig. 2). Therefore, an appropriate dosage may possibly resolve the toxicity challenge and ensure the safe delivery of QDs (yet in the unbound state) to the desired point. This fact needs to be taken into account when devising clinical diagnostic applications.
Conclusions
Our work represents the first example of systematic investigation on the interaction of QDs and serum proteins using CE-ICP-MS. As shown above, this hyphenated instrumental platform satisfies many expectations for a simple, highly sensitive and high-throughput monitoring tool for the characterization of the QD–protein conjugates in blood compartments. The binding response can be obtained within minutes after direct sample introduction, virtually unaffected by the analytical system, and specifically for different core–shell metals, as well as metal-containing proteins. Despite revealing some unusual binding features, such as nanoparticle disintegration and the conjugation of the released core and shell with minor serum protein(s), our measurements fit the general picture of QD behavior within biological media. Furthermore, there is every reason to believe that CE-ICP-MS can be applied to portraying other types of QDs, variable in core/shell chemistry, functionalization, and size, that are interacting with proteins.However, to become a conventional nano-bio approach that will attract the interest of a growing number of nanoscientists, the CE-ICP-MS methodology requires a number of add-ons to help meet new challenges. Of several key problems that need to be addressed, the problem of providing more detailed information on the composition of biomolecular corona ranks first. In this regard, the implementation of molecular-specific detectors, directly or following a shotgun proteomic procedure, will be our next task.
Acknowledgements
Financial support provided by the National Science Centre, Poland (PRELUDIUM, grant no. 2013/11/N/ST4/01480) and the Russian Foundation of Basic Research (grant no. 16-03-00492) is gratefully acknowledged.References
- A. M. Coto-García, E. Sotelo-González, M. T. Fernández-Argüelles, R. Pereiro, J. M. Costa-Fernández and A. Sanz-Medel, Anal. Bioanal. Chem., 2011, 399, 29–42 CrossRef PubMed.
- H.-C. Huang, S. Barua, G. Sharma, S. K. Dey and K. Rege, J. Controlled Release, 2011, 155, 344–357 CrossRef CAS PubMed.
- J. Li and J.-J. Zhu, Analyst, 2013, 138, 2506–2515 RSC.
- T. R. Pisanic, Y. Zhang and T. H. Wang, Analyst, 2014, 139, 2968–2981 RSC.
- A. B. Chinen, C. M. Guan, J. R. Ferrer, S. N. Barnaby, T. J. Merkel and C. A. Mirkin, Chem. Rev., 2015, 115, 10530–10574 CrossRef CAS PubMed.
- B. Wang, W. Feng, Y. Zhao and Z. Chai, Metallomics, 2013, 5, 793–803 RSC.
- Q. Mu, G. Jiang, L. Chen, H. Zhou, D. Fourches, A. Tropsha and B. Yan, Chem. Rev., 2014, 114, 7740–7781 CrossRef CAS PubMed.
- Y. Choi, K. Kim, S. Hong, H. Kim, Y.-J. Kwon and R. Song, Bioconjugate Chem., 2011, 22, 1576–1586 CrossRef CAS PubMed.
- K. E. Sapsford, K. M. Tyner, B. J. Dair, J. R. Deschamps and I. L. Medintz, Anal. Chem., 2011, 83, 4453–4488 CrossRef CAS PubMed.
- Z. W. Lai, Y. Yan, F. Caruso and E. C. Nice, ACS Nano, 2012, 6, 10438–10448 CrossRef CAS PubMed.
- A. L. Capriotti, G. Caracciolo, C. Cavaliere, V. Colapicchioni, S. Piovesana, D. Pozzi and A. Laganà, Chromatographia, 2014, 77, 755–769 CAS.
- F. Morgner, S. Stufler, D. Geißler, I. L. Medintz, W. R. Algar, K. Susumu, M. H. Stewart, J. B. Blanco-Canosa, P. E. Dawson and N. Hildebrand, Sensors, 2011, 11, 9667–9984 CrossRef CAS PubMed.
- C. Hao, Adv. Mater. Res., 2012, 457–458 Search PubMed.
- Z. Chen, H. Chen, H. Meng, G. Xing, X. Gao, B. Sun, X. Shi, H. Yuan, C. Zhang, R. Liu, F. Zhao, Y. Zhao and X. Fang, Toxicol. Appl. Pharmacol., 2008, 230, 364–371 CrossRef CAS PubMed.
- S. S. Aleksenko, A. Y. Shmykov, S. Oszwałdowski and A. R. Timerbaev, Metallomics, 2012, 4, 1141–1148 RSC.
- M. Matczuk, K. Anecka, F. Scaletti, L. Messori, B. K. Keppler, A. R. Timerbaev and M. Jarosz, Metallomics, 2015, 7, 1364–1370 RSC.
- I. Grabowska-Jadach, M. Haczyk, M. Drozd, A. Fischer, M. Pietrzak, E. Malinowska and Z. Brzózka, Electrophoresis, 2016, 37, 425–431 CrossRef CAS PubMed.
- T. Tang, J. Deng, M. Zhang, G. Shi and T. Zhou, Talanta, 2016, 146, 55–61 CrossRef CAS PubMed.
- X. Huang, J. Weng, F. Sang, X. Song, C. Cao and J. Ren, J. Chromatogr., A, 2006, 1113, 251–254 CrossRef CAS PubMed.
- A. R. Timerbaev, K. Pawlak, S. S. Aleksenko, L. S. Foteeva, M. Matczuk and M. Jarosz, Talanta, 2012, 102, 164–170 CrossRef CAS PubMed.
- J. Wang, J. Li, Y. Teng, W. Hu, H. Chai, J. Li, C. Wang, L. Qui and P. Jiang, J. Nanopart. Res., 2014, 16, 1–7 Search PubMed.
- P. T. Gomme, K. B. McCann and J. Bertolini, Drug Discovery Today, 2005, 10, 267–273 CrossRef CAS PubMed.
- A. R. Timerbaev, L. S. Foteeva, K. Pawlak and M. Jarosz, Metallomics, 2011, 3, 761–764 RSC.
- E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh and V. Puntes, ACS Nano, 2010, 4, 3623–3632 CrossRef CAS PubMed.
- M. Lindquist, J. Stigler, T. Cedervall, T. Berggard, M. B. Flanagan, I. Lynch, G. Elia and K. A. Dawson, ACS Nano, 2011, 5, 7503–7509 CrossRef PubMed.
- J. Lazarovits, Y. Y. Chen, E. A. Sykes and W. C. W. Chan, Chem. Commun., 2015, 51, 2756–2767 RSC.
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an00276e |
| This journal is © The Royal Society of Chemistry 2016 |