
Surfactant modulated aggregation induced enhancement of emission (AIEE)—a simple demonstration to maximize sensor activity†
Rahul
Bhowmick
a,
Abu
Saleh Musha Islam
a,
Atul
Katarkar
b,
Keya
Chaudhuri
b and
Mahammad
Ali
*a
aDepartment of Chemistry, Jadavpur University, 188, Raja Subodh Chandra Mallick Rd, Kolkata, West Bengal 700032, India. E-mail: m_ali2062@yahoo.com
bDepartment of Molecular & Human Genetics Division, CSIR-Indian Institute of Chemical Biology, 4 Raja S.C. Mallick Road, Kolkata-700032, India
First published on 5th November 2015
Abstract
A new type of easily synthesized rhodamine-based chemosensor L3, with potential NO2 donor atoms, selectively and rapidly recognizes Hg2+ ions in the presence of all biologically relevant metal ions and toxic heavy metals. A very low detection limit (78 nM) along with cytoplasmic cell imaging applications with no or negligible cytotoxicity indicate good potential for in vitro/in vivo cell imaging studies. SEM and TEM studies reveal strongly agglomerated aggregations in the presence of 5 mM SDS which turn into isolated core shell microstructures in the presence of 9 mM SDS. The presence of SDS causes an enhanced quantum yield (φ) and stability constant (Kf) compared to those in the absence of SDS. Again, the FI of the [L3–Hg]2+ complex in an aqueous SDS (9 mM) medium is unprecedentedly enhanced (∼143 fold) compared to that in the absence of SDS. All of these observations clearly manifest in the enhanced rigidity of the [L3–Hg]2+ species in the micro-heterogeneous environment significantly restricting its dynamic movements. This phenomenon may be ascribed as an aggregation induced emission enhancement (AIEE). The fluorescence anisotropy assumes a maximum at 5 mM SDS due to strong trapping (sandwiching) of the doubly positively charged [L3–Hg]2+ complex between two co-facial laminar microstructures of SDS under pre-miceller conditions where there is a strong electrostatic interaction that causes an improved inhibition to dynamic movement of the probe-mercury complex. On increasing the SDS concentration there is a phase transition in the SDS microstructures and micellization starts to prevail at SDS ≥ 7.0 mM. The doubly positively charged [L3–Hg]2+ complex is trapped inside the hydrophobic inner core of the micelle which is apparent from the failure to quench the fluorescence of the complex on adding 10 equivalents of H2EDTA2− solution but in the absence of SDS it is quenched effectively.
Introduction
In the nature, there is a continuous drive of a web of chemical reactions of elements, ions and molecules.1 Living organisms and their environment interact with each other through a diverse array of reactions spanning from covalent to non-covalent interactions like electrostatic, hydrophobic, van der Waals, hydrogen bonding etc.2,3 and significant progress in the design and fabrication of appealing supramolecular assemblies has been achieved by chemists through bottom-up approaches by elegant utilization of these non-covalent interactions, which led to the successful fabrication of optical materials and advanced nano-devices. It involves coupling of structurally different building blocks (ionic pairs) by electrostatic interactions.3 Various combinations between polyelectrolytes, peptides, surfactants and extended rigid organic scaffolds could be employed for creating new material via ionic self-assembly.4–9 Another promising feature of these supramolecular assemblies concerns their emissive behavior in solution and solid states. The assemblies formed thereof may exhibit either aggregation caused quenching (ACQ) or aggregation induced emission enhancement (AIE or AIEE);10–17 the latter shows enhanced fluorescence emission efficiency in an aggregated state as compared to that in a solution state. In this context, the AIEE phenomenon became a key point in developing materials for fluorescent sensors, optoelectronic devices, and cell imaging applications. Up to now, the AIEE mechanism has been observed in silole derivatives,18 1,1,2,2-tetraphenylethene (TPE),19–22 and 1-cyano-trans-1,2-bis-(4-methylphenyl) ethylene (CN-MBE),23,24 which were claimed to be stimuli responsive materials capable of detecting volatile organic vapor, biological polymers, pH changes, explosives, metal ions etc.Fluorescence efficiency greatly depends on a number of factors that maximize the stability of a fluorophore in the excited state and minimize various non-radiative decay processes such as intermolecular interaction,25 intramolecular charge transfer,26,27 and intramolecular torsional and rotational motions.28–30 One of the ways to achieve this is to confine a probe by changing the outside environment which significantly deactivates these non-radiative decay processes and enhances the fluorescence efficiency – a way to develop organic light-emitting diodes for practical applications.31,32 Thus, a number of fluorescent molecules exhibit aggregation-induced emission (AIE) properties by restricting the intramolecular vibrational, rotational, and torsional motion resulting in a turned on or enhanced fluorescence.33 An obvious choice is to use ionic surfactants where electrostatic interactions with a charged fluorescent probe suppress the non-radiative decay and hence enhance the fluorescence emission efficiency. Surfactants generally form micelles, vesicles, and other kinds of aggregates,34 and therefore could be a good choice to modulate the aggregation of ionic fluorescent dyes in water and to adjust the fluorescence intensity. All of these have found applications in biochemistry, analytical chemistry, and photosensitization.35–37 However, only a few reports are available where induced fluorescence enhancement of dye molecules by surfactants occurs.38–42
Again, pollution due to heavy metals like mercury arises due to their wide scale applications in agriculture and industry. It is also widespread in air, water and soil by oceanic and volcanic eruptions, combustion of fossil fuels, gold mining and solid waste incineration and seems to be inescapable.43–45 An example of a highly toxic and widespread pollutant is the water-soluble Hg2+ ion, which can damage the brain, nervous system, kidneys, and endocrine system. In living organisms Hg2+ deactivates many enzymes due to its strong affinity for sulfur in –SH groups which stops or alters metabolic processes.46,47 As a result of the accumulation of Hg2+ in the body, various diseases like prenatal brain damage, serious cognitive and motion disorders, Minamata etc. are common.48
There are several sophisticated analytical instruments like atomic absorption and emission spectroscopy,49 inductively coupled plasma mass spectroscopy (ICP-MS),50 inductively coupled plasma atomic emission spectrometry (ICP-AES),51 voltammetry52etc. which are used for Hg2+ detection, however, most of these methods are either very costly or time-consuming and not suitable for performing assays. However, quick response times, high sensitivity and good selectivity make the fluorescence technique superior to others and has attracted tremendous attention from chemists, biologists and environmentalists.53–59
Special structural features along with good photostability, a high molar extinction coefficient and a longer emission wavelength (550 nm) make rhodamine a good choice for the construction of OFF–ON fluorescent chemosensors51 to avoid background fluorescence below 500 nm.53,57,60
Here, a new rhodamine-based probe with potential NO2 donor atoms has been synthesized and successfully employed for the selective and rapid recognition of toxic Hg2+ ions (Scheme 1) in a 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (H2O:MeCN) medium. It exhibits very rapid chromo- and fluorogenic OFF–ON responses through metal-induced opening of the spirolactam ring. Not only that, in aqueous SDS medium, it was found to exhibit aggregation induced emission enhancement (AIEE).
:2 (H2O:MeCN) medium. It exhibits very rapid chromo- and fluorogenic OFF–ON responses through metal-induced opening of the spirolactam ring. Not only that, in aqueous SDS medium, it was found to exhibit aggregation induced emission enhancement (AIEE).
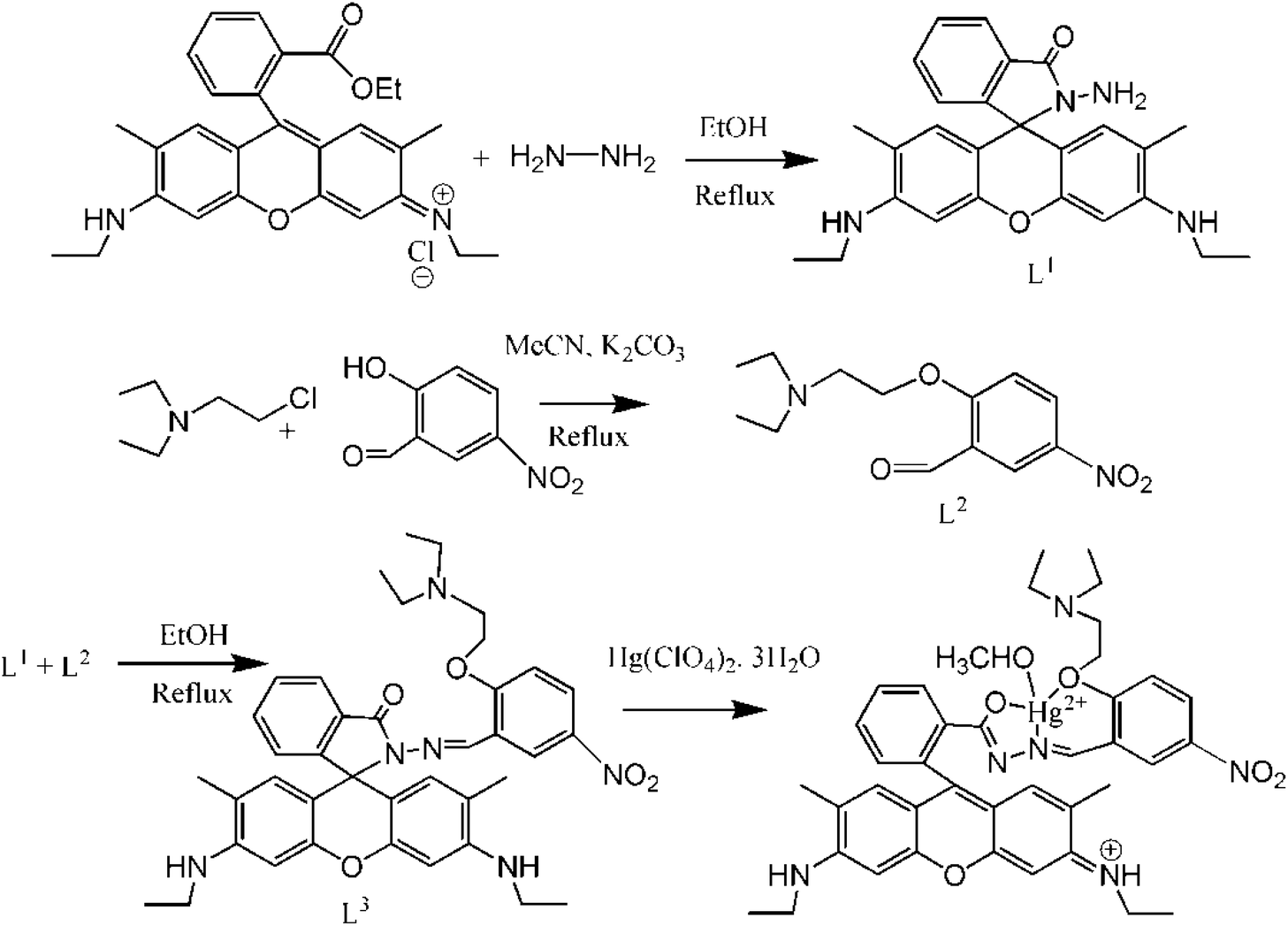 | ||
| Scheme 1 | ||
Experimental section
Materials and instruments
Rhodamine 6G hydrochloride, 2-chloro-N,N-diethylethylamine hydrochloride and metal salts such as perchlorates of Na+, K+, Ca2+, Fe2+, Co2+, Ni2+, Zn2+, Pb2+, Cd2+, Hg2+ and Cu2+ and anions such as SO42−, NO3−, PO43−, S2−, Cl−, F−, Br−, OAc−, H2AsO4−, N3−, ClO4−, PPi, S2O42−, HCO3−, SCN−, CO32−, P2O74− and NO2− were purchased from Sigma-Aldrich and used as received. All solvents used for synthetic purposes were of reagent grade (Merck) unless otherwise mentioned. For spectroscopy (UV/Vis and fluorescence) studies HPLC-grade MeCN and deionized water from MiliQ Millipore were used.UV/Vis absorption spectra were recorded on an Agilent 8453 diode array spectrophotometer. Steady-state fluorescence studies were carried out with a PTI (QM-40) spectrofluorimeter. NMR spectra were recorded on a Bruker spectrometer at 300 MHz. The ESI-MS+ spectra were recorded on a Waters XEVO G2QTof mass spectrometer.
Preparation of Rhodamine 6G hydrazide (L1)
Rhodamine-6G hydrazide was prepared according to a method in the literature.61Preparation of 2-[2-(diethylamino)-ethoxy]-5-nitro-benzaldehyde (L2)
In a typical procedure 5-nitro salicylaldehyde (5 mmol, 0.835 g) was dissolved in dry MeCN (30 mL) to which K2CO3 (6 mmol, 0.3312 g) was added, and the mixture was heated at reflux for 40 min. 2-Chloro-N,N-diethylethylamine hydrochloride (6 mmol, 1.032 g) was then added and reflux was continued for another 5 h. It was then cooled to room temperature and filtered. The filtrate was evaporated to one third of its initial volume and diluted with 40 mL water. The pH of the resulting solution was then adjusted to 4 by the addition of 1 M HCl and extracted with dichloromethane (DCM; 2 × 40 mL). The pH of the aqueous solution was further adjusted to 8 by the addition of 4.0 M Na2CO3 solution and again extracted with DCM (3 × 40 mL). The combined organic phase was dried over anhydrous Na2SO4 and then evaporated to dryness under reduced pressure to yield a reddish yellow solid. The solid product was recrystallized from MeCN/DCM (8:2, v/v) to give L2 as an amorphous solid.
Preparation of L3
2-[2-(Diethylamino)-ethoxy]-5-nitro-benzaldehyde (L2) (1.10 mmol, 0.266 g) in MeOH (10 mL) was added dropwise to a methanolic solution (30 mL) of L1 (1 mmol, 0.464 g) containing 1 drop of acetic acid under hot (50–60 °C) conditions over 30 min and was then stirred for about 6 h at room temperature whereupon a yellow precipitate formed and was collected by filtration. The residue was washed thoroughly with cold methanol to isolate L3 in pure form at 78% yield. The detailed synthetic steps are illustrated in Scheme 1.Analysis
1H NMR (DMSO-d6): δ = 8.83(s, 1 H), 8.31 (s, 1 H), 8.04 (s, 1 H), 7.92 (s, 1 H), 7.58 (m, 2 H), 7.03(d, 1-H), 6.92 (d, 1 H), 6.30 (s, 2 H), 6.16 (s, 2 H), 5.07 (t, 2 H), 3.11(t, 4 H), 2.48 (s, 14 H), 1.82 (s, 6H), 1.18 (t, 6H) ppm (Fig. S1†). IR:![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 1722 cm−1 (spirolactam amide-keto), 1622 cm−1 (–C
= 1722 cm−1 (spirolactam amide-keto), 1622 cm−1 (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) (Fig. S2†). MS (ES+): m/z = 677.4009 [L + H+] (Fig. S3†).
N) (Fig. S2†). MS (ES+): m/z = 677.4009 [L + H+] (Fig. S3†).
Preparation of complex L3–Hg2+
Hg(ClO4)2 (0.272 g, 0.6 mmol) was added to a 10 mL MeCN solution of L3 (0.338 g, 0.5 mmol) and the mixture was stirred for about 30 minutes. It was then filtered and allowed to evaporate slowly at ambient temperature to obtain the crystalline solid product.Analysis: 1H NMR (DMSO-d6): δ = 8.83(s, 1 H), 8.29 (d, 1H), 8.05 (d, 1 H), 8.03 (d, 1 H), 7.60 (m, 2 H), 7.03 (d, 1 H), 6.92 (d, 1 H), 6.35 (s, 2 H), 6.18 (s, 2 H), 5.45 (s, 1 H), 3.12 (m, 4 H), 2.48 (s, 14 H), 1.83 (s, 6 H), 1.18 (t, 6 H) ppm (Fig. S4†). IR: = 1633 cm−1 (spirolactam ring open), 1608 cm−1 (–CN) (Fig. S2†). MS (ES+): m/z = 532.5052 (L3 + Hg2+ + ClO4 + H2O + CH3OH + CH3CN) and 629.4355 (L3 + Hg2+ + (ClO4)3 + (H2O)3 + Li2) (Fig. S5†).
Solution preparation for UV-Vis and fluorescence studies
For both UV-Vis and fluorescence studies, a 1.0 × 10−3 M stock solution of L3 was prepared by dissolving the required amount of ligand in 2 ml MeCN and finally the volume was adjusted to 10 ml by deionized water. In a similar way, a 1.0 × 10−3 M stock solution of Hg2+ was prepared in deionised H2O. A 250 mL 10 mM HEPES buffer solution in 8:2 H2O:MeCN (v/v) was prepared and pH was adjusted to 7.2 using HCl and NaOH. 2.5 ml of this buffer solution was pipetted out into a cuvette to which the required volume of the 1.0 × 10−3 M probe was added to achieve 20 μM and 10 μM final concentrations for UV-Vis and fluorescence titration, respectively. At a regular interval a volume of Hg2+ ions was added incrementally and UV-Vis and fluorescence spectra were recorded for each solution. Cuvettes with a 1 cm path length were used for absorption and emission studies. Fluorescence measurements were performed using a 2 nm × 2 nm slit width.
Preparation of samples for SEM and TEM studies
We prepared 10 mL 1 mM stock solution of the ligand and 10 ml 0.10 M stock solution of SDS. Now, 25 μL of the ligand solution was added to (a) 125 μL and (b) 225 μL SDS solution in 2.5 mL deionized water with stirring for 5 minutes. The concentrations of SDS/L3 in the resulting solutions were (a) 5.0/0.10 mM and (b) 9.0/0.10 mM respectively. The use of higher concentrations of SDS (up to 20 mM) does not show any formation of microstructures. We have prepared another set of solutions (a) and (b) with an appropriate amount of ligand (25 μL) and to each solution 1 equivalent of Hg2+ was added. All of the solutions were aged overnight at room temperature before characterization. The samples in both cases were found to give the best microstructure as analyzed using SEM and TEM.Methods of characterization
The morphologies of the synthesized nano/microstructures were studied using scanning electron microscopy (SEM) with a ZEOL, JSM 8360 operated at an accelerating voltage of 5 kV. Before SEM studies, the samples were vacuum dried on a glass plate and deposited as a thin layer of carbon. Transmission electron microscopy (TEM) studies were performed on a JEOL JEM 2100 HR with EELS operating between accelerating voltages of 80 kV to 200 kV. The sample was deposited on a copper grid and vacuum dried.Steady-state fluorescence and fluorescence anisotropy were measured with a PTI QM-40 spectrofluorometer. Fluorescence anisotropy (r) is defined as:
| r = (IVV − GIVH)/(IVV + 2GIVH) | (1) |
| G = IHV/IHH | (2) |
Cell culture
Human hepatocellular liver carcinoma (HepG2) cell lines were procured from NCCS (Pune). The cells were cultured in DMEM (penicillin-100 μg ml−1, 10% FBS, streptomycin-50 μg ml−1) at 37 °C in a 95% air and 5% CO2 incubator.Cell viability assay
To determine % cell viability, a colorimetric MTT assay of ligand L3 was performed by a method reported earlier.62Cell imaging study
1 × 105 cells were cultured and incubated on a coverslip in a 35 × 10 mm culture dish for 24 h at 37 °C. The cells were treated with 10 μM L3 (prepared by dissolving L3 in a mixed solvent of DMSO:water = 1:9 v/v) and were then allowed to incubate for 45 min at 37 °C. After incubation cells were incubated with MitoTracker® Green FM (Invitrogen) for 30 min to track the cytoplasm followed by washing thrice with 1× PBS. The cells were then counterstained by DAPI (2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride, 4′,6-diamidino-2-phenylindole, used for nuclear staining, Sigma). Fluorescence images of HepG2 cells were taken using a fluorescence microscope (Leica DM3000, Germany) with an objective lens of 40× magnification. Additionally, fluorescence images of HepG2 cells were taken where cells were pre-incubated with 2 μM, 4 μM, 8 μM, 16 μM, or 20 μM Hg2+ for 3 h in three different culture dishes at 37 °C followed by washing thrice with 1× PBS and then incubated with an equimolar amount of ligand L3 (2 μM, 4 μM, 8 μM, 16 μM, or 20 μM respectively) for 30 min at 37 °C. After incubation, cells were washed with 1× PBS three times and fluorescence images were taken. L3 shows intracellular cytoplasmic red fluorescence by forming a complex with Hg2+ against the nuclear counterstain DAPI (blue color) and cytoplasmic stain MitoTracker Green (green color).
Results and discussion
A Schiff base condensation between L1 and L2 in methanol (Scheme 1) under refluxing conditions yields L3, which was thoroughly characterized by 1H-NMR, IR and ESI-MS+ spectroscopy.Absorption and steady-state emission studies
UV-Vis titration with a fixed concentration of L3 (20 μM) and variable concentrations of Hg2+ (0–25.0 μM) at 25 °C in aqueous MeCN (8:2, v/v, HEPES buffer, pH 7.2) showed a gradual development of a new absorption band around 533 nm on addition of Hg2+ (Fig. 1a).
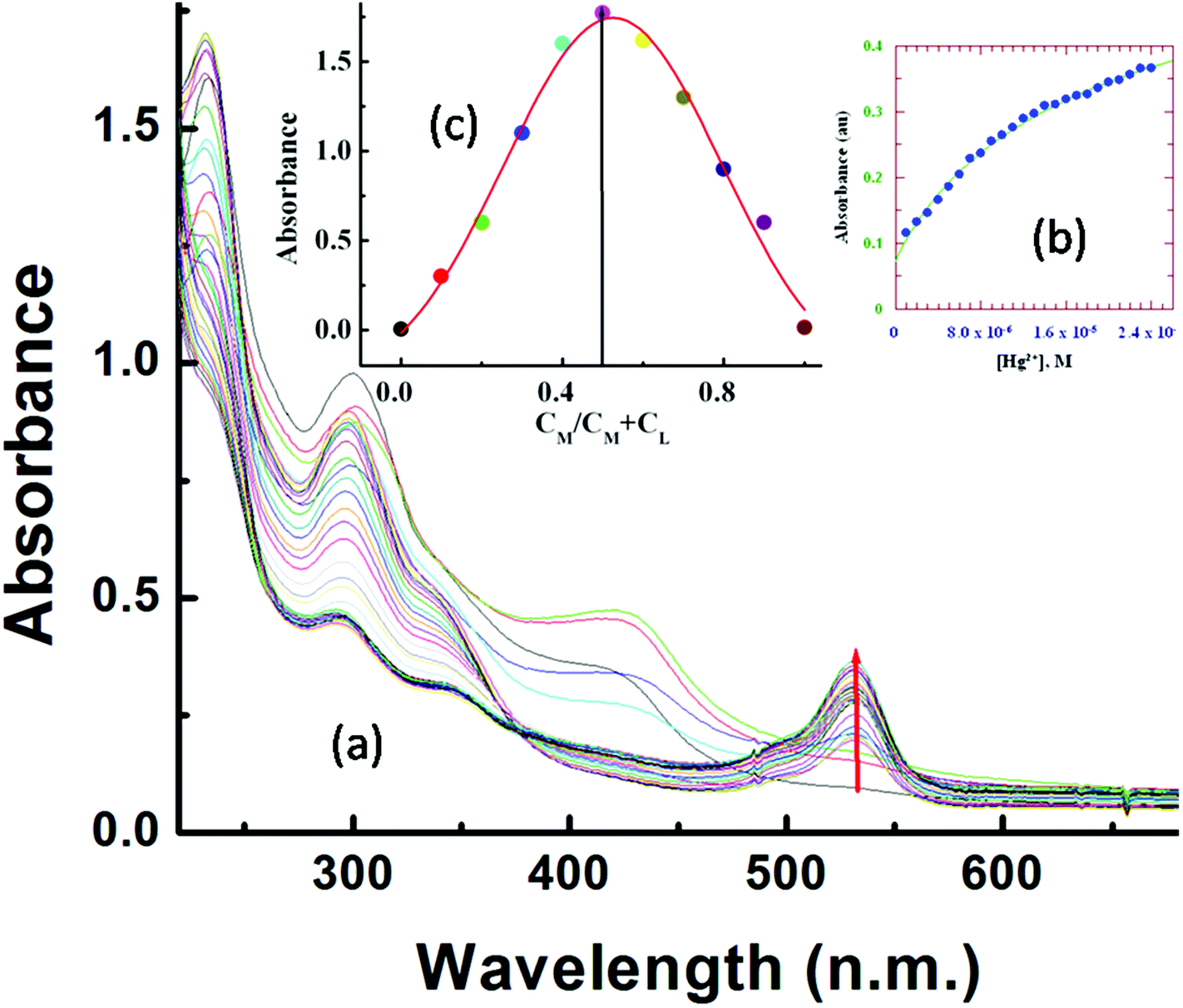 | ||
| Fig. 1 (a) Absorption titration of L3 (20.0 μM) with Hg2+ in MeCN–H2O (2:8, v/v) in HEPES buffer (10 mM) at pH 7.2; (b) Non-linear fit of absorbance vs. [Hg2+] plot at λ = 533 nm; (c) Job's plot for the determination of the composition of the L3–Hg2+ complex. | ||
A plot of absorbance vs. [Hg2+] gives a non-linear curve (Fig. 1b) with decreasing slope and can be analyzed using eqn (3)63 below. The binding constant of the formed L3–Hg2+ complex was determined to be Kf = (2.39 ± 0.49) × 104 M−1.
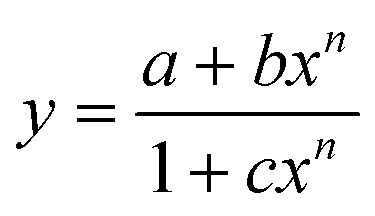 | (3) |
Job's method was employed to determine the composition of the complex which was found to be 1:1 (Fig. 1c). The emission spectra of L3 and its fluorescence titration with Hg2+ were performed in water:MeCN (8:2, v/v) solution with a fixed concentration of L3 (10 μM) (10 mM HEPES buffer, pH 7.2; Fig. 2). On gradual addition of Hg2+ (0–56.0 μM) to the non-fluorescent solution of L3 (10.0 μM), a 316-fold enhancement in fluorescence intensity at 553 nm was observed following excitation at 510 nm, which also suggests an opening of the spirolactam ring in L3 on coordination to the Hg2+ ion.
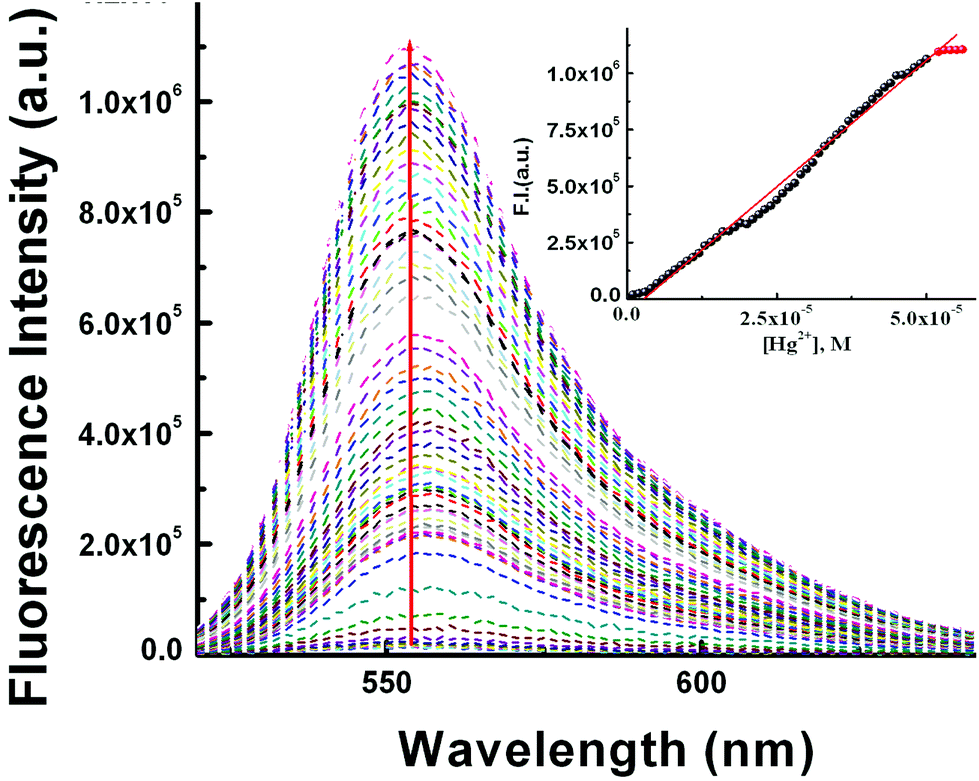 | ||
| Fig. 2 Fluorescence titration of L3 (10.0 μM) in MeCN–H2O (2:8, v/v) in HEPES buffer at pH 7.2 by the gradual addition of Hg2+ with λex = 510 nm and λem = 553 nm. Inset: linear fit plot of FI vs. [Hg2+]. | ||
A plot of FI vs. [Hg2+] gives a straight line up to 50 μM where eqn (3) becomes y = a + b × c × x under the conditions 1 ≫ c × x and the linear dependence of such a plot gives a slope = b × c, where b = fluorescence maximum (Fmax) and c = Kf′ = apparent formation constant. So, slope/Fmax gives Kf′ = (2.02 ± 0.03) × 104 M−1. It is interesting to note that the values of Kf′ obtained separately from absorbance and fluorescence titrations are very close to each other and clearly indicate the internal consistency of our results.
The detection of Hg2+ was not affected by the presence of biologically abundant metal ions like Na+, K+, Ca2+ and Mg2+. Likewise, under identical reaction conditions no significant color or spectral change was observed for transition-metal ions, namely Cr3+, Mn2+, Fe2+, Fe3+, Co2+, Cu2+, Ni2+ and Zn2+, and heavy-metal ions, like Cd2+and Pb2+ and also anions like SO42−, NO3−, PO43−, S2−, Cl−, F−, Br−, OAc−, H2AsO4−, N3−, ClO4−, PPi, S2O42−, HCO3−, SCN−, CO32−, P2O74− and NO2− (Fig. 3).
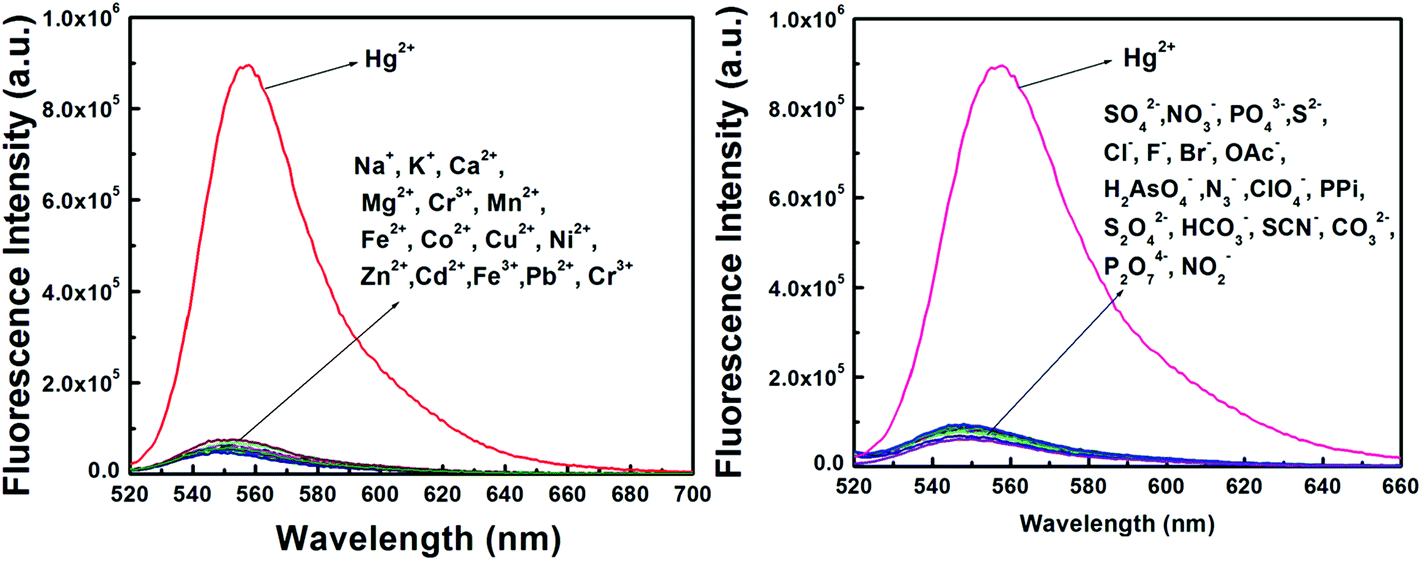 | ||
| Fig. 3 Fluorescence emission induced by different cations and anions. | ||
We have also checked the thiophilicity of Hg2+ ions using cysteine under extracellular conditions. It was observed that the probe undergoes complete fluorescence quenching on addition of thiol (10 μM) to an ensemble of 10 μM L3–Hg2+. This clearly indicates that the probe could be used to detect Hg2+ as well as thiols to a concentration of at least 10 μM which are best viewed in Fig. S6.† The quantum yield (φ) of the free ligand and the L3–Hg2+ complex in pure water in the absence and presence of 9 mM SDS were determined and were found to be: 0.0029 (L3), 0.11 (L3 in the presence of 9 mM SDS), 0.009 (L3 in the presence of 1.2 equivalents of Hg2+) and 0.48 (L3 + 9 mM SDS + 1.2 equivalents of Hg2+ in pure water). The lifetime of the free ligand in the absence and presence of SDS (9 mM) is 0.41 and 3.33 ns, respectively, while that of the L3–Hg2+ complex is 4.51 and 3.31 ns, respectively.
pH stability check
The pH-titration over a wide range of pH (2–12) reveals no obvious fluorescence emission of L3 between pH 4 and 12. However in the presence of Hg2+ it becomes fluorescent between pH 6.5–12 suggesting a convenient application of this probe under physiological conditions (Fig. 4).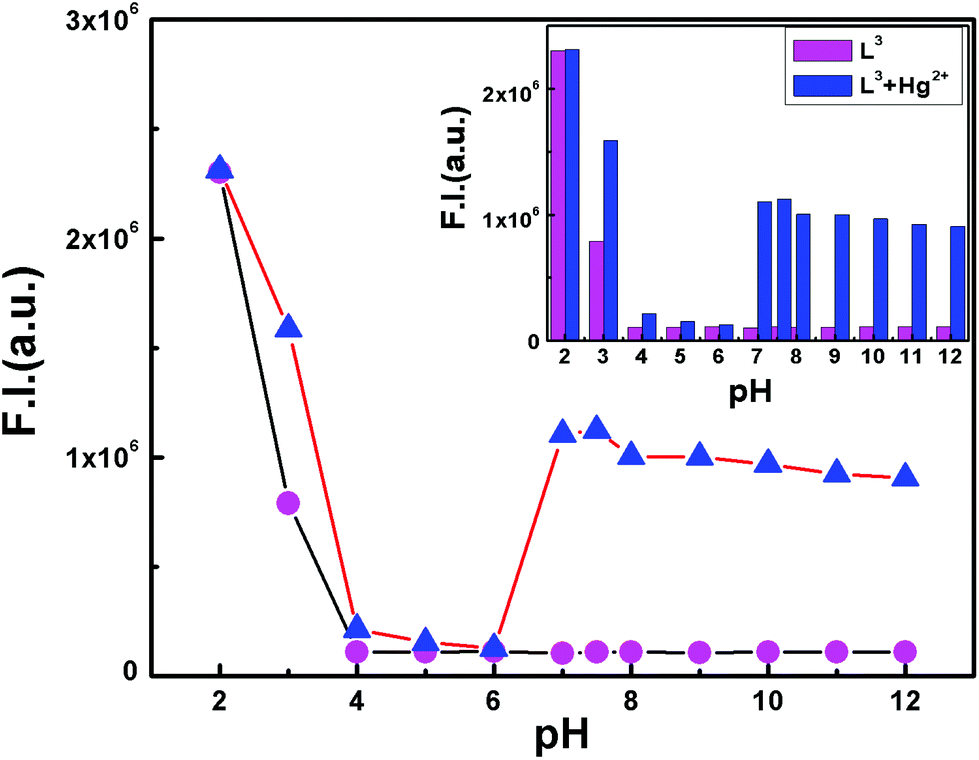 | ||
| Fig. 4 pH dependent FI of free ligand L3 (magenta) and the L3–Hg2+ complex with L3:Hg2+ = 1:1.05 (blue) in the MeCN/H2O (2:8 v/v) solvent system with λex = 510 nm. The inset shows the histogram plot. | ||
Determination of LOD
The 3σ method was adopted to determine the limit of detection (LOD) of Hg2+ and it was found to be as low as 78 nM (Fig. 5) which indicates that L3 is an ideal chemosensor for Hg2+ ions.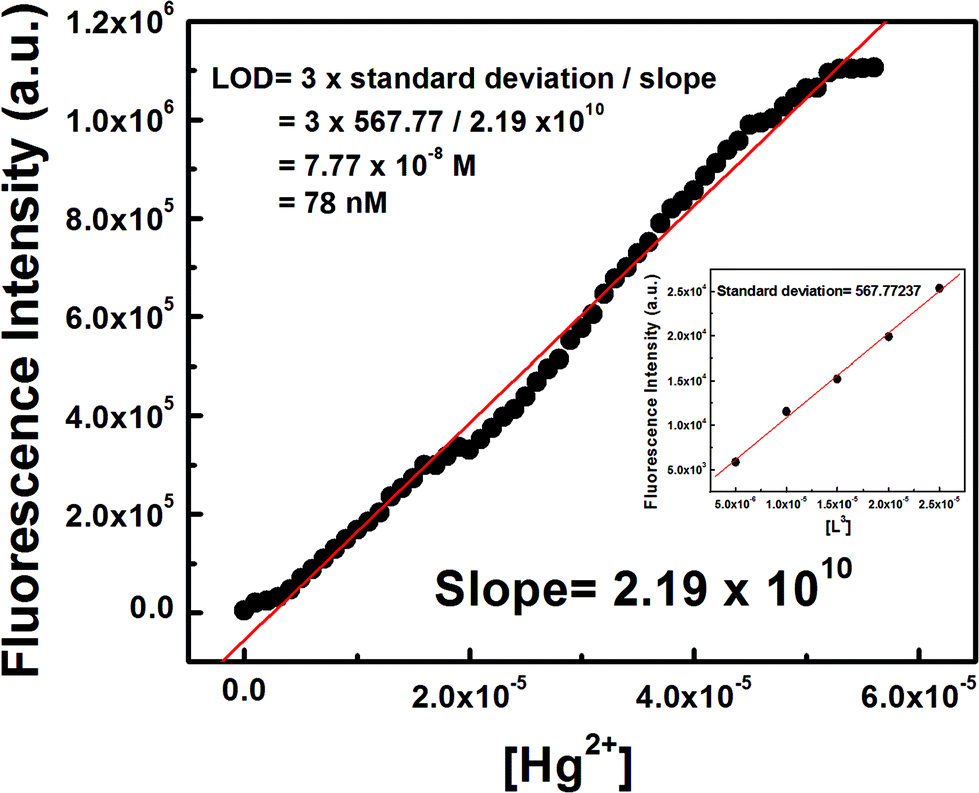 | ||
| Fig. 5 Determination of the limit of detection (LOD) of Hg2+ by L3 from the slope of the plot of FI vs. [Hg2+] and the standard deviation of the blank (intercept) determined from the plot of FI vs. [L3] utilizing the 3σ method. | ||
Mechanism of ring opening
The characteristic stretching frequency of the amidic “CO” bond of the rhodamine moiety at 1722 cm−1 is shifted to a lower wave number (1633 cm−1) in the presence of 1.2 equiv. of Hg2+ (Fig. S2†) indicating a strong polarization of the CO bond upon efficient binding to the Hg2+ ion and, in fact, indicates the cleavage of the N–C bond in the spirolactam ring. Also, the 1H NMR spectra showed an up-field shift for the azomethine proton mainly due to an increase in electron density arising from the opening of the spirolactam ring.
Steady-state fluorescence studies in the presence of SDS
Steady state fluorescence studies were also carried out in the presence of SDS under two experimental conditions. Firstly, the SDS concentration was kept fixed at 9 mM and [Hg2+] was varied between 0 and 14 μM keeping [L3] = 10 μM (Fig. 6a). A plot of FI vs. [Hg2+] resulted in a non-linear curve (Fig. 6b) and was solved by adopting eqn (3). The evaluated Kf value (4.19 ± 0.02) × 105 M−1 was found to be about one order of magnitude higher than that obtained in the absence of SDS. In order to get the SDS dependent Kf values we have varied [Hg2+] at different concentrations of SDS and thereby evaluated Kf values were plotted against the concentration of SDS. It was interesting to observe that there is a slow but gradual increase in Kf values with [SDS] up to 6 mM and further increasing [SDS] leads to a steep rise in the Kf value up to 9 mM (Fig. 7).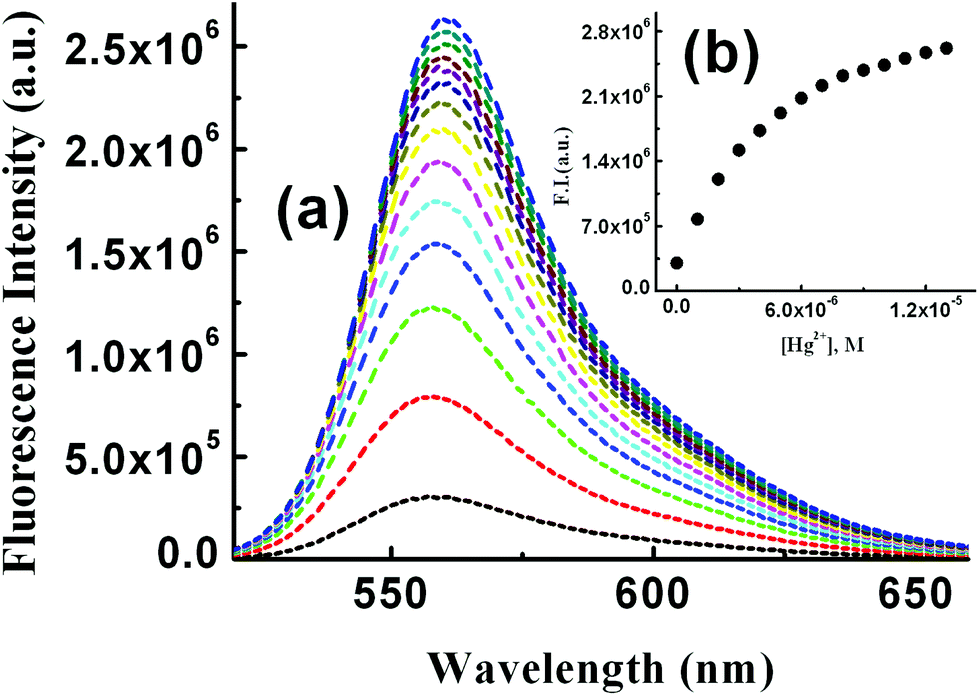 | ||
| Fig. 6 (a) Fluorescence titration of L3 as a function of [Hg2+] at [L3] = 10 μM and [SDS] = 9.0 mM, in pure water. (b) Plot of FI vs. [Hg2+]. | ||
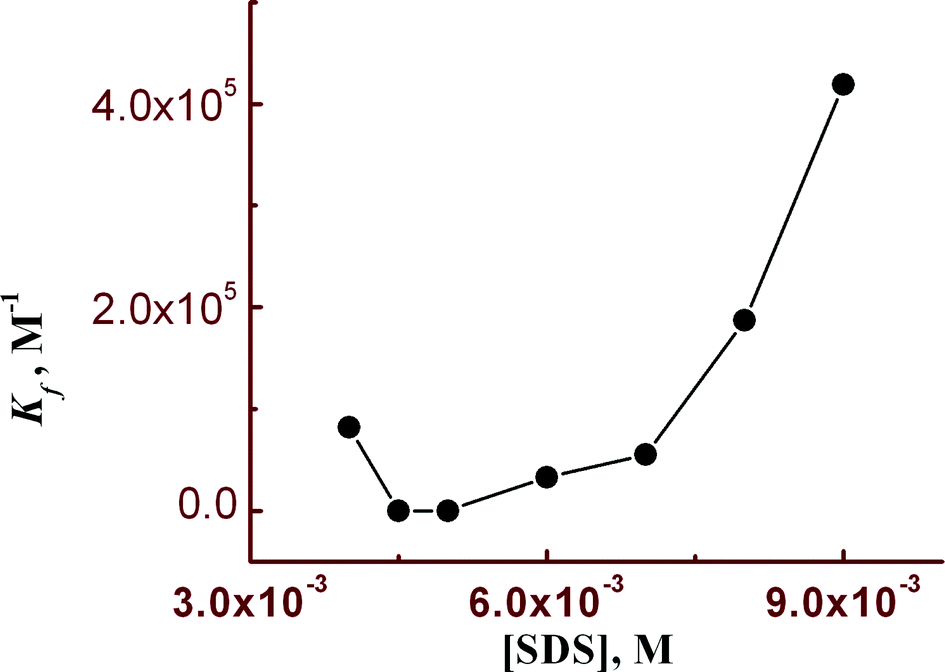 | ||
| Fig. 7 A plot of Kf as a function of SDS concentration. | ||
This enhanced stability constant value may be attributed to the restriction to the free dynamic movement of the doubly positively charged [L3–Hg]2+ complex by the strong electrostatic attraction with the negatively charged head groups of SDS.
Secondly, both [L3] and [Hg2+] were kept fixed at 10 μM and [SDS] was varied between 0 and 10 mM which yielded a non-linear curve on plotting FI vs. [SDS] showing a plateau at [SDS] ∼ >9.0 mM with a fluorescence enhancement of ∼143 fold with respect to the complex in the absence of SDS (Fig. 8). The appearance of a plateau at ∼9 mM SDS clearly indicates the critical micellar concentration (CMC) of SDS is ∼9.0 mM under the experimental conditions. The increase in FI with [SDS] demonstrates aggregation induced enhancement (AIE) of fluorescence. The binding of L3–Hg2+ to anionic sulfonic acid groups via electrostatic interactions was confirmed by the failure to observe any change in fluorescence intensity in the presence of cetyltrimethylammonium bromide (CTAB) – a cationic surfactant.
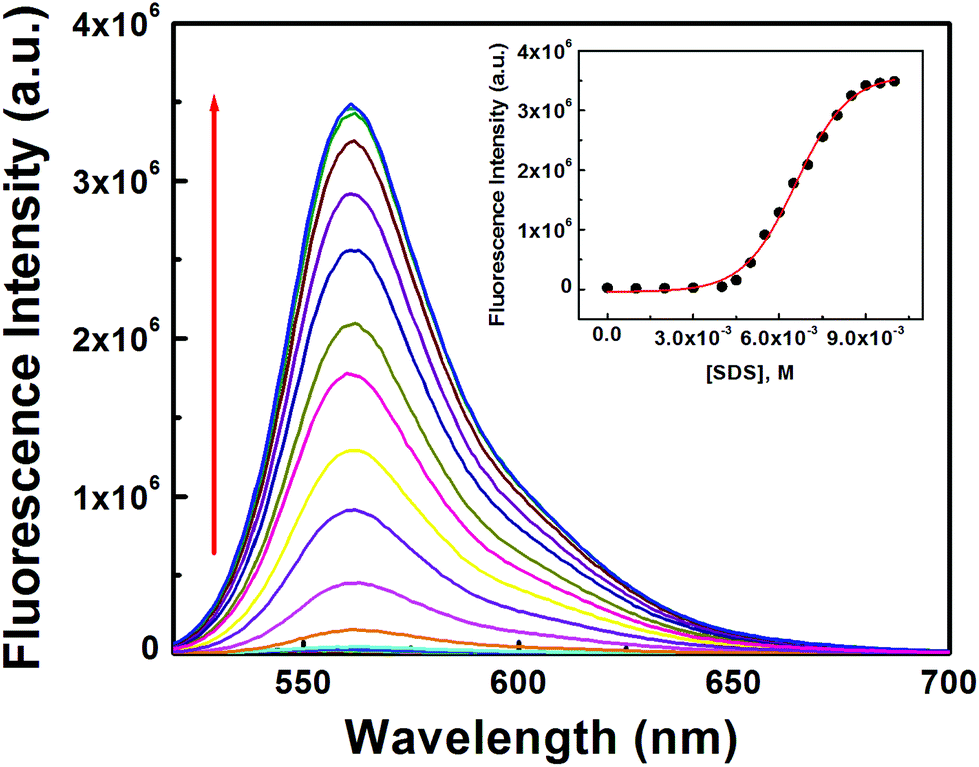 | ||
| Fig. 8 Fluorescence titration as a function of [SDS] at [L3] = 10 μM and [Hg2+] = 10 μM, in pure water. Inset is the plot of FI vs. [SDS]. | ||
On the basis of these observations, it is concluded that the intermolecular electrostatic interaction between [L3–Hg]2+ and SDS may effectively lower the dynamic motion and decrease the non-radiative decay process and hence increase the fluorescence intensity, quantum yield (φ) and stability constant (Kf) values compared to the respective values in the absence of SDS.
Steady-state fluorescence anisotropy measurements
Steady-state fluorescence anisotropy can be exploited to get motional information in the micro-heterogeneous environments.62 We have monitored the fluorescence anisotropy (r) as a function of SDS concentration (0–10 mM) at a fixed concentration of L3 and Hg2+ (10 μM each) at 553 nm. A plot of r vs. [SDS] showed a gradual increase in r with [SDS], reaches a maximum at ∼5 mM and then decreases up to 5.5 and again increases with [SDS] and becomes constant at [SDS] ≥ 7.0 mM. This unusual dependence of r on SDS concentration can be rationalized by considering the fact that the initial increase in r is due to trapping of the bi-positive [L3–Hg]2+ complex between two co-facial laminar layers which mostly prevailed within 3.0–5.5 mM concentration of SDS (Scheme 2). The decrease in r after 5.0 mM may arise due to phase transition and the second increase in r may be due to the formation of spherical aggregates upon micellization at ∼7.0 mM of SDS and after that the r values become almost constant. The variation of fluorescence anisotropy (r) as a function of SDS concentration is presented in Fig. 9.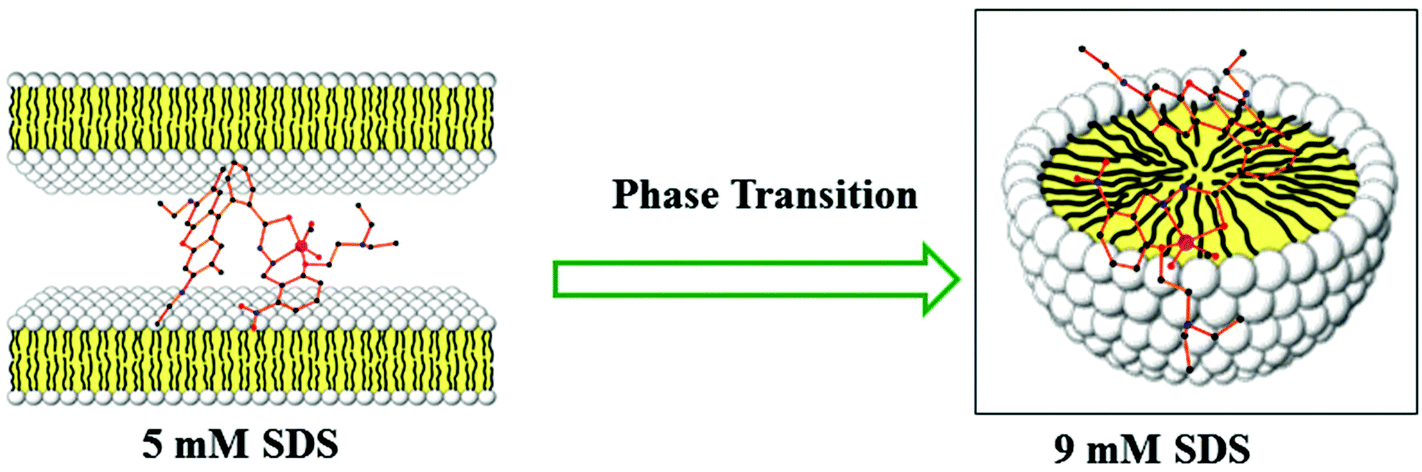 | ||
| Scheme 2 Schematic representation of the interactions of L3–Hg2+ with the laminar microstructure and micellar microstructure of SDS (adopted from http://fig.cox.miami.edu/~cmallery/255/255chem/mcb2.20.micelle.jpg). | ||
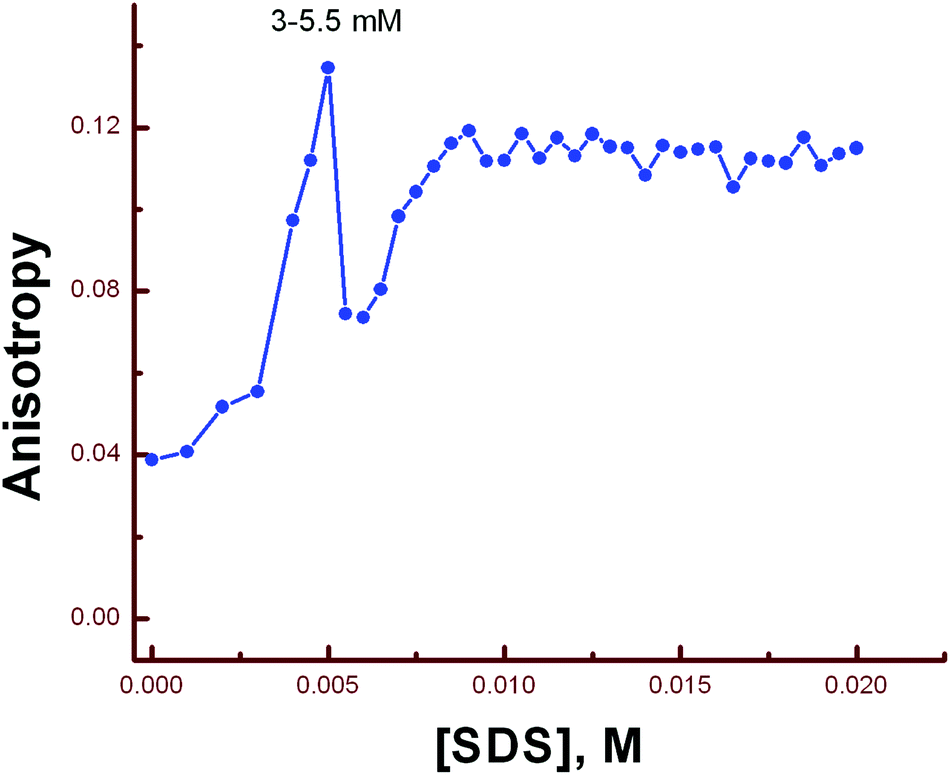 | ||
| Fig. 9 A plot of anisotropy (r) as a function of SDS concentration. | ||
To verify whether the probe is trapped inside the hydrophobic core of the micelle or remains in the Stern–Volmer layer we have carried out a fluorescence quenching experiment using EDTA in the presence of SDS at 5 mM and 9 mM concentration and also in the absence of SDS. It was interesting to note that there is no practical quenching of fluorescence intensity of the L3–Hg2+ complex on adding 10 equivalents of H2EDTA2− in the presence of 9 mM SDS; however it undergoes a fluorescence quenching by ∼50% of its original value in the presence of 5 mM SDS and 100% quenching occurs when there is practically no SDS in the solution (Fig. S7†). All of these observations indicate that the non-accessibility of the L3–Hg2+ complexes towards H2EDTA2− is surely due to trapping of L3–Hg2+ in the hydrophobic core of the spherical micelle when the SDS concentration is 9 mM. But partial availability of L3–Hg2+ towards H2EDTA2− occurs when it is sandwiched in the hydrophilic region between two co-facial laminar layers. We have also carried out SEM and TEM studies on the aggregates at 5.0 mM and 9.0 mM of SDS to support the above proposition and indeed there is some positive indication (Fig. 10 and 11).
 | ||
| Fig. 10 SEM images of L3 (10 μM)–Hg2+(10 μM) in the presence of (A) 5.0 mM SDS showing an agglomerated microstructure with no particular shape and (B) 9.0 mM SDS with a spherical microstructure. | ||
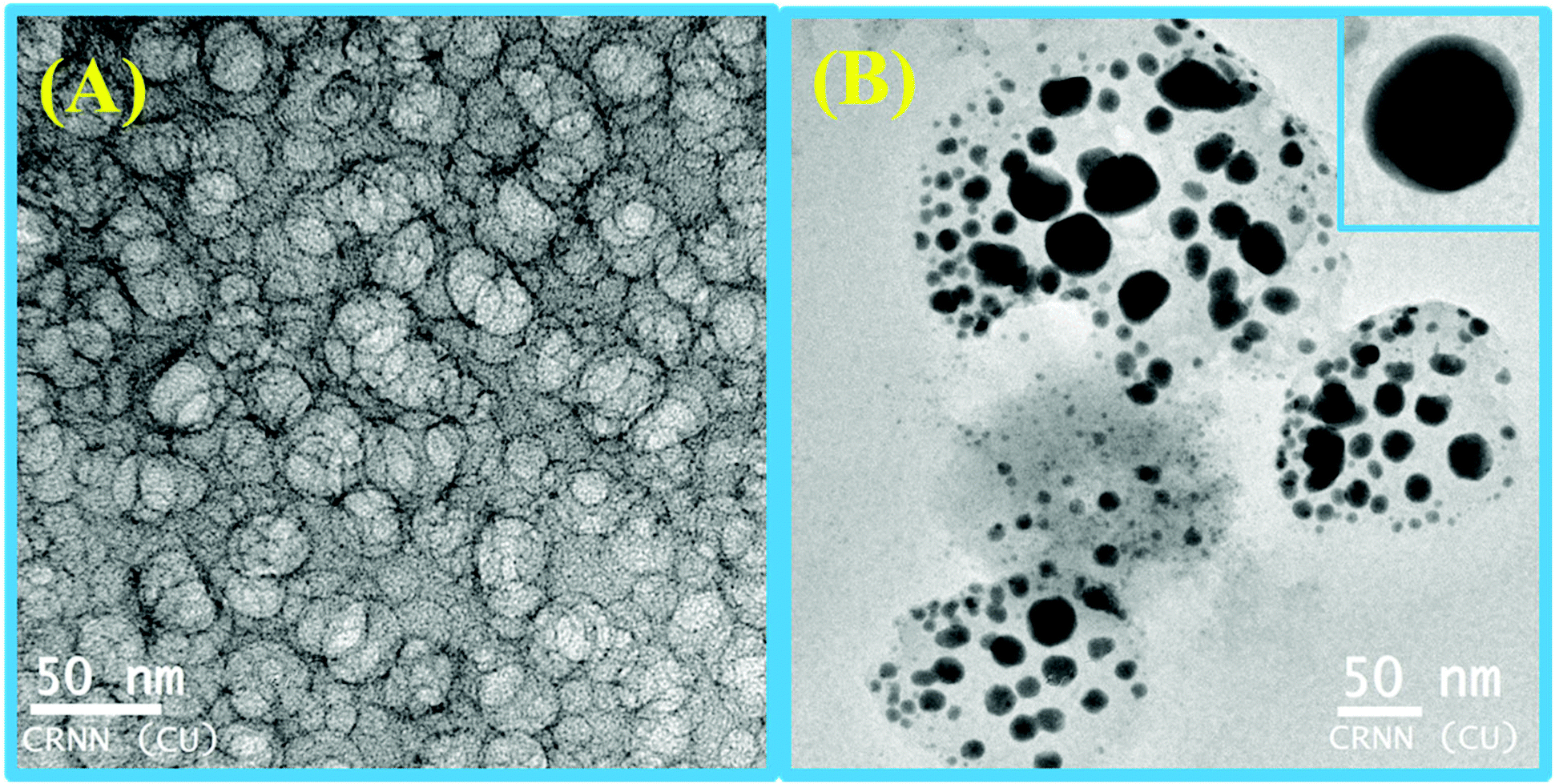 | ||
| Fig. 11 TEM images of L3–Hg2+ in the presence of (A) 5.0 mM SDS and (B) 9.0 mM SDS. | ||
SEM and TEM studies
At higher and lower magnifications of the SEM images at 9 mM SDS, a core–shell structure is quite apparent, in which the metallic part is embedded with the ligand and SDS in a definite spherical Janus like structure (Fig. 10). This core–shell structure can also be explained by the TEM studies; the inner core (dark black) of the particle is comprised of the metallic zone (Fig. 11) and the outer shell is the ligand along with SDS which are in agglomerated conditions due to the hydrophilic interactions. However, in the presence of 5 mM SDS the probe undergoes extensive aggregation with no definite shape which is also prevailing from both the SEM and TEM studies.Cell studies
Mitochondria are cytoplasmic organelles in the eukaryotic cell and produce ATP (95%) required for the cell. In nerve cells mitochondrial function has a very crucial role to meet the high, long term and specific energy demand to regulate brain functions. The basic mechanism for mercury toxicity emerges from the direct and indirect damage of mitochondria through reduction of glutathione due to extreme ROS (reactive organic species) generation and increased mitochondrial membrane permeability. Hence, there is a certain need to detect Hg2+ in its permissible limits to avoid its toxicity and also to apply a chelator based treatment to reduce Hg2+ poisoning in humans. In the present study we evaluate the chemosensing capability of L3 to detect Hg2+. The cytotoxic effects of L3 were determined by a cell viability assay in HepG2 cells. Up to 50 μM shows less than 30% cytotoxicity for L3 and more than 87% of cells are viable up to a dose of 20 μM (Fig. S8†). Hence no significant cytotoxicity was observed up to 20 μM. Furthermore, we have assessed the Hg2+ ion detection capability of L3 using in vitro fluorescence analysis in HepG2 cells (Fig. 12). Intracellular imaging of HepG2 cells treated with the L3 ligand (10 μM) reveals no intracellular fluorescence. Yet, live cell imaging of HepG2 cells treated with equimolar amounts of Hg2+ and the L3 ligand shows excellent intracellular cytoplasmic red fluorescence (Fig. 12).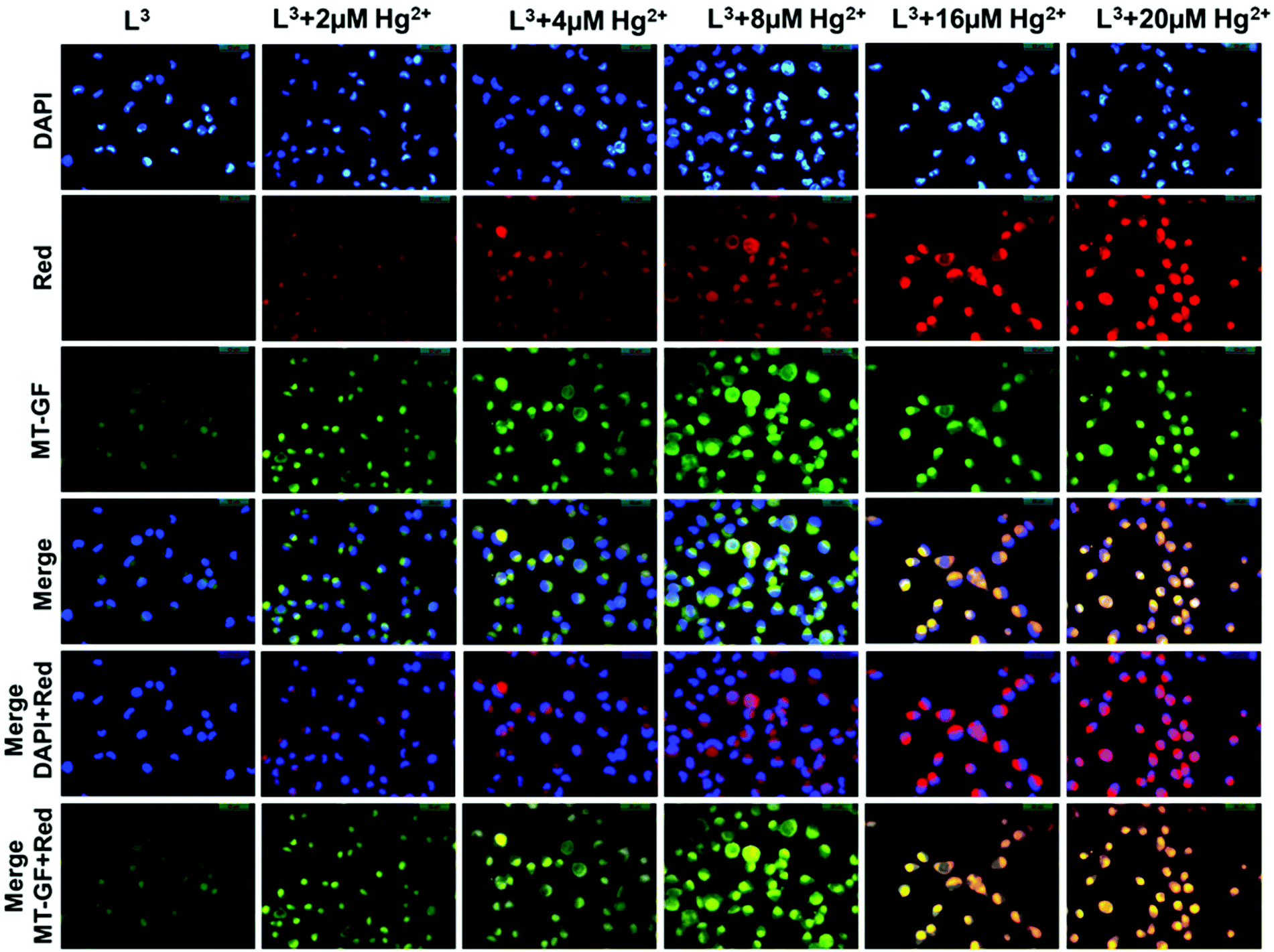 | ||
| Fig. 12 Cell imaging studies of Hg2+ ions with L3. The fluorescence images of the HepG2 cells were captured (40×) after incubation with 10 μM L3 for 30 min at 37 °C and pre-incubation with (2, 4, 8, 16 and 20 μM) Hg2+ for 3 h at 37 °C followed by washing thrice with 1× PBS. The images show strong cytoplasmic red florescence when L3 complexes with Hg2+ (red). The merged images show that the L3–Hg2+ fluorescence is cytoplasmic and not in the nucleus. Cytoplasmic complex formation was confirmed using DAPI (nuclear stain, blue) and MitoTracker Green FM (MT-GF, cytoplasm stain, green). | ||
A concentration dependent increase in the red fluorescence was noted indicating L3 as an excellent cytoplasmic Hg2+ tracker. We have observed that L3 can sense low concentrations of Hg2+ ions present in the cytoplasm (2–8 μM). We didn't see any nuclear binding of Hg2+–L3; this may be due to the restricted permeability of L3 to the cytoplasm (Fig. 12). Hence the presented ligand has low cytotoxicity and is biocompatible for cellular cytoplasmic Hg2+ ion detection and can be used for Hg2+ ion detection in biological samples.
Conclusion
A new type of easily synthesized rhodamine-based chemosensor with potential NO2 donor atoms showed selective and rapid recognition of toxic Hg2+ ions. The binding stoichiometry of the sensor with Hg2+ was established by the combined Job's and HRMS (m/z) methods. All biologically relevant metal ions as well as toxic heavy metal ions and anions did not interfere with the detection of Hg2+ ions. The detection limit of Hg2+ calculated using the 3σ method gives a value of 78 nM. Bio-compatibility and good solubility in a mostly aqueous medium (8:2, H2O:MeCN) along with its cell permeability with no or negligible cytotoxicity indicate good potential for in vitro/in vivo cell imaging studies which showed cytoplasmic recognition of Hg2+ ions. Under an optical microscope, a heavily agglomerated microstructure of L3–Hg2+ in 5 mM SDS changes to a spherical shape in 9 mM SDS. Not only that, we have also carried out fluorescence titrations in the presence of SDS and also the SDS concentration was varied at a fixed concentration of the receptor and the guest. The presence of SDS causes an enhanced quantum yield (φ) and more importantly the stability constant (Kf) by ∼an order of magnitude compared to those in the absence of SDS. Again, the FI of the [L3–Hg]2+ complex is enhanced by 143 fold compared to that in the absence of SDS. So an attempt was made to get some idea of the dependence of Kf on SDS concentration. It was interesting to see that a plot of Kf as a function of [SDS] showed an initial slow increase in Kf up to 6 mM of SDS and then it picked up rapidly indicating the occurrence of aggregation induced emission enhancement (AIEE). All of these observations clearly manifest in an enhanced rigidity of the [L3–Hg]2+ complex in the heterogeneous micro-environment restricting the dynamic movement of the [L3–Hg]2+ complex. The change in fluorescence anisotropy (r) with [SDS] is best described by a change in the morphology of the SDS microstructure from laminar to a spherical shape; the former being more efficient to provide better rigidity to the [L3–Hg]2+ complex and hence higher r values at ∼5 mM of SDS.
Acknowledgements
Financial support from the Department of Science and Technology (DST), Government of India, New Delhi (ref. number SR/S1/IC-20/2012) is gratefully acknowledged. Instrumental facilities FE-SEM from the Department of Physics (DST-FIST) Jadavpur University and TEM from the CRNN, University of Calcutta are gratefully acknowledged.Notes and references
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- L. Wang, L. L. Li, H. L. Ma and H. Wang, Chin. Chem. Lett., 2013, 24, 351–358 CrossRef CAS.
- T. L. Greaves and C. J. Drummond, Chem. Soc. Rev., 2013, 42, 1096–1120 RSC.
- T. Y. Ma, H. Li, Q. F. Deng, L. Liu, T. Z. Ren and Z. Y. Yuan, Chem. Mater., 2012, 24, 2253–2255 CrossRef CAS.
- C. Lv, G. Xu and X. Chen, Chem. Lett., 2012, 41, 1201–1203 CrossRef CAS.
- A. M. Percebom, J. Janiak, K. Schillén, L. Piculell and W. Loh, Soft Matter, 2013, 9, 515–526 RSC.
- Y. N. Guo, Y. Li, B. Zhi, D. Zhang, Y. Liu and Q. Huo, RSC Adv., 2012, 2, 5424–5429 RSC.
- L. G. Chen and H. Bermudez, Langmuir, 2012, 28, 1157–1162 CrossRef CAS PubMed.
- S. L. de-Rooy, S. Das, M. Li, B. El-Zahab, A. Jordan, R. Lodes, A. Weber, L. Chandler, G. A. Baker and I. M. Warner, J. Phys. Chem. C, 2012, 116, 8251–8260 CAS.
- Y. Liu, X. Feng, J. Shi, J. Zhi, B. Tong and Y. Dong, Chin. J. Polym. Sci., 2012, 30, 443–450 CrossRef CAS.
- Y. Ren and T. Baumgartner, Inorg. Chem., 2012, 51, 2669–2678 CrossRef CAS PubMed.
- X. Chen, Y. Xiang, P. Song, R. Wei, Z. Zhou, K. Li and A. Tong, J. Lumin., 2011, 131, 1453–1459 CrossRef CAS.
- B. Xu, Z. Chi, X. Li, H. Li, W. Zhou, X. Zhang, C. Wang, Y. Zhang, S. Liu and J. Xu, J. Fluoresc., 2011, 21, 433–441 CrossRef CAS PubMed.
- E. Lucenti, C. Botta, E. Cariati, S. Righetto, M. Scarpellini, E. Tordin and R. Ugo, Dyes Pigm., 2013, 96, 748–755 CrossRef CAS.
- C. Shi, Z. Guo, Y. Yan, S. Zhu, Y. Xie, Y. S. Zhao, W. Zhu and H. Tian, ACS Appl. Mater. Interfaces, 2013, 5, 192–198 CAS.
- P. Guo, S. Yan, Y. Zhou, C. Wang, X. Xu, X. Weng and X. Zhou, Analyst, 2013, 138, 3365–3367 RSC.
- W. Z. Yuan, Z. Q. Yu, P. Lu, C. Deng, J. W. Y. Lam, Z. Wang, E. Q. Chen, Y. Mad and B. Z. Tang, J. Mater. Chem., 2012, 22, 3323–3326 RSC.
- G. Yu, S. Yin, Y. Liu, J. Chen, X. Xu, X. Sun, D. Ma, X. Zhan, Q. Peng, Z. Shuai, B. Z. Tang, D. Zhu, W. Fang and Y. Luo, J. Am. Chem. Soc., 2005, 127, 6335–6346 CrossRef CAS PubMed.
- X. Gu, J. Yao, G. Zhang, C. Zhang, Y. Yan, Y. Zhao and D. Zhang, Chem. – Asian J., 2013, 8, 2362–2369 CrossRef CAS PubMed.
- J. Luo, X. Wang, X. Wang and W. Su, Chin. J. Chem., 2012, 30, 2488–2494 CrossRef CAS.
- X. Zhou, H. Li, Z. Chi, B. Xu, X. Zhang, Y. Zhang, S. Liu and J. Xu, J. Fluorsc., 2012, 22, 565–572 CrossRef CAS PubMed.
- W. Wu, S. Ye, R. Tang, L. Huang, Q. Li, G. Yu, Y. Liu, J. Qin and Z. Li, Polymer, 2012, 53, 3163–3171 CrossRef CAS.
- B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- S. J. Li, B. K. An, S. D. Jung, M. A. Chung and S. Y. Park, Angew. Chem., Int. Ed., 2004, 43, 6346–6350 CrossRef PubMed.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley, London, 1970 Search PubMed.
- X. Cao, R. W. Tolbert, J. L. McHale and W. D. Edwards, Dye J. Phys. Chem. A, 1998, 102, 2739–2748 CrossRef CAS.
- R. Badugu, J. R. Lakowicz and C. D. Geddes, J. Am. Chem. Soc., 2005, 127, 3635–3641 CrossRef CAS PubMed.
- Y. Ren, J. W. Y. Lam, Y. Dong, B. Z. Tang and K. S. Wong, J. Phys. Chem. B, 2005, 109, 1135–1140 CrossRef CAS PubMed.
- H. Tong, Y. Dong, Y. Hong, M. Haussler, J. W. Y. Lam, H. H. Y. Sung, X. Yu, J. Sun, I. D. Williams, H. S. Kwok and B. Z. Tang, J. Phys. Chem. C, 2007, 111, 2287–2294 CAS.
- Z. Li, Y. Dong, B. Mi, Y. Tang, M. Haussler, H. Tong, Y. Dong, J. W. Y. Lam, Y. Ren, H. H. Y. Sung, K. S. Wong, P. Gao, I. D. Williams, H. S. Kwok and B. Z. Tang, J. Phys. Chem. B, 2005, 109, 10061–10066 CrossRef CAS PubMed.
- L. S. Hung and C. H. Chen, Mater. Sci. Eng., 2002, 39, 143–222 Search PubMed.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS.
- J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535–1546 CrossRef CAS.
- M. Rosoff, Ed. Vesicles, Dekker, New York, 1996 Search PubMed.
- C. F. J. Faul, Adv. Mater., 2003, 15, 673–683 CrossRef CAS.
- P. Bilski, R. N. Holt and C. F. Chignell, J. Photochem. Photobiol., A, 1997, 110, 67–74 CrossRef CAS.
- P. Bilksi and C. F. Chignell, J. Photochem. Photobiol., A, 1994, 77, 49–58 CrossRef.
- H. Hachisako and R. Murakami, Chem. Commun., 2006, 1073–1075 RSC.
- H. Hachisako, N. Ryu and R. Murakami, Org. Biomol. Chem., 2009, 7, 2327–2337 CAS.
- A. K. Chibisov, V. I. Prokhorenko and H. Görner, Chem. Phys., 1999, 250, 47–60 CrossRef CAS.
- A. K. Mandal and M. K. Pal, Chem. Phys., 2000, 253, 115–124 CrossRef CAS.
- L. Tang, J. Jin, S. Zhang, Y. Mao, J. Sun, W. Yuan, H. Zhao, H. Xu, A. Qin and B. Tang, Sci. China, Ser. B: Chem., 2009, 52, 755–759 CrossRef CAS.
- Y.F. Zhang, Q. Yuan, T. Chen, X. B. Zhang, Y. Chen and W. H. Tan, Anal. Chem., 2012, 84, 1956–1962 CrossRef CAS PubMed.
- E. S. Childress, C.A. Roberts, D. Y. Sherwood, C. L. M. LeGuyader and E. J. Harbron, Anal. Chem., 2012, 84, 1235–1239 CrossRef CAS PubMed.
- L. Deng, X. Y. Ouyang, J. Y. Jin, C. Ma, Y. Jiang, J. Zheng, J. S. Li, Y. H. W. Li, H. Tan and R. H. Yang, Anal. Chem., 2013, 85, 8594–8600 CrossRef CAS PubMed.
- P. B. Tchounwou, W. K. Ayensu, N. Ninashvili and D. Sutton, Environ. Toxicol., 2003, 18, 149–175 CrossRef CAS PubMed.
- V. D. Bittner Jr., J. S. Echeverria, H. V. Woods, C. Aposhian, M. D. Naleway, R. K. Martin, N. J. Mahurin and M. Cianciola, Neurotoxicol. Teratol., 1998, 20, 429–439 CrossRef.
- F. Di Natale, A. Lancia, A. Molino, M. Di Natale, D. Karatzaand and D. Musmarra, J. Hazard. Mater., 2006, 132, 220 CrossRef CAS PubMed.
- Y. Gao, S. De Galan, A. DeBrauwere, W. Baeyens and M. Leermakers, Talanta, 2010, 82, 1919–1923 CrossRef CAS PubMed.
- F. Moreno, T. Garcia-Barrera and J. L. Gomez-Ariza, Analyst, 2010, 135, 2700–2705 RSC.
- X. Chai, X. Chang, Z. Hu, Q. He, Z. Tu and Z. Li, Talanta, 2010, 82, 1791–1796 CrossRef CAS PubMed.
- X. Fu, X. Chen, Z. Guo, C. Xie, L. Kong, J. Liu and X. Huang, Anal. Chim. Acta, 2011, 685, 21–28 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006, p. 67 Search PubMed.
- R. P. Haugland, The Handbook: A Guide to Fuorescent Probes and Labeling Technologies, Invitrogen Corp., Karlsbad, CA, 10th edn, 2005 Search PubMed.
- H. Y. Lee, K. M. K. Swamy, J. Y. Jung, G. Kim and J. Yoon, Sens. Actuators, B, 2013, 182, 530–537 CrossRef CAS.
- F. Wanga, S. W. Nama, Z. Guoa, S. Parka and J. Yoona, Sens. Actuators, B, 2012, 161, 948–953 CrossRef.
- X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910–1956 CrossRef CAS PubMed.
- K. Bera, A. K. Das, M. Nag and S. Basak, Anal. Chem., 2014, 86, 2740–2746 CrossRef CAS PubMed.
- V. Dujols, F. Ford and A. W. Czarnik, J. Am. Chem. Soc., 1997, 119, 7386–7387 CrossRef CAS.
- R. P. Haugland, The Handbook: A Guide to Fuorescent Probes and Labeling Technologies, Invitrogen Corp., Karlsbad,CA, 10th edn, 2005 Search PubMed.
- L. Huang, X. Wang, G. Xie, P. Xi, Z. Li, M. Xu, Y. Wu and D. Baib, Dalton Trans., 2010, 39, 7894–7896 RSC.
- R. Bhowmick, R. Alam, T. Mistri, D. Bhattacharya, P. Karmakar and M. Ali, Appl. Mater. Interfaces, 2015, 7, 7476–7485 CrossRef CAS PubMed.
- C. R. Lohani, J. M. Kim, S. Y. Chung, J. Yoon and K. H. Lee, Analyst, 2010, 135, 2079–2084 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and corresponding characterization data for compound L3. See DOI: 10.1039/c5an01916h |
| This journal is © The Royal Society of Chemistry 2016 |