
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic DNA-controlled “stop-and-go” assembly of well-defined protein domains on RNA-scaffolded TMV-like nanotubes†
Angela
Schneider
,
Fabian J.
Eber
,
Nana L.
Wenz
,
Klara
Altintoprak
,
Holger
Jeske
,
Sabine
Eiben
* and
Christina
Wege
*
Department of Molecular Biology and Plant Virology, Institute of Biomaterials and Biomolecular Systems, University of Stuttgart, Pfaffenwaldring 57, 70569 Stuttgart, Germany. E-mail: christina.wege@bio.uni-stuttgart.de; sabine.eiben@bio.uni-stuttgart.de; Tel: +49-711-685-65073 Tel: +49-711-685-65595
First published on 9th November 2016
Abstract
A DNA-based approach allows external control over the self-assembly process of tobacco mosaic virus (TMV)-like ribonucleoprotein nanotubes: their growth from viral coat protein (CP) subunits on five distinct RNA scaffolds containing the TMV origin of assembly (OAs) could be temporarily blocked by a stopper DNA oligomer hybridized downstream (3′) of the OAs. At two upstream (5′) sites tested, simple hybridization was not sufficient for stable stalling, which correlates with previous findings on a non-symmetric assembly of TMV. The growth of DNA-arrested particles could be restarted efficiently by displacement of the stopper via its toehold by using a release DNA oligomer, even after storage for twelve days. This novel strategy for growing proteinaceous tubes under tight kinetic and spatial control combines RNA guidance and its site-specific but reversible interruption by DNA blocking elements. As three of the RNA scaffolds contained long heterologous non-TMV sequence portions that included the stopping sites, this method is applicable to all RNAs amenable to TMV CP encapsidation, albeit with variable efficiency most likely depending on the scaffolds’ secondary structures. The use of two distinct, selectively addressable CP variants during the serial assembly stages finally enabled an externally configured fabrication of nanotubes with highly defined subdomains. The “stop-and-go” strategy thus might pave the way towards production routines of TMV-like particles with variable aspect ratios from a single RNA scaffold, and of nanotubes with two or even more adjacent protein domains of tightly pre-defined lengths.
Introduction
Efficient procedures to adjust the size range and shape of technically promising nanoparticles have been essential prerequisites before several nanoparticle species have found their way into present applications, e.g. in electronics,1,2 data storage and magnetic separation technology,3 catalysis,4,5 diagnostics,6–9 chemical processing, healthcare and cosmetics, as reviewed in detail.10 Among these, elongated nanoparticles are of special interest as scaffolding, stabilizing or conducting fibers, often applied in composite materials making use of inorganic, synthetic or carbon-based nanotubes. Their fabrication and post-processing, however, mostly demands for harsh conditions with temperatures up to 4000 °C under non-ambient pressure, and costly instrumentation.11 Harmful by-products or toxicity of the nanotubes themselves12,13 may constitute further drawbacks.By contrast, bionanotubes and building blocks thereof are produced under ambient conditions in vivo, may be re-organized into altered shapes and compositions through mild environmental changes in many cases, and can be used as versatile organic scaffolds. This enables templating of inorganic materials,14 and also the preparation of nanostructured carrier scaffolds for the immobilization of functional molecules,15–18 thereby obviating the aforementioned drawbacks. A special advantage of biogenic nanostructures lies in the availability of precisely positioned, genetically programmable and selectively addressable functional groups19–21 exposed at high densities. In several supramolecular biological systems these groups are arranged in complexes of a highly organized architecture and defined dimensions, some of them with a high aspect ratio.22–24 However, bionanoparticles, -filaments and -tubes often come along with poor stability, low expression and purification yields and are pathogenic in the case of many viruses or amyloid peptide fibers. Furthermore, for protein or peptide nanotubes, precise and rational length control is difficult to achieve.25,26 In contrast, elongated nucleoprotein complexes of certain plant viruses and bacteriophages lack most of these disadvantages: they are non-infectious for animals and humans, can be produced in planta or in bacterial and yeast cell cultures, and there exist species with high thermal and organic solvent stability.27–30 Amongst these, tobacco mosaic virus (TMV) has the advantages of unequaled yield, an assembly mechanism which does not rely on a living host, and the possibility of genetic adaptations. Hence tube-like TMV bionanoparticles have been employed successfully, for example, as additives in ferrofluids,31 scaffolding networks in electronic components,32,33 as nanoelectrical memory devices34 and templates for the surface design of battery electrodes or antireflective current collectors.35–37 Recent studies point at particular advantages of TMV and TMV-like particles (TLPs) applied as carrier templates for bioactive molecules,18 with different biosensor layouts profiting from plant viral nanorod coatings densely equipped with analyte-recognizing enzymes38,39 or antibody capture tags,40 as reviewed in detail.18
As with synthetic nanoparticles, length control and extensive tunability of bionanostructures are desirable prerequisites in order to adjust their physical or (bio-)chemical properties and, putatively, activities.31,41,42 In the case of TMV, the particle shape and size are determined by a scaffolding genomic RNA, naturally 6395 nucleotides (nts) long, which directs the helical arrangement of 2130 identical coat proteins (CP). Concomitantly, the RNA becomes encapsidated and protected by a tubular proteinaceous coat of 300 nm length, and inner and outer diameters of 4 and 18 nm, respectively. Since an RNA-guided self-assembly of TMV particles is possible also in vitro, the process has been studied extensively.43
How can an RNA molecule direct the assembly of proteins into higher-order architectures in vitro? The clue seems to lie in the selective interaction of a secondary stem–loop structure located internally in the TMV RNA (i.e. the TMV origin of assembly, OAs) with a certain TMV CP oligomer, thereby initiating a series of intricate conformational changes and rearrangements which are mutually affected and induced by both the protein and the nucleic acid partners. The nucleation of the process most likely involves a ring-shaped 34-meric CP aggregate (a two-layered holey “disk”). Upon contacting the OAs, a conformational transition of the “disk” into a helical “lockwasher” structure incorporates a first stretch of the RNA and threads an RNA “travelling loop” into the central hole.43and references therein Serial binding of further CP disks to the nascent tube's pole exposing the “travelling loop” results in their helical reorganization one after the other, rapidly incorporating the complete RNA 5′ portion between the protein layers concomitantly.44 Its back-folded end is pulled up through the central channel until it is fully embedded inside the protein helix. The 3′ portion of the RNA is packaged more slowly by the addition of CP monomers, trimers and other small aggregates (see Fig. 2 in the Results and discussion section). The RNA encapsidation speed is thus dependent on the relative position of the OAs between the RNA stretches up- (3′) or downstream (5′).45 The rates reported for the bidirectional, entropy-driven tube assembly correspond to an average addition of ∼6 CP subunits per second through cooperative attachment in the 5′ direction (using disks), and ∼1.6 subunits per second in the 3′ orientation (using smaller aggregates), respectively.43,46 However, both initiation and elongation rates depend on the CP concentration.47
Such an RNA-directed self-assembly allows a combination of engineered nucleic acid templates and molecular biology techniques to influence the formation of nanotubular nucleoprotein complexes by rational approaches, an issue only rarely investigated in bionanotechnology in the past. We thus have started a series of studies exploring both prospects and limits of selected strategies for the design of TMV-based architectures. Simple changes of the RNA scaffold length were sufficient to alter the length distribution of the resulting TLPs accordingly, and thereby their characteristics as beneficial ferrofluid additives31 or tumor-targeting colloids.42 Against the background of the bidirectional assembly process described above, not only freely dispersed components, but also RNAs 3′-terminally immobilized on flat supports or metal cores, respectively, which indeed gave rise to carrier stick arrays on planar surfaces48 or “nanostars” with multiple arms were tested.49 If two or more OAs sequences were combined in single RNA scaffolds, non-linear “nanoboomerangs” or branched nanotube systems up to tetrapods were generated predictably, with kinks occurring at sites where the growing ends of adjacent nanohelices collided with each other.46,50 This RNA-based toolbox was combined with the use of distinct TMV CP variants, exposing selectively addressable surface groups enabling the binding of functionalities such as nanoparticles,31 fluorescent dyes,51–53 peptides42,54 or enzymes.38,39 An integration of more than a single CP type into TMV-deduced assemblies allows the fabrication of bi- or multifunctional objects. This may be realized by way of CP blends employed over the whole particle surface.55 A further extended range of applications, however, seems possible if specific functionalities can be spatially confined in distinct rod domains and thus ordered on the nano- to mesoscale. Our first approach towards this goal used a serial, stepwise assembly strategy, with a substoichiometric amount of a first CP added to an RNA scaffold prior to completing rod growth with a second CP species.56 The resulting nanoparticles exhibited two to three domains of at the maximum two CP variants, depending on the relative amount of CP educts over OAs-containing RNA molecules. Despite a convincingly reproducible overall arrangement of the distinct CP species, however, the method did not achieve tight control over the final domain lengths, as it would be desirable e.g. for optimizing the spacing of enzyme groups in cooperative biocatalytic systems.
Hence, it is tempting to find out if the major assembly-directing handle employed so far, i.e. the intramolecular OAs of the RNA scaffold, may be complemented by further tools of nucleic acid nanotechnology to direct and tune the outcome of the assembly process, with respect to size and arrangement of bionanotube subdomains. Previous in vitro studies have shown that TMV CPs may also package heterologous RNAs equipped with the viral OAs into TLPs,50,57–63 whereupon, however, encapsidation may be slowed down or blocked by extensive and stable secondary structures within the RNA.57,59 We aimed to modulate the nanotubes’ assembly progress in trans – i.e. in a way that allows an external, scaffold site-specific steering of the CP–RNA encapsidation process by tertiary nucleic acid interactions. The sequence specificity of nucleic acid strand hybridization together with automated DNA synthesis has led to striking artificial 3D nanostructures with the DNA origami technique64 being the most prominent, and to nanoactuation methods “fueled” by the free energy released upon base pairing.65 The underlying strategies seemed suitable to us to provide tools that might give rise to a “stop-and-go” method for controlling the growth of TMV-like nanotubes dynamically.
Switchable changes in nucleic acid structures are typically achieved by the displacement of an oligonucleotide strand (DNA oligomer 1) from its hybridization target strand by means of a second “fuel” strand (DNA oligomer 2). Strand displacement becomes possible because the DNA oligomer 1 is only partially complementary to the target; it contains a non-complementary and thereby overhanging stretch, the “toehold”. DNA oligomer 2, in turn, is fully complementary to oligomer 1, allowing the initial binding to the target-hybridized oligomer 1 via its toehold sequence. As base pairing proceeds, DNA oligomer 2 competes with the target for hybridization to oligomer 1, which is eventually displaced from the target sequence through branch migration, making it available to undergo further reactions.66 The powerful toehold-release technique has been exploited for the serial polymerization of DNA elements triggered by an initiator molecule (hybridization chain reaction HCR)67–70 and to create programmable, complex dynamic DNA3,65,71–74 and gold nanoparticle assemblies75 fueled by short target DNA sequences in e.g. colorimetric detection assays.76,77
The following study expands the dynamic DNA technology into a further hierarchical dimension: the externally directed modification of ribonucleic acid scaffold functionality, which in turn will govern a stepwise self-organization of compact supramolecular nucleoprotein complexes. Specifically, we have assessed whether the RNA-directed self-assembly process of TMV CPs may be controlled by means of a blocking DNA oligomer hybridized to the RNA, and its toehold-assisted release via strand displacement. “Stopper” DNA oligomers targeting different sites of TMV and TMV-derived RNA scaffolds as well as OAs-containing heterologous RNAs (hRNAs) were applied and their capabilities to stall the assembly of nucleoprotein complexes was investigated by transmission electron microscopy (TEM) and agarose gel electrophoresis. After successful blocking (“stop”), reconstitution of assembly was induced (“go”) by the addition of a “release” DNA oligomer capable of displacing the stopper. To fathom the method's potential for fabricating a soft-matter template species not accessible so far, i.e. nanotubes with distinct highly defined subdomains, a protocol enabling completion of enriched partial rods with a selectively addressable second CP species was developed. Coupling of large biomolecules allowed TEM visualization of the corresponding domains of the bulk products, and thus judging the benefit of the approach.
Experimental section
Preparation of RNA scaffolds
The wt-RNA of TMV was prepared from plant-derived virus particles28 on the basis of a previous protocol described by Chapman (1998),78 as follows: 0.8 mL of a TMV preparation (10 mg mL−1) in 10 mM sodium potassium phosphate buffer (SPP, pH 7.4) was mixed with 0.2 mL 5× extraction buffer (0.5 M NaCl, 5 mM EDTA, 5% SDS, 0.1 M Tris pH 8). 1 mL of phenol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :chloroform:isoamylalcohol (PCI, 25:24:1) was added and vortexed briefly. The emulsion was centrifuged for 1 min at 20000g at room temperature. The aqueous phase was collected and the PCI extraction was repeated twice. The third aqueous phase was extracted with 1 mL CI (24:1) and centrifuged under the same conditions. To precipitate the RNA, 0.1 volume of 3 M sodium acetate (pH 5.3) and 2.5 volumes of ethanol were added, the sample was incubated for 15 min at −20 °C and centrifuged for 15 min at 20000g at 4 °C. The pellet was washed with 100 μL 70% ethanol and resuspended in 100 μL dimethyl dicarbonate (DMDC)-treated H2O. The RNA concentration was determined using a NanoDrop UV spectrophotometer (Thermo Electron Scientific Instruments LLC, Madison, USA) by assuming an A260 of 1 for 40 μg mL−1 RNA.
:chloroform:isoamylalcohol (PCI, 25:24:1) was added and vortexed briefly. The emulsion was centrifuged for 1 min at 20000g at room temperature. The aqueous phase was collected and the PCI extraction was repeated twice. The third aqueous phase was extracted with 1 mL CI (24:1) and centrifuged under the same conditions. To precipitate the RNA, 0.1 volume of 3 M sodium acetate (pH 5.3) and 2.5 volumes of ethanol were added, the sample was incubated for 15 min at −20 °C and centrifuged for 15 min at 20000g at 4 °C. The pellet was washed with 100 μL 70% ethanol and resuspended in 100 μL dimethyl dicarbonate (DMDC)-treated H2O. The RNA concentration was determined using a NanoDrop UV spectrophotometer (Thermo Electron Scientific Instruments LLC, Madison, USA) by assuming an A260 of 1 for 40 μg mL−1 RNA.
The DNA template for the in vitro transcription of RNA 2253 was plasmid pGEM/TMV4407–619149 linearized with PciI. RNA was generated by the use of a MEGAscript® T7 High Yield Transcription Kit (Ambion, Austin, USA) according to the manufacturer's instructions.
In addition to these solely TMV-based RNA scaffolds, three types of OAs-containing RNA species with heterologous (non-TMV) portions were created and named hRNA I–III, with distinct pro- and eukaryotic coding and non-coding sequences (see ESI Fig. S1† for the respective constructs, and S10† for the images of hRNA I–III after electrophoretic separation in denaturing agarose gels). To obtain the underlying plasmid construct I, a cDNA containing the major sequence portion of the TMV OAs44 (nts 5350–5531) was inserted into pGEM®-T Easy vector (Promega, Mannheim, Germany) replacing positions 24 and 199 of the plasmid sequence by common cloning procedures.79 The plasmid was linearized with DraIII-HF (NEB, Ipswich, USA) prior to in vitro transcription by T7 RNA polymerase as described above. Due to a T7 RNA polymerase transcription terminator region80 at positions 1262–1301 of the pGEM® plasmid sequence (TTTTTTTGTTTGCAAGCAGCAGATTACGCGCAGAAAAAAA; see Promega Corporation product information sheet # 9PIP256, revised version 8/13, Madison, USA), in vitro transcription yielded a mixture of two RNA fragments: the pre-maturely terminated product of 1262 nts, and a read-through RNA of 2748 nts. The preparation is referred to as hRNA I henceforth.
The DNA template for hRNA II is based on a clone kindly provided by Tatjana Kleinow, Stuttgart, and was synthesized as follows. The full-length DNA A genome component of the plant DNA begomovirus Abutilon mosaic virus (AbMV; EMBL accession number X15983) was cloned via HindIII restriction sites into the vector pLITMUS28i (New England Biolabs, NEB, Ipswich, USA) in its reverse-complementary orientation (with respect to the subsequent linearization, see below). The cDNA of the TMV OAs (comprising TMV nts 5211–5584) was amplified by PCR from p843pe35TMVr.1 (56) with primers producing an NdeI restriction site 5′ and a SacII site 3′, and inserted via NdeI/SacII-HF (NEB) restriction into the AbMV sequence replacing a fragment from the position 4247 to 4395. This final template construct was linearized with SacI-HF (NEB) and in vitro transcribed by T7 RNA polymerase to generate hRNA II of 2982 nts.
The template DNA for hRNA III was created from a partial TMV cDNA (comprising TMV nts 4253–6398 including the OAs) by EcoRI-HF/KpnI-HF (NEB) restriction of p843pe35TMVr.1. This was inserted into EcoRI/KpnI restricted pBlueScript II SK(+) (Agilent Technologies, Santa Clara, USA) between the plasmid positions 657 and 702. The resultant template construct was linearized with ScaI-HF as above, and subjected to in vitro transcription by T3 RNA polymerase by the use of a MEGAscript® T3 High Yield Transcription Kit. The resulting hRNA III was 3303 nts in length.
Stopper design
Guidelines for the design of stopper DNA oligomers (named “stoppers” henceforth irrespective of whether they worked efficiently or not) were the same as that for PCR primers.81 They were designed to avoid secondary structures (with a minimum of 4 base pairs assumed for stable hairpin formation) and self-dimerization (≥5 self-complementary bases).82 The melting temperatures for RNA/DNA hybrids were estimated using the nearest-neighbor two-state-model and thermodynamic parameters.83 Salt correction was performed.84,85 Overhangs for toehold-release of 8 nts were found to be optimal for rapid strand displacement.86 Different from those stoppers used with the two TMV-derived RNA scaffolds, wt-RNA and RNA 2253 (see Table 1 and Fig. 1 for details), all stoppers applied with hRNA constructs addressed different non-TMV sequence portions (see Fig. S1†). Identical toehold overhangs of all DNA oligomers, and their complementary portions in the different release (“fuel”) oligomers were used in all the evaluated experiments. Oligonucleotides purified by high performance liquid chromatography were bought from Biomers (Ulm, Germany).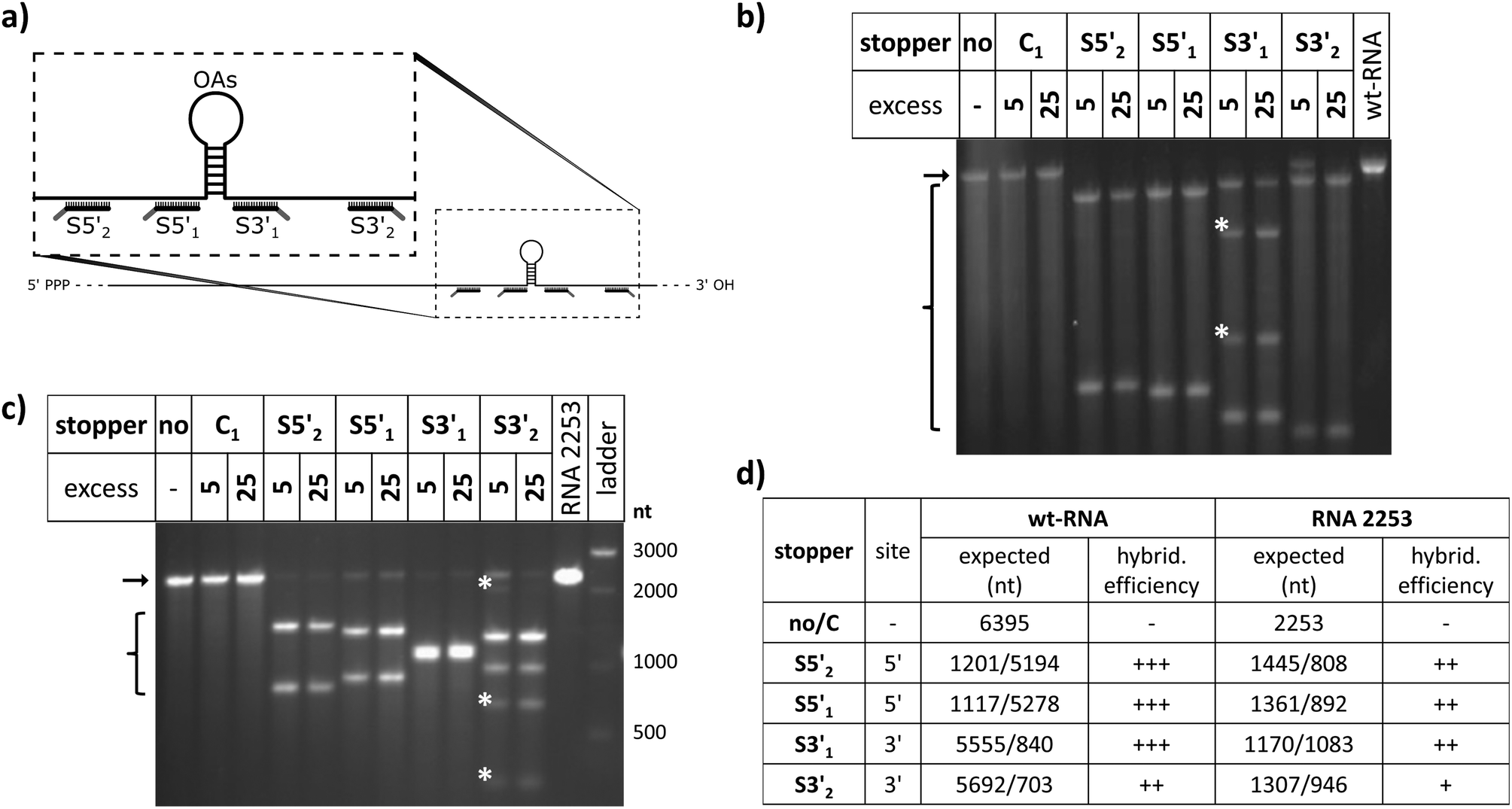 | ||
| Fig. 1 Hybridization of stopper DNA oligomers to wt-TMV-RNA and TMV-deduced RNA 2253 both containing the OA sequence. (a) Scheme of the binding sites of the different stoppers “S” on both RNAs in relation to the OAs position (symbolized as a hairpin). The black lines represent the stopper portions complementary to the RNA, and the grey lines toehold overhangs. (b) and (c) Agarose gel electrophoretic separation of the products after RNase H cleavage of DNA/RNA hybridization products of wt-RNA (b) or RNA 2253 (c). Fragments were separated on 1% or 2% agarose gels, respectively, under denaturing conditions. Stoppers were hybridized to the RNAs in 5- and 25-fold molar excess, prior to RNase H treatment. The arrows denote the bands of undigested RNA, and brackets the range of fragments expected after digestion. The white asterisks label additional fragments occurring upon RNase H incubation. (d) Expected RNA fragment lengths and apparent hybridization efficiencies of the various stoppers deduced from the RNase H assay (+++ → +: decreasing efficiency). The hybridization efficiency is reflected by the amount of cleaved RNA in relation to the non-cleaved RNA. | ||
| Task | Sitea | Name | Sequence (5′>3′) |
T
mb |
wt-RNA | RNA 2253 |
|---|---|---|---|---|---|---|
| Expected tube lengthc (nm) | Expected tube lengthc (nm) | |||||
| *Calculation with parameters for DNA–DNA hybrids,94 salt correction with 75 mM Na+, oligodeoxyribonucleotide concentration estimated with 7 μM.a Binding site on the TMV-derived RNA relative to the OAs position.b Nearest neighbor calculation93 with parameters for RNA–DNA hybrids,83 salt correction84,85 with 75 mM Na+ and 1.5 mM Mg2+, oligodeoxyribonucleotide concentration estimated with 700 nM.c For RNA as a scaffold in the presence of the respective stopper or control DNA.d C1 and C2 could also be applied to release S5′2 and S5′1, respectively. | ||||||
| Stopper DNA oligomers | 5′ | S5′2 | GAGTGGCCTCGTCGGCTCTTTC![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) ![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) ![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) |
74 | 56 | 68 |
| 5′ | S5′1 | GGTGGTTATAGCATAATTGGGA |
58 | 52 | 64 | |
| 3′ | S3′1 | CTCTTTTTCCCGGTTCGAGATCG |
64 | 261 | 55 | |
| 3′ | S3′2 | CGGCGACAGTAGCCTCCGAATC |
64 | 267 | 61 | |
| Controld | — | C1 | GAAGTCAGGAAAGAGCCGACGAGGCCACTC | — | 300 | 106 |
| — | C2 | GAAGTCAGTCCCAATTATGCTATAACCACC | — | 300 | 106 | |
| Release DNA oligomer | — | R3′1 | CGATCTCGAACCGGGAAAAAGAG |
69* | 300 | 106 |
Hybridization of stoppers, RNase H assays and denaturing electrophoresis
Hybridization was performed in 75 mM SPP buffer pH 7.2 supplemented with 1.5 mM MgCl2. Unless otherwise stated, 700 nM DNA oligomers were hybridized to 140 nM RNA (corresponding to a 5-fold molar excess of stopper over RNA; 1 μg RNA in a total volume of 3.14 μl or 9.6 μl, in the case of wt-RNA or RNA 2553, respectively.)The hybridization procedure included an initial denaturation for 5 min at 65 °C with a subsequent temperature gradient (1 °C s−1) to 30 °C and a subsequent storage on ice until further use. Hybridization success was evaluated with a modified RNase H assay for wt-TMV and RNA 2253, respectively.87 Briefly, 1 μg and 0.5 μg of wt-RNA and RNA 2253, respectively, with or without hybridized DNA (corresponding to 3.1 μl (wt-RNA), 4.8 μl (RNA 2253) of the hybridization mixtures) were treated with 0.3 U of RNase H (Roche Applied Science, Mannheim, Germany) for 20 min at 37 °C. The enzyme was inactivated at 65 °C for 20 min. RNA fragments were analyzed in 0.3 M formaldehyde-containing 1% or 2% agarose gels for wt-RNA or RNA 2553, respectively, in 1 × 3-(N-morpholino)propanesulfonic acid (MOPS) buffer (10× MOPS: 200 mM MOPS, 50 mM NaOAc × 3H2O, 10 mM EDTA pH 7.0) with buffer circulation according to standard protocols.79 Gels were pre-incubated at 3.5 V cm−1 for 15 min before use. RNA samples (1 μg or 0.5 μg of wt-RNA or RNA 2253, respectively) were incubated in 15 μl denaturing solution (1.7 μl 10× MOPS, 9.9 μl deionized formamide, 2.9 μl 37% formaldehyde, 0.5 μl DMDC-treated H2O, 0.05 μl 1% EtBr) for 15 min at 65 °C, supplemented with 1.5 μl loading buffer (1 mM EDTA, 0.4% bromophenol blue, 50% glycerol) and separated electrophoretically at 2.3 V cm−1 for 15 min and at 3.2 V cm−1 for the remaining time (around 4 h). An ssRNA molecular weight marker (N0362S, New England Biolabs Inc.) served as the calibration standard.
Nucleoprotein tube assembly with or without stoppers and subsequent RNase H assays
TMV CP (from wt-TMV) was prepared from virus particles isolated from infected plants as reported.88 In order to induce 20 S disk formation,89 CPs were incubated in 75 mM SPP buffer at pH 7.2 for at least 48 h at room temperature (RT). For RNA-directed in vitro assembly, 0.2 μg μl−1 (final concentration, f.c) of RNA incubated with stopper or control oligomers (see above) were incubated with 3.8 μg μl−1 (f.c.) of TMV CPs at 25 °C for 3 h (RNA:CPs w/w ratio: 5/95). Reactions were stopped by cooling on ice and stored at 4 °C for further experiments. For RNase H assays, aliquots were supplied with 0.3 U of enzyme and digestion was allowed for 20 min at 37 °C. The enzyme was inactivated at 65 °C for 20 min. Nucleic acids were purified from these samples by using an RNeasy MinElute Cleanup Kit (Qiagen) and examined electrophoretically in formaldehyde-containing agarose gels as described above. Further aliquots were subjected to native agarose gel electrophoresis and TEM analysis. The preparation of TMV-like nanorods with subdomains of wt-CP and CPLys-Bio56 is described below.
Toehold-release of stopper S3′1
For toehold-release of stopper S3′1 after its hybridization to RNA (see above) and optionally after 3 h of partial nanotube reconstitution at 25 °C, a 10-fold molar excess of the release DNA oligomer R3′1 over the stopper S3′1 was added and incubation performed at 25 °C for 4 h unlike otherwise stated. The samples in the absence of CPs after the hybridization reaction but with R3′1 added served as controls and were analyzed by denaturing agarose gel electrophoresis after an RNase H assay as described above. The samples of partially and fully assembled nanotubes were prepared for TEM or native agarose gel electrophoresis.“Stop-and-go” fabrication of TMV-like nanotubes with biomolecule-fashioned subdomains (domain-TLPs)
The “stop-and-go” technology was extended to the production of nanotubes with longitudinal domains of distinct TMV CP chemistry, allowing selective coupling of molecules to a single domain only. This enabled the visualization of the domains obtained by the use of wt-RNA and S3′1 in combination with R3′1. To this end, the TMV wt-CP was used in the first assembly step (to yield the long 260 nm domain), and the second (short 40 nm) domain made use of a TMV mutant, TMVLys, exposing an accessible amino group on the surface of every CPLys subunit.56 These amino functions were covalently coupled to succinimidyl-(NHS-) PEG12-biotin-linkers (EZ-Link® NHS-PEG12-Biotin, Thermo Scientific, Rockford, USA) using the manufacturer's protocol. CPLys-Bio was prepared from the resulting biotin-functionalized TMVLys-Bio particles as described above.88Hybridization of stopper S3′1 to the wt-RNA scaffold and the subsequent partial assembly with wt-CP was performed as detailed above. Stalled nanotubes in the presence of S3′1 were sedimented via ultracentrifugation (Beckman Coulter Optima L90K, 45 Ti rotor) at 38000 rpm (≈113000g) and 4 °C for 2 h, to pellet assemblies with sedimentation coefficients above ≈95 S. Thereby, particles stopped precisely at the stopper site were enriched and, in turn, free wt-CP and pre-maturely stopped small nanotubes below 95 S removed. R3′1 and CPLys-Bio (1.5-fold assembly excess in relation to the remaining 13% unencapsidated RNA directing further 40 nm tube growth) were added to the resuspended partial nanotubes and incubated at 25 °C for 16 h. To remove free CPLys-Bio after completion of the assembly, the resulting domain-TLPs were sedimented via ultracentrifugation (Beckman Coulter Optima L90K, 45 Ti rotor) at 34000 rpm (≈90500g) and 4 °C for 2 h. These products were subjected to bioaffinity coupling of avidin (Sigma-Aldrich, Saint Louis, USA) followed by another ultracentrifugation step as above, to enable the visualization of avidin-decorated subdomains by TEM.
Native agarose gel electrophoresis
Native agarose gel electrophoresis was performed following a variation of a published protocol.55,90 About 20 μg of products arose under the conditions promoting nucleoprotein tube growth (corresponding to 1 μg of scaffold RNA) were applied to a 1.0% agarose gel for wt-RNA or a 1.5% agarose gel for RNA 2553 as well as the hRNAs, prepared in 50 mM SPP buffer pH 7.2, and run for 19 h at 1.1 V cm−1 with buffer circulation. Buffer and gel contained EtBr to visualize the uncoated RNA. Proteins were detected by post-electrophoretic staining with 0.1% Coomassie Brilliant Blue R250 (Serva Electrophoresis, Heidelberg, Germany) for 1.5 h and extensive washing with destain solution.79Transmission electron microscopy
The presence and lengths of nanotubes, as well as the spatial organization of domain-TLPs, were analyzed by TEM. The samples were adsorbed to Formvar©-carbon-coated copper grids, stained with a solution of 1% uranyl acetate and 0.25 mg ml−1 bacitracin91 and analyzed using a Tecnai G2 Sphera transmission electron microscope (FEI, Eindhoven, Netherlands) at 200 kV with a 16 megapixel camera (TemCam-F416R, TVIPS, Gauting, Germany). The images were evaluated by software Image J.92Statistical evaluation of products generated through the “stop-and-go” methodology
For quantification and statistical analyses, the lengths of randomly selected nanotubes readily separated from each other on the coated grid surface were measured by the help of Image J software92 and classified (10 nm class size). Three independent assembly experiments for each of the four statistically evaluated RNA scaffolds were conducted statistically, to determine the inter-experimental variation and to scrutinize the consistency between repetitions using the non-parametric Mann–Whitney Rank Sum Test. These data are presented as boxplots (lines: median values, box boundaries: 25/75% quartiles, whiskers: 10/90% percentiles, dots: 5/95% percentiles). The length class distributions were averaged for every separate length class, and are displayed in histograms with their standard deviations. The histograms were compiled using Microsoft Excel. For statistical analyses and boxplots, SigmaStat version 4.0.0.37 (Systat Software, Inc.) was used.Results and discussion
To evaluate the effect of DNA stoppers hybridized to RNA scaffolds guiding the self-assembly of TMV-like nucleoprotein tubes, four 30- or 31-meric oligodeoxyribonucleotides were designed. They possess portions complementary to distinct sites 5′ or 3′ of the OAs of both wt-TMV RNA and a shortened RNA deduced thereof, respectively (Table 1, Fig. 1a). These 22- to 23-nucleotide scaffold binding portions were 5′- or 3′-terminally extended by an 8-nucleotide stretch non-complementary to the RNA (underlined in Table 1), to serve as the toehold sequence enabling their removal via a fuel oligomer. Nanotube lengths expected to assemble in the presence of the different stably bound stoppers are shown in Table 1 for both wt-RNA and RNA 2253, respectively. The latter is an in vitro transcribed OAs-containing RNA 2253 nts in length, derived from a TMV cDNA subclone (see the Experimental section). Non-complementary DNA oligomers (“C”) with lengths comparable to the stoppers were included in the experiments as negative controls. R3′1 served as a release DNA oligomer for the stopper S3′1, triggering the strand-displacement reaction by its portion reverse-complementary to the stopper's toehold (underlined in Table 1).The binding capabilities of the distinct stoppers to the nanotube-scaffolding RNAs were tested using an RNase H assay87 with two different ratios of stoppers to both RNAs. Hybridization is indicated by the formation of two RNA fragments representing portions up- and downstream the recognition sites, and concomitant disappearance of the original template band; hence the hybridization efficiency correlates with the amount of cleaved RNA in relation to non-cleaved RNA. In this way, the assay detected the saturation of both RNA constructs with stoppers at a 5-fold molar oligodeoxyribonucleotide excess, except for stopper S3′2, which exhibited a substantially lower apparent hybridization efficiency (Fig. 1b and c). It demanded for an at the minimum 25-fold molar excess in combination with both RNA templates (Fig. 1b and c) to mediate complete target degradation. Additional non-expected RNA fragments obtained in the presence of S3′1 and wt-RNA, as well as S3′2 and RNA 2253 (asterisks in Fig. 1b and c) might result from non-specific DNA/RNA binding events occurring during RNase H incubation in these stopper/scaffold combinations.
To find out how the RNA-directed in vitro self-organization of TMV-like nucleoprotein tubes was influenced by the different DNA stoppers, assembly-competent CP preparations containing pre-formed disk aggregates89,95 were added to the RNA/stopper complexes (schematically shown in Fig. 2b) and to control mixtures, and were incubated at 25 °C for 3 h. This resulted in a self-assembly of stable nanotubular products in the presence of all DNA oligomer/RNA combinations analyzed, as detailed henceforth. Whether or not the resulting tube lengths corresponded with the positions of the assembly-blocking stoppers on the RNAs (Fig. 2c, d and Table 1 for the expected product lengths) was determined by a combined analysis of the bulk products employing native agarose gel electrophoresis and TEM. Consistent results were obtained for both RNAs and are shown here for wt-RNA (and for RNA 2253 in the ESI†).
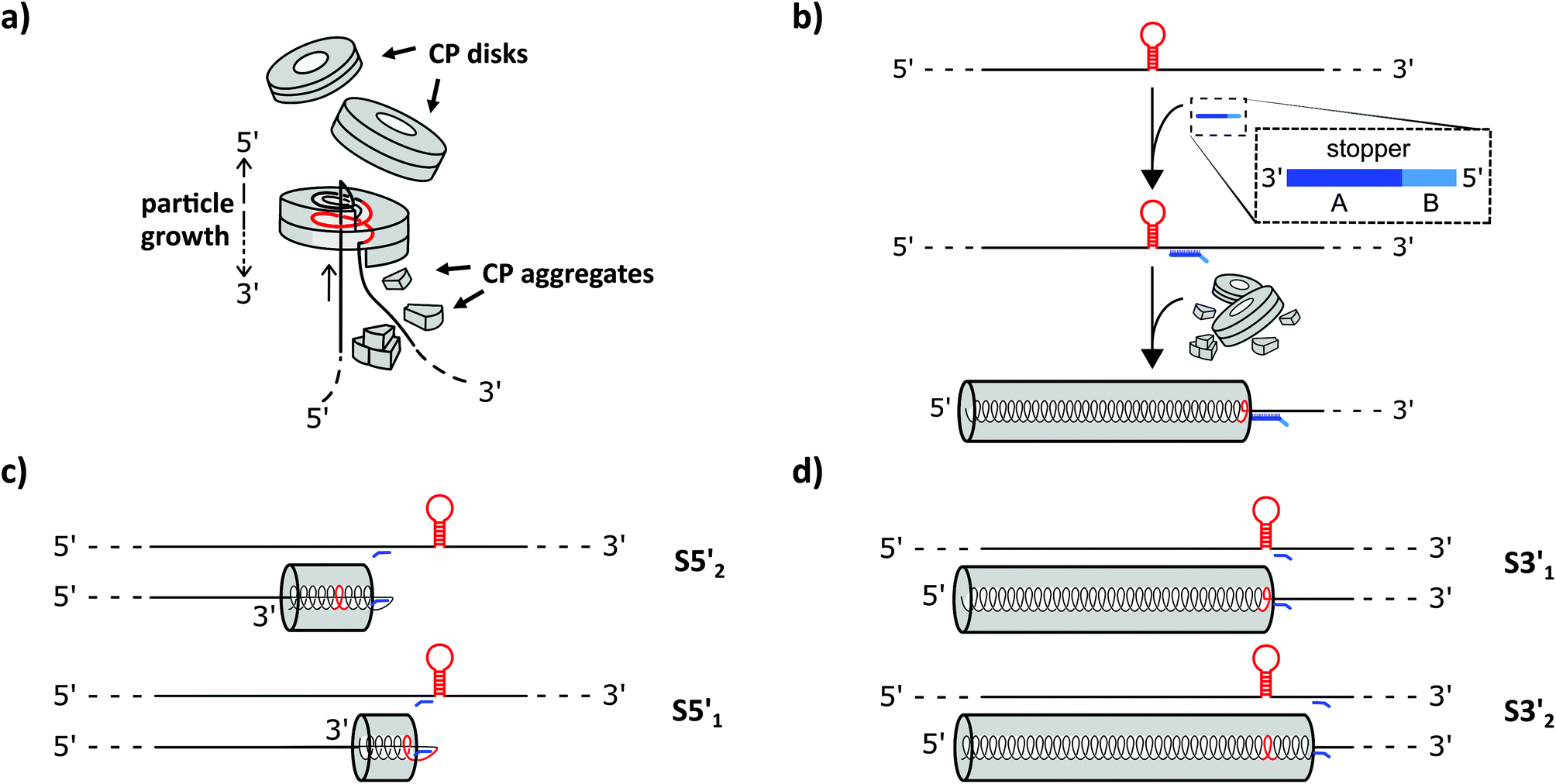 | ||
| Fig. 2 Mechanistic details assumed for the assembly of TMV-like nanotubes in vitro: initiation at the OAs structure, and putative stalling in the presence of effective DNA blocking elements targeting distinct RNA sites. (a) TMV assembly initiation through the interaction of a double-layered CP disk with the OAs (red) in the viral RNA, followed by a conformational switch into a helical complex. Thereby, an RNA “travelling loop” is formed at the growing 5′ nanotube end, which is accompanied by a foldback of the 5′-RNA portion that is threaded into the nanotube channel, with its 5′ RNA terminus protruding from the channel adjacent to the 3′ RNA portion. The latter undergoes packaging into small CP aggregates, whereas the 5′-stretch is pulled towards the “travelling loop” upon its spiralization and sandwiching between the short CP helices serially added to the 5′ tube end. (b) Scheme of partial ribonucleoprotein tube assembly, initiated at the OAs, arrested by a stopper “S” (blue) containing a 3′ portion that hybridizes to the RNA scaffold (A), and a 5′ non-complementary portion serving as a toehold (B, potentially accessible to base-pairing with a third nucleic acid strand). (c+d) Cartoons illustrating the distinct types of stalled nanotubes with a non-encapsidated RNA portion expected to arise upon effective blocking of their assembly by different stoppers, hybridized to sites 5′ (c, upstream) or 3′ (d, downstream) of the OAs on wt-RNA. | ||
TMV-like tubes with partial RNA encapsidation blocked at the DNA/RNA hybrid sites (Fig. 2c, d and S2†) will not only be shorter than their fully assembled counterparts, they will also exhibit a protruding ssRNA portion and thus a higher RNA/protein ratio with an accordingly increased negative net charge. This results in a higher electrophoretic mobility in native gel systems in dependence of both particle size and charge.55,56,90Fig. 3 and S3† indicate that out of the four oligodeoxyribonucleotides tested, stopper S3′1 exerted the strongest and most efficient impact on the RNA-directed self-assembly of TMV CPs. Compared to negative controls without (no), or with non-binding DNA oligomer (C1), the products of assembly with S3′1/wt-RNA resulted in dominant Coomassie Brillant Blue-stained particle bands of increased electrophoretic mobility upon application of ≥5-fold molar stopper excess (Fig. 3, right, asterisks). Ethidium bromide staining yielded co-localized signals (Fig. 3, left, asterisks) indicative of a protruding non-encapsidated RNA stretch. Hence it could be concluded that stopper S3′1 was able to block nanotube assembly in the 3′ direction efficiently, giving rise to partially assembled TMV-like particles accumulating in a single major electrophoretic fraction. With RNA 2253, S3′1 had the strongest effect of all four stoppers as well, although the electrophoresis data suggested a lower proportion of incomplete tubes than that with wt-RNA (Fig. S2†).
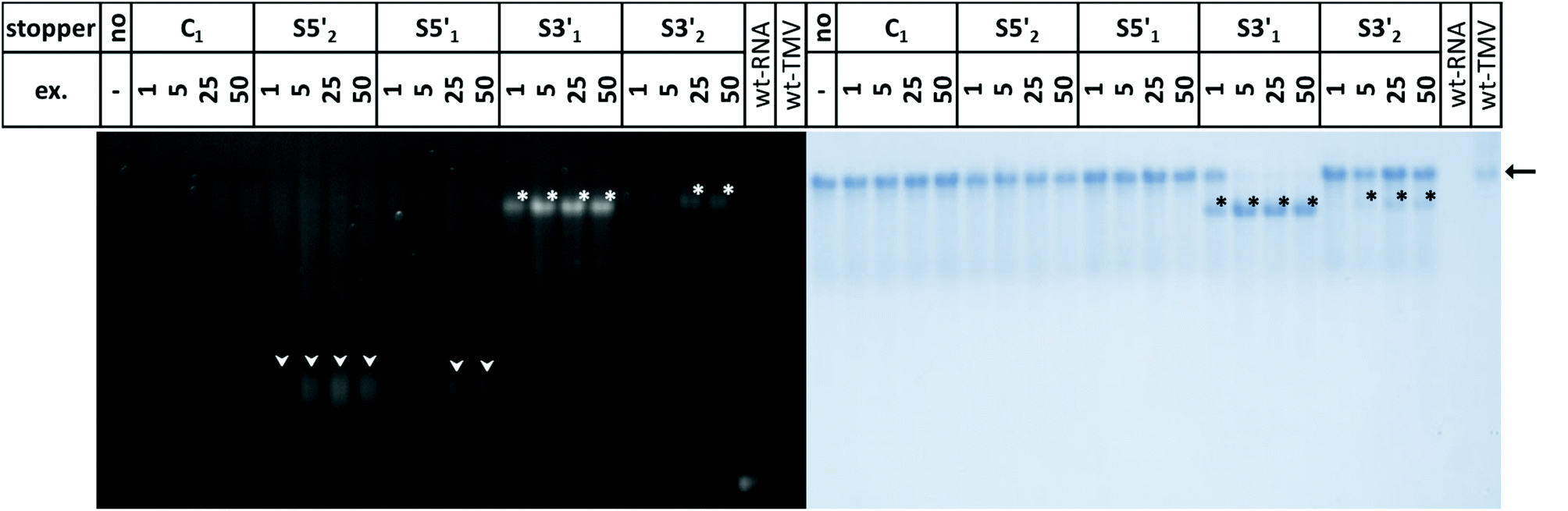 | ||
| Fig. 3 “Stop”: the influence of distinct stoppers on the self-assembly of TMV CP with wt-RNA, as visualized after nanoparticle separation in a native agarose gel in comparison to control reaction products. Two different control samples were prepared: “no”, i.e. fully assembled nanotubes scaffolded by RNA devoid of stoppers, and “C1”, i.e. fully assembled nanotubes obtained in the presence of a DNA oligomer with no complementarity to the RNA. Hybridization was performed with variable molar excesses (ex.) of stoppers over RNA for 5 min at 65 °C, cooling to 30 °C with a rate of 1 °C s−1, followed by assembly for 7 h at 25 °C. wt-TMV and wt-RNA serve as markers. The gel was first stained with ethidium bromide (left), followed by Coomassie Brilliant Blue-staining (right). The arrow indicates bands of fully assembled tubes as confirmed by the wt-TMV particles. The black/white asterisks (*) label the bands with higher electrophoretic mobility. The white triangles indicate a faint ethidium bromide signal corresponding to these bands, and residual free RNA; their Coomassie Brilliant Blue signal is below the visualization limit. | ||
By means of the second DNA oligomer (S3′2) addressing a site 3′ (downstream) of the OAs of both RNA scaffolds, substantially smaller fractions of particles exhibiting increased electrophoretic mobility were obtained, with a need of at least 25-fold molar stopper excess for well-detectable bands (Fig. 3 and S3†). The most prominent bands of samples “S3′2/RNA” in these gels represent a high proportion of fully assembled nanotubes, despite the presence of the stopper. These findings are in agreement with its low hybridization efficiency indicated by RNase H analysis. As its Tm equals to that of the potent stopper S3′1, short- or long-range secondary structures of the RNA templates, or intermolecular tertiary interactions are likely to compromise the accessibility of the S3′2 target sequences.
For the two stoppers addressing sites 5′ of the OAs (S5′2 and S5′1), with significantly higher or lower Tm value than the 3′ stoppers, respectively (see Table 1), the electrophoretic analysis did not reveal any impact on the TMV CP assembly with wt-RNA (Fig. 3), and only negligible effects in combination with RNA 2253 (Fig. S3†), although both stoppers had been shown to hybridize to their target sites effectively (Fig. 1). RNA incubated with a control DNA oligomer (C1 in Fig. 3, with no complementarity to the RNA scaffolds) directed the formation of tubes with the same mobility as the full-length particles assembled with free wt-RNA (“no”), and wt-TMV particles isolated from plants.
Taken together, these electrophoretic analyses indicated that the stopper hybridized efficiently to 3′ of the OAs (S3′1) imparted an effective blocking of TMV-like tube assembly. In contrast, two stoppers binding to 5′ of the OAs with equal efficiencies were obviously not able to stall tube growth, even though the melting temperature of S5′2 was ∼10 °C above that of the potent stopper S3′1.
TEM analysis substantiated these conclusions and revealed straight tubes in all samples containing RNA and TMV CPs (Fig. 4 and S4,† and data not shown). In the presence of stopper S3′1 and wt-RNA, the majority of particles were about 30–40 nm shorter than the complete rods in the control samples (Fig. 4). Also with RNA 2253, S3′1 arrested nanotube growth efficiently (i.e. much better than anticipated from the electrophoretic analyses; Fig. S4†). The length reductions met the expectations for an S3′1 binding site-specific blockage of the assembly in combination with both RNA scaffolds (Table 1). RNase H assays with such TMV-like particles confirmed this partially assembled state by producing the same fragment patterns as that with bare RNAs after hybridization of stopper S3′1, and the expected product lengths (Fig. S5†).
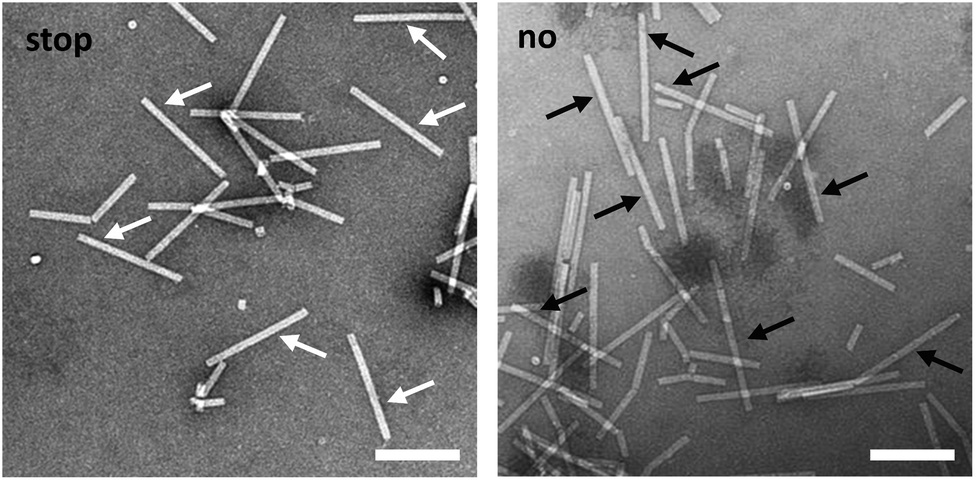 | ||
| Fig. 4 Visualizing the “stop” state in the presence of stopper S3′1 hybridized to wt-RNA during the self-assembly with TMV CPs. TEM analysis of the respective nanotubular products. “stop”: partial, stalled assembly directed by S3′1/RNA. The white arrows show the resulting particles with lengths in the range of 260 to 270 nm. “no”: Assembly directed by RNA without any stopper. The black arrows show fully assembled nanotubes with lengths between 300 and 310 nm. Scale bars: 200 nm. | ||
To investigate whether an interrupted assembly could be “switched on” again on demand, we conducted a series of “stop-and-go” experiments (schematically depicted in Fig. 5a) and characterized the products before and after removal of stopper S3′1via toehold-release. The aliquots were subjected to native agarose gel and quantitative TEM analysis. The initial tests in this context confirmed full functionality of the stopper/release oligodeoxyribonucleotide pair S3′1/R3′1 (see Table 1) on RNA in the absence of TMV CPs. Complete displacement of S3′1 from the S3′1/RNA heteroduplex by using a ten-fold excess of release strand R3′1 was verified by the RNase H assay for both RNA scaffolds after four hour incubation at 25 °C (ESI Fig. S5†).
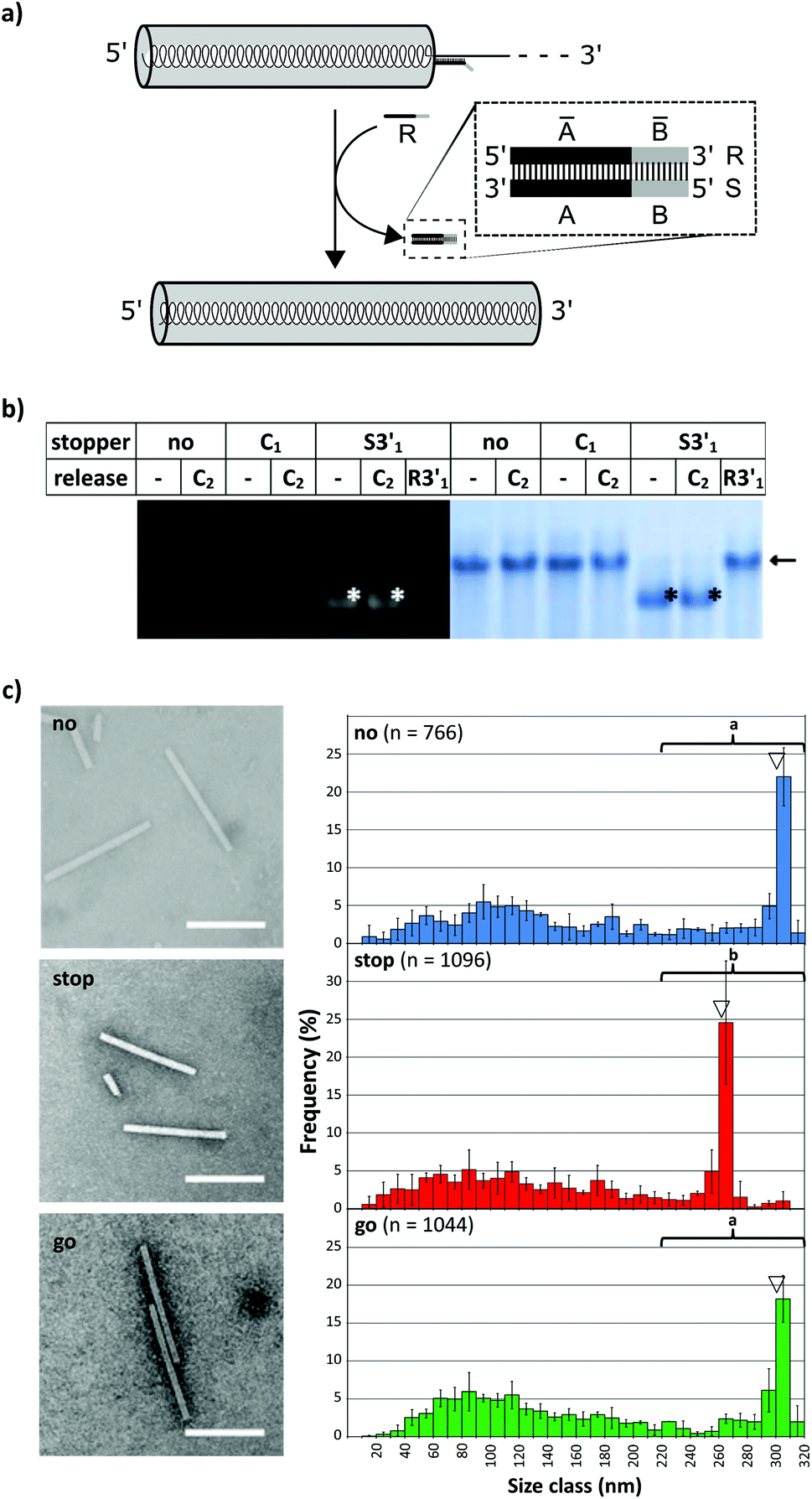 | ||
| Fig. 5 “Stop-and-go”: controlled stop of the RNA-directed self-assembly of TMV CPs by hybridization of stopper S3′1 to wt-RNA, and its subsequent displacement by toehold-release with the DNA “fuel” oligomer R3′1. (a) Scheme of the “stop-and-go” principle. (b) Native agarose gel of products in the “stop”, “release” and “control” reaction states, i.e. after incubation at 25 °C to allow the assembly of nucleoprotein tubes in the absence (no) or presence (S3′1) of the stopper, or after its subsequent release by a suitable oligomer (R3′1). The arrow indicates the bands of fully assembled tubes as confirmed by comparison with wt-TMV particles. The black/white asterisks (*) label the bands of increased electrophoretic mobility, reflecting stalled nanotubes. (c) Analysis of the resulting nanotube length distributions. “no”: Assembly directed by RNA without any stopper; “stop”: temporarily stopped assembly directed by S3′1/RNA; “go”: assembly directed by S3′1/RNA first, after subsequent addition of R3′1. Left: TEM images; scale bars: 200 nm. Right: Corresponding histograms with n structures analyzed. The triangles indicate the expected nanotube lengths. The frequencies of three independent experiments were averaged for every length class, error bars show the standard deviations. The cumulated length data under the brackets were tested for significant differences (Mann–Whitney Rank Sum Test, p < 0.001) between the applications. The same lower case alphabetic characters indicate no significant difference. | ||
The DNA-steered stepwise “stop-and-go” assembly of TLPs was set up under the same conditions. Following hybridization of stopper S3′1 to either RNA in a temperature gradient, an assembly of nanotube domains upstream (i.e. 5′) of the stopper binding sites was achieved through the addition of an excess amount of TMV CPs and incubation at 25 °C for 3 h (see Fig. 2). Reaction mixtures then were divided into several aliquots and a ten-fold excess of release DNA oligomer R3′1 over the stopper added to a subset of them (“release”), but not to the others (“stop”; Fig. 5). Controls were generated with the same RNA scaffolds in the presence of DNA strands lacking complementarity to the RNAs (“C”), to direct the assembly of complete nanotubes after otherwise being treated as the “stopper”-supplemented samples.
Native agarose gels indicated that re-initiation of stalled nanotube growth indeed took place with both RNAs efficiently (Fig. 5b). The nucleoprotein complexes generated upon toehold-mediated displacement of stopper S3′1 by oligomer R3′1 in the presence of excess TMV CPs in the reaction mixture had gained the same electrophoretic mobility as that of the completely encapsidated scaffold RNA (arrows in Fig. 5b and S6†). The particle length distributions, as revealed by electron microscopy, represented the different assembly stages: the electron micrographs (Fig. 5c and S7†) show exemplary particles, and the histograms depict the lengths of nanotubes for wt-RNA or RNA 2253, respectively, after uninterrupted (no), stopped (stop), and re-started assembly (go). Compared to the particles grown continuously on wt-RNA (Fig. 5c), with a major fraction of about 300 nm length (no), the ‘stopped’ reaction yielded predominant tube lengths of 260–280 nm. Subsequent toehold-release of the DNA blocking element (go) reconstituted a size distribution corresponding to that of complete particles (no). Congruent results were obtained with RNA 2253 (Fig. S7†): in both cases, the largest fractions of stalled particles exhibited tube lengths in good agreement with the position of the stopper DNA oligomer on the RNA scaffold, and could be re-activated to eventually reach full-length through stopper displacement. The presence of additional TMV-like tubes of heterogeneous size in all assembly reactions (Fig. 5 and S6†) reflects a background of diverse CP-containing complexes typically received upon reconstitution of TMV, including prematurely stalled nucleoprotein tubes due to e.g. unfavorable RNA conformations and stacked RNA-free CP assemblies; compare e.g.ref. 31, 43, 46, 49 and 56. “Oversized” particles result from head-to-tail aggregates with RNA-scaffolded tubes.43
The major nanotube length classes in all the cases corresponded to the RNA portion accessible between the 5′-end and either the DNA blocking element, or the RNA scaffold's 3′-terminus. The data represent measurements of several hundred particles per experiment, as depicted in the histograms. The differences between the repeated assembly experiments per treatment were not significant (Fig. S8†) and the values could therefore be cumulated for the comparison of different treatments (Fig. 5c). In conclusion, the stepwise assembly approach seems to be both robust and predictable with respect to the resulting nanotube population. Furthermore, repeated testing over extended time periods revealed that partially assembled particles equipped with a DNA stopper (S3′1/wt-RNA) had remained stalled after 12 days of storage at 4 °C as well as at 20 °C, but were fully accessible to reconstitution of the assembly process upon using excess of TMV CPs in the reaction mixture simply by addition of R3′1 (data not shown).
To find out if the “stop-and-go” procedure is also amenable to non-TMV RNA and thus to a broadly applicable technology, three further scaffold constructs based on the sequences of distinct sources were generated and analyzed for their compatibility with the approach. The heterologous RNAs (named hRNAs I to III henceforth), transcribed from the corresponding plasmids, all include the TMV OAs, but embedded in distinct RNA segments derived from variable sources (see Fig. S1†). They contain no, a short, or a longer additional OAs-surrounding TMV RNA context, and substantial portions of prokaryotic (bacterial or phage in hRNAs I and III), or eukaryotic (plant viral in hRNA II) genomes consisting of coding and non-coding stretches, respectively (see the ESI†). Stopper sequences were designed to target non-TMV portions downstream (i.e. 3′) of the OAs in all the cases. After having identified a well-binding DNA oligomer out of four to five variants tested for every RNA (not shown), full “stop-and-go” experiments were carried out in combination with the respective complementary release oligomers applying the same conditions as the native TMV RNA scaffolds. The products of repeated independent preparations (Fig. S9†) representing the distinct stages of the assembly (Fig. 6), as well as control reactions lacking a stopper (Fig. S10†), were subjected to qualitative and quantitative electron microscopy analyses, comparing the observed particle length distributions with the predicted sizes of intermediate (stop) and completed (go) nanotubes.
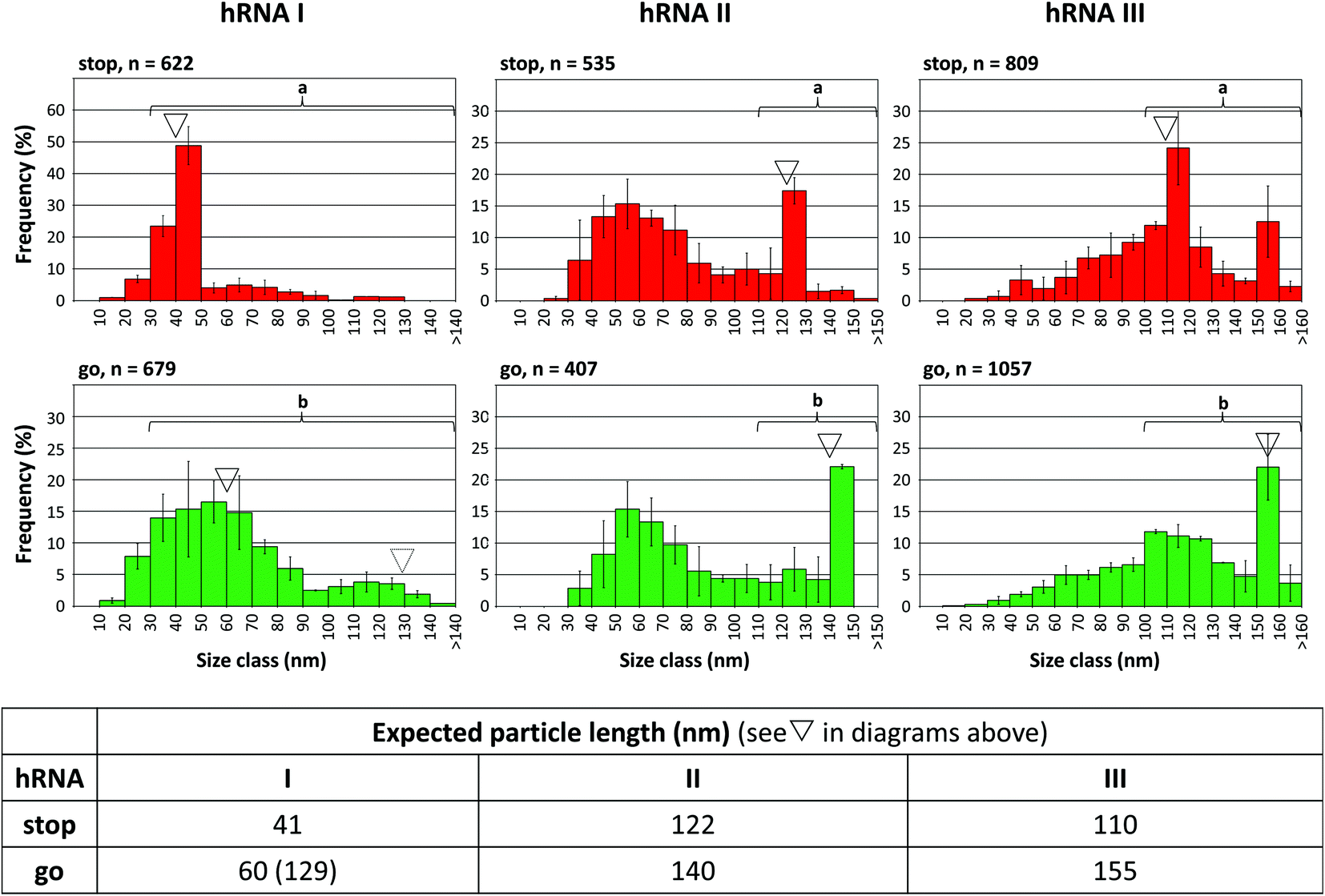 | ||
| Fig. 6 “Stop-and-go” employing heterologous non-TMV RNA scaffold sequences: controlled “stop” of the RNA-directed self-assembly of TMV CPs by annealing suitable stoppers to three hRNAs, and their subsequent displacement by toehold-release with the DNA “fuel” oligomer: “go”. The histograms represent n structures analyzed. The frequencies of three independent experiments were averaged, error bars show the standard deviations. The triangles indicate the expected nanotube lengths. Statistic comparisons as described for Fig. 5. | ||
All of the RNA/stopper/release oligomer combinations met the expectations: the length classes containing the stalled (see ∇ in Fig. 6), or the fully assembled products, respectively, were the most prominent fractions consistently, before and after DNA-directed release of the stoppers (for details, refer to the figure legend and the ESI†). For the hRNA I preparation containing two RNA subspecies (Fig. S1 and S10,† and the Experimental section above), the major class of stopped particles is in good agreement with the expected ∼41 nm length, showing that the stopping process worked effectively on both RNAs in the mixture. After the release of the stopper, encapsidation of the longer 2748 nt RNA obviously was less homogeneous than that of the 1262 nt species, as no second peak at a tube length of 130 nm arose (Fig. 6, dotted triangle), which is in accordance with the comparable results obtained in the absence of a stopper (Fig. S10†).
Although some variation in the reaction efficiencies is obvious, these tests verified that an externally DNA-guided “stop-and-go” control over the OAs-nucleated RNA encapsidation by TMV CPs is possible in a predictable and technically simple fashion with heterologous non-TMV sequences. It might be feasible to further improve their packaging efficiencies by optimizing the reaction conditions for the different stages of the “stop-and-go” procedure individually for each RNA. Here, all experiments with the hRNAs were conducted exactly as adjusted for wt-RNA.
Taken together, applying native TMV and also foreign RNA sequences, we have shown that it is possible to arrest the RNA-scaffolded self-assembly of TMV CPs site-selectively with the help of mere RNA/DNA base pairing, without covalent cross-linkage of the DNA element to the RNA as done in an earlier study on the TMV assembly in vitro.96 Interestingly, solely particle growth in the 3′ direction was blocked efficiently by the hybridization of 23-meric oligodeoxynucleotide stretches, yielding major tube fractions of the expected lengths. DNA bound with similar efficiency or even substantially higher DNA/RNA melting temperature to the RNA domain 5′ of the OAs was not suited to stall nanotube extension correspondingly.
We attribute this domain-dependent blocking amenability of the RNA to the different mechanisms of up- versus downstream assembly of TMV CP helices.45,97,98 If “disks” add to the nascent nanotubes in the 5′ direction of the OAs serially, 102 nucleotides are incorporated at once per “disk”, which corresponds to about 10 times the amount of nucleotides packaged per step in the 3′ direction with the predominant addition of small CP oligomers to the RNA. Therefore, upon 5′-progressing assembly, a lot more free energy is released per time unit than that in the opposite direction. Concomitantly, growth in this direction proceeds about 4 to 5 times faster than 3′ assembly, as shown in our46 and others’ previous experimental approaches.43,45and references therein This seems to suffice for the displacement of 5′ hybridized DNA oligomers from the RNA by the nascent nucleoprotein tube under the conditions applied.
The site-selective blockage of nucleoprotein tube growth imparted by a DNA stopper hybridized 3′ of their OAs to the RNA scaffolds was fully reversible: toehold-mediated release of the blocking elements after addition of an inverse-complementary “fuel” DNA restored the RNA-guided assembly, attaining complete encapsidation of the scaffolds’ 3′-domains. The process exhibits analogies to the postulated pausing of TMV virion assembly at certain RNA-internal secondary hairpin structures: this has been proposed as one reason for the formation of a characteristic 70 nm long tube fraction during the in vitro reconstitution of wt-TMV, which could be converted into full-length products by means of additional CPs added.99 Upon packaging of heterologous non-viral RNA sequences in vitro, substantial proportions of shortened tubes in distinct size classes were obtained and attributed to stable internal base-pairing obstructing the elongation of TMV-like tubes at specific sites.57,59 Both studies, however, have neither been extended to the identification of the exact blocking positions, nor to methods enforcing completion of the prematurely arrested products in the reaction mixtures by external intervention.
Finally, we sought to find out if the “stop-and-go” technology enables the fabrication of nanotubes with tightly defined, differently addressable longitudinal domains. To get access to enriched preparations of such domain-TLPs, with subdomains containing distinct CP species, an intermediate purification of partially assembled CP nanotubes without degradation of the protruding RNA is essential. Hence, a protocol for the stepwise “stop-and-go” assembly of wt-RNA with two CP types was developed, with wt-CP used for the first 5′-terminal domain. Ultracentrifugation was optimized to remove free CPs and by-products of small, inefficiently assembled tubes from sedimented particles stalled at the intended “stop” positions. Upon release of the stopper DNA, nanotube growth was retaken and completed with a second CP variant exposing a biotin moiety on a NHS-PEG12-biotin linker coupled to a lysine residue on the outer CP surface (CPLys-Bio). Bioaffinity coupling decorated the CPLys-Bio-domains with avidin, which was then detectable as a fuzzy corona by electron microscopy (Fig. 7). The envisaged domain-TLP structures were indeed efficiently generated, with smooth wt-CP domains of the expected 260 nm length, and avidin-fashioned CPLys-Bio-domains measuring 45 nm, of which the extra 5 nm can be attributed to the size of the linker-installed avidin.
 | ||
| Fig. 7 Electron micrograph of TMV-like nanotubes with two subdomains of distinct CP variants. These domain-TLPs were prepared through the stepwise, externally controlled “stop-and-go” procedure. Ultracentrifugation firstly enriched stalled nanotubes exposing wt-CP, and – after stopper release and nanotube completion – fully assembled two-domain nanotubes. Avidin was coupled to the second, functionalized CP species (CPLys-Bio). The dotted arrow denotes a smooth wt-CP domain, the small continuous arrow the adjacent avidin decorated CPLys-Bio domain. Scale bar: 200 nm. | ||
Conclusions
The dynamic DNA technology-based strategy applied here for the first time to achieve tight external dimensional and temporal control over the assembly of ribonucleoprotein nanotubes introduces a novel steering handle for the fabrication and adaptation of TMV-based building blocks. The use of externally added stopper DNA oligomers has allowed the tuning of the tubes’ aspect ratio via the target site of hybridization. It thus may rationalize and economize the production of soft-matter nanotubes for technical and biomedical purposes in which a single RNA type can guide the growth of size-modulated particles, which were shown to exhibit selective rheology-regulative31 or tissue-targeting42 capacities. Post-fabrication separation of the desired major length fraction according to the particles’ sedimentation coefficient has been demonstrated to work efficiently.99Particularly, the stalling mechanism is shown to be reversible on demand, as the subsequent addition of an appropriate release DNA re-initiated the elongation of the nucleoprotein helix upon displacement of the blocking element, up to complete encapsidation of the RNA scaffold. This “stop-and-go” strategy provides the opportunity to transform nanotubes of distinct lengths stepwise into products of increased size with altered physical characteristics. By repetitive “stop-and-go” procedures, utilizing a number of site-selectively hybridized DNA stoppers and their sequential sequence-dependent release, it seems feasible to generate “striped” TMV-like particles with two or more longitudinal domains, and even barcode-like patterns. This has been realized for a first proof-of-concept product species through the successive use of two CPs with different functionalities exposed, resulting in nanotubular templates with selectively addressable surface regions. We could show that the dimensions of such domains are much better defined than those previously achievable by a serial addition of limited portions of distinct TMV CPs one after the other to a uniform RNA scaffold.56 Eventually, the new technology might lead to novel types of rigid soft-matter nanotools, enabling a spatially precise arrangement of functionalities on a scale most interesting for various fundamental analyses of molecular cross-talk.
Acknowledgements
We owe thanks to our gardeners, Annika Allinger, Marvin Müller and Diether Gotthardt, for taking great care of the plants, Sigrid Kober for isolating TMV particles from plants, Conny Kocher for support with TEM grid preparation, and PD Dr Tatjana Kleinow for providing the plasmid pLitmus28i_AbMV A for further cloning steps. Many thanks to Rebecca Hummel for preparing RNA and CPs from TMV particles as well as for endless hours of reproducing experiments and taking electron micrographs. We also like to thank PD Dr Michael Schweikert for his introduction into the operation and his great support with knowledge of the transmission electron microscope. We are grateful to the Baden-Wuerttemberg-Stiftung, Network of Competence “Functional Nanostructures”, the Carl-Zeiss-Stiftung and the University of Stuttgart (Projekthaus NanoBioMater), and the DFG SPP 1569 for financial support.References
- T. Markussen, A.-P. Jauho and M. Brandbyge, Phys. Rev. Lett., 2009, 103, 55502 CrossRef PubMed.
- Z. L. Wang, Adv. Mater., 2003, 15, 432–436 CrossRef CAS.
- Y. Chu, J. Hu, W. Yang, C. Wang and J. Z. Zhang, J. Phys. Chem. B, 2006, 110, 3135–3139 CrossRef CAS PubMed.
- O. M. Wilson, M. R. Knecht, J. C. Garcia-Martinez and R. M. Crooks, J. Am. Chem. Soc., 2006, 128, 4510–4511 CrossRef CAS PubMed.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981–983 CrossRef CAS PubMed.
- X. Gao and H. Matsui, Adv. Mater., 2005, 17, 2037–2050 CrossRef CAS.
- A. Guerrero-Martínez, S. Barbosa, I. Pastoriza-Santos and L. M. Liz-Marzán, Curr. Opin. Colloid Interface Sci., 2011, 16, 118–127 CrossRef.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–7248 CrossRef CAS PubMed.
- K.-S. Lee and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 19220–19225 CrossRef CAS PubMed.
- W. Stark, P. Stoessel, W. Wohlleben and A. Hafner, Chem. Soc. Rev., 2015, 44, 5793–5805 RSC.
- G. D. Nessim, Nanoscale, 2010, 2, 1306–1323 RSC.
- C. W. Lam, J. T. James, R. McCluskey and R. L. Hunter, Toxicol. Sci., 2004, 77, 126–134 CrossRef CAS PubMed.
- M. Simko and M. O. Mattsson, Part. Fibre Toxicol., 2010, 7, 42 CrossRef CAS PubMed.
- S. Sotiropoulou, Y. Sierra-Sastre, S. S. Mark and C. A. Batt, Chem. Mater., 2008, 20, 821–834 CrossRef CAS.
- D. Cardinale, N. Carette and T. Michon, Trends Biotechnol., 2012, 30, 369–376 CrossRef CAS PubMed.
- S. Cuenca, C. Mansilla, M. Aguado, C. Yuste-Calvo, F. Sánchez, J. M. Sánchez-Montero and F. Ponz, Front. Plant Sci., 2016, 7, 464 Search PubMed.
- J. Besong-Ndika, M. Wahlsten, D. Cardinale, J. Pille, J. Walter, T. Michon and K. Mäkinen, Front. Plant Sci., 2016, 7, 89 Search PubMed.
- C. Koch, F. J. Eber, C. Azucena, A. Förste, S. Walheim, T. Schimmel, A. M. Bittner, H. Jeske, H. Gliemann, S. Eiben, F. Geiger and C. Wege, Beilstein J. Nanotechnol., 2016, 7, 613–629 CrossRef CAS PubMed.
- Y. Leng, H. P. Wei, Z. P. Zhang, Y. F. Zhou, J. Y. Deng, Z. Q. Cui, D. Men, X. Y. You, Z. N. Yu, M. Luo and X. E. Zhang, Angew. Chem., Int. Ed., 2010, 49, 7243–7246 CrossRef CAS PubMed.
- S. Werner, S. Marillonnet, G. Hause, V. Klimyuk and Y. Gleba, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17678–17683 CrossRef CAS PubMed.
- M. L. Smith, J. A. Lindbo, S. Dillard-Telm, P. M. Brosio, A. B. Lasnik, A. A. McCormick, L. V. Nguyen and K. E. Palmer, Virology, 2006, 348, 475–488 CrossRef CAS PubMed.
- A. M. Bittner, Naturwissenschaften, 2005, 92, 51–64 CrossRef CAS PubMed.
- N. F. Steinmetz, K. C. Findlay, T. R. Noel, R. Parker, G. R. Lomonossoff and D. J. Evans, ChemBioChem, 2008, 9, 1662–1670 CrossRef CAS PubMed.
- K. Gerasopoulos, M. McCarthy, P. Banerjee, X. Fan, J. N. Culver and R. Ghodssi, Nanotechnology, 2010, 21, 055304 CrossRef CAS PubMed.
- C. Valery, F. Artzner and M. Paternostre, Soft Matter, 2011, 7, 9583–9594 RSC.
- M. Ueda, A. Makino, T. Imai, J. Sugiyama and S. Kimura, J. Pept. Sci., 2011, 17, 94–99 CrossRef CAS PubMed.
- L. Olofsson, J. Ankarloo, P. O. Andersson and I. A. Nicholls, Chem. Biol., 2001, 8, 661–671 CrossRef CAS PubMed.
- G. V. Gooding and T. T. Hebert, Phytopathology, 1967, 1285 Search PubMed.
- J. M. Alonso, M. L. Gorzny and A. M. Bittner, Trends Biotechnol., 2013, 31, 530–538 CrossRef CAS PubMed.
- A. M. Bittner, J. M. Alonso, M. L. Górzny and C. Wege, in Structure and physics of viruses: an integrated textbook, ed. M. G. Mateu, Springer Science+Business Media, Dordrecht, 2013, vol. 68, pp. 667–702 Search PubMed.
- Z. Wu, A. Mueller, S. Degenhard, S. E. Ruff, F. Geiger, A. M. Bittner, C. Wege and C. E. Krill 3rd, ACS Nano, 2010, 4, 4531–4538 CrossRef CAS PubMed.
- P. Atanasova, D. Rothenstein, J. J. Schneider, R. C. Hoffmann, S. Dilfer, S. Eiben, C. Wege, H. Jeske and J. Bill, Adv. Mater., 2011, 23, 4918–4922 CrossRef CAS PubMed.
- P. Atanasova, N. Stitz, S. Sanctis, J. H. M. Maurer, R. C. Hoffmann, S. Eiben, H. Jeske, J. J. Schneider and J. Bill, Langmuir, 2015, 31, 3897–3903 CrossRef CAS PubMed.
- R. J. Tseng, C. L. Tsai, L. P. Ma and J. Y. Ouyang, Nat. Nanotechnol., 2006, 1, 72–77 CrossRef CAS PubMed.
- J. N. Culver, A. D. Brown, F. Zang, M. Gnerlich, K. Gerasopoulos and R. Ghodssi, Virology, 2015, 479–480C, 200–-212 CrossRef PubMed.
- E. Royston, A. Ghosh, P. Kofinas, M. T. Harris and J. N. Culver, Langmuir, 2008, 24, 906–912 CrossRef CAS PubMed.
- C.-Y. Chiang, J. Epstein, A. Brown, J. N. Munday, J. N. Culver and S. Ehrman, Nano Lett., 2012, 12, 6005–6011 CrossRef CAS PubMed.
- C. Koch, K. Wabbel, F. J. Eber, P. Krolla-Sidenstein, C. Azucena, H. Gliemann, S. Eiben, F. Geiger and C. Wege, Front. Plant Sci., 2015, 6, 1137 Search PubMed.
- M. Baecker, C. Koch, S. Eiben, F. Geiger, F. J. Eber, H. Gliemann, A. Poghossian, C. Wege and M. J. Schoening, Sens. Actuators, B, 2017, 238, 716–722 CrossRef CAS.
- X. Z. Fan, L. Naves, N. P. Siwak, A. Brown, J. Culver and R. Ghodssi, Nanotechnology, 2015, 26, 205501 CrossRef CAS PubMed.
- R. Chapman, K. A. Jolliffe and S. Perrier, Polym. Chem., 2011, 2, 1956–1963 RSC.
- S. Shukla, F. J. Eber, A. S. Nagarajan, N. A. DiFranco, N. Schmidt, A. M. Wen, S. Eiben, R. M. Twyman, C. Wege and N. F. Steinmetz, Adv. Healthcare Mater., 2015, 4, 874–882 CrossRef CAS PubMed.
- P. J. G. Butler, Philos. Trans. R. Soc. London, Ser. B, 1999, 354, 537–550 CrossRef CAS PubMed.
- D. Zimmern, EMBO J., 1983, 2, 1901–1907 CAS.
- P. J. G. Butler and G. P. Lomonossoff, Biophys. J., 1980, 32, 295–312 CrossRef CAS PubMed.
- F. J. Eber, S. Eiben, H. Jeske and C. Wege, Nanoscale, 2015, 7, 344–355 RSC.
- P. J. G. Butler, J. Mol. Biol., 1974, 82, 343–353 CrossRef CAS PubMed.
- A. Mueller, F. J. Eber, C. Azucena, A. Petershans, A. M. Bittner, H. Gliemann, H. Jeske and C. Wege, ACS Nano, 2011, 5, 4512–4520 CrossRef CAS PubMed.
- F. J. Eber, S. Eiben, H. Jeske and C. Wege, Angew. Chem., Int. Ed., 2013, 52, 7203–7207 CrossRef CAS PubMed.
- D. R. Gallie, K. A. Plaskitt and T. M. A. Wilson, Virology, 1987, 158, 473–476 CrossRef CAS PubMed.
- C. Cremer, R. Kaufmann, M. Gunkel, F. Polanski, P. Müller, S. Degenhard, C. Wege, M. Hausmann and U. J. Birk, Histochem. Cell Biol., 2014, 142, 43–59 CrossRef CAS PubMed.
- C. Cremer, R. Kaufmann, M. Gunkel, S. Pres, Y. Weiland, P. Mueller, T. Ruckelshausen, P. Lemmer, F. C. Geiger, S. Degenhard, C. Wege, N. A. W. Lemmermann, R. Holtappels, H. Strickfaden and M. Hausmann, Biotechnol. J., 2011, 6, 1037–1051 CrossRef CAS PubMed.
- A. Harder, M. Dieding, V. Walhorn, S. Degenhard, A. Brodehl, C. Wege, H. Milting and D. Anselmetti, Beilstein J. Nanotechnol., 2013, 4, 510–516 CrossRef PubMed.
- K. Altintoprak, A. Seidenstücker, A. Welle, S. Eiben, P. Atanasova, N. Stitz, A. Plettl, J. Bill, H. Gliemann, H. Jeske, D. Rothenstein, F. Geiger and C. Wege, Beilstein J. Nanotechnol., 2015, 6, 1399–1412 CrossRef CAS PubMed.
- S. Eiben, N. Stitz, F. Eber, J. Wagner, P. Atanasova, J. Bill, C. Wege and H. Jeske, Virus Res., 2014, 180, 92–96 CrossRef CAS PubMed.
- F. C. Geiger, F. J. Eber, S. Eiben, A. Mueller, H. Jeske, J. P. Spatz and C. Wege, Nanoscale, 2013, 5, 3808–3816 RSC.
- D. E. Sleat, P. C. Turner, J. T. Finch, P. J. G. Butler and T. M. A. Wilson, Virology, 1986, 155, 299–308 CrossRef CAS PubMed.
- D. R. Turner, C. J. McGuigan and P. J. Butler, J. Mol. Biol., 1989, 209, 407–422 CrossRef CAS PubMed.
- J. P. Gaddipati and A. Siegel, Virology, 1990, 174, 337–344 CrossRef CAS PubMed.
- P. Maharaj, J. Mallajosyula, G. Lee, P. Thi, Y. Zhou, C. Kearney and A. McCormick, Int. J. Mol. Sci., 2014, 15, 18540–18556 CrossRef CAS PubMed.
- P. Lam, N. M. Gulati, P. L. Stewart, R. A. Keri and N. F. Steinmetz, Sci. Rep., 2016, 6, 23803 CrossRef CAS PubMed.
- M. L. Smith, T. Corbo, J. Bernales, J. A. Lindbo, G. P. Pogue, K. E. Palmer and A. A. McCormick, Virology, 2007, 358, 321–333 CrossRef CAS PubMed.
- B. Cooper, Genome Biol., 2014, 15, R67 CrossRef PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- B. Yurke, A. J. Turberfield, A. P. Mills, F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605–608 CrossRef CAS PubMed.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS PubMed.
- H. M. T. Choi, J. Y. Chang, L. A. Trinh, J. E. Padilla, S. E. Fraser and N. A. Pierce, Nat. Biotechnol., 2010, 28, 1208–1214 CrossRef CAS PubMed.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS PubMed.
- Y. Xu and Z. Zheng, Biosens. Bioelectron., 2016, 79, 593–599 CrossRef CAS PubMed.
- H. G. Chen, W. Ren, J. Jia, J. Feng, Z. F. Gao, N. B. Li and H. Q. Luo, Biosens. Bioelectron., 2016, 77, 40–45 CrossRef PubMed.
- J. Bath and A. J. Turberfield, Nat. Nanotechnol., 2007, 2, 275–284 CrossRef CAS PubMed.
- R. A. Muscat, J. Bath and A. J. Turberfield, Nano Lett., 2011, 11, 982–987 CrossRef CAS PubMed.
- B. Groves, Y.-J. Chen, C. Zurla, S. Pochekailov, J. L. Kirschman, P. J. Santangelo and G. Seelig, Nat. Nanotechnol., 2016, 11, 287–294 CrossRef CAS PubMed.
- L. Qian and E. Winfree, Science, 2011, 332, 1196–1201 CrossRef CAS PubMed.
- P. Hazarika, B. Ceyhan and C. M. Niemeyer, Angew. Chem., Int. Ed., 2004, 43, 6469–6471 CrossRef CAS PubMed.
- R. Duan, B. Wang, F. Hong, T. Zhang, Y. Jia, J. Huang, A. Hakeem, N. Liu, X. Lou and F. Xia, Nanoscale, 2015, 7, 5719–5725 RSC.
- J. Chen, J. Wen, L. Zhuang and S. Zhou, Nanoscale, 2016, 8, 9791–9797 RSC.
- S. N. Chapman, Methods Mol. Biol., 1998, 81, 123–129 CAS.
- M. R. Green and J. Sambrook, Molecular cloning : a laboratory manual, Cold Spring Harbor Laboratory Press, New York, 4th edn, 2012 Search PubMed.
- S. T. Jeng, J. F. Gardner and R. I. Gumport, J. Biol. Chem., 1992, 267, 19306–19312 CAS.
- M. Jansohn and A. Aigner, Gentechnische Methoden eine Sammlung von Arbeitsanleitungen für das molekularbiologische Labor, Elsevier, Spektrum, München, 4. Aufl edn, 2007 Search PubMed.
- W. A. Kibbe, Nucleic Acids Res., 2007, 35, W43–W46 CrossRef PubMed.
- N. Sugimoto, S. Nakano, M. Katoh, A. Matsumura, H. Nakamuta, T. Ohmichi, M. Yoneyama and M. Sasaki, Biochemistry, 1995, 34, 11211–11216 CrossRef CAS PubMed.
- R. Owczarzy, Y. You, B. G. Moreira, J. A. Manthey, L. Huang, M. A. Behlke and J. A. Walder, Biochemistry, 2004, 43, 3537–3554 CrossRef CAS PubMed.
- R. Owczarzy, B. G. Moreira, Y. You, M. A. Behlke and J. A. Walder, Biochemistry, 2008, 47, 5336–5353 CrossRef CAS PubMed.
- B. Yurke and A. P. Mills, Gen. Program. Evolv. Machines, 2003, 4, 111–122 CrossRef.
- H. Donis-Keller, Nucleic Acids Res., 1979, 7, 179–192 CrossRef CAS PubMed.
- H. Fraenkel-Conrat, Virology, 1957, 4, 1–4 CrossRef CAS PubMed.
- P. J. G. Butler, J. Mol. Biol., 1972, 72, 25–35 CrossRef CAS PubMed.
- R. Hogue and A. Asselin, Can. J. Bot., 1984, 62, 457–462 CrossRef CAS.
- A. Mueller, A. Kadri, H. Jeske and C. Wege, J. Virol. Methods, 2010, 166, 77–85 CrossRef CAS PubMed.
- W. S. Rasband, ImageJ, US National Institutes of Health, Bethesda, Maryland, USA, 1997–2011 Search PubMed.
- K. J. Breslauer, R. Frank, H. Blocker and L. A. Marky, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 3746–3750 CrossRef CAS.
- H. T. Allawi and J. SantaLucia, Biochemistry, 1997, 36, 10581–10594 CrossRef CAS PubMed.
- W. K. Kegel and P. van der Schoot, Biophys. J., 2006, 91, 1501–1512 CrossRef CAS PubMed.
- L. Fairall, J. T. Finch, C. F. Hui, C. R. Cantor and P. J. G. Butler, Eur. J. Biochem., 1986, 156, 459–465 CrossRef CAS PubMed.
- G. Lebeurier, A. Nicolaieff and K. E. Richards, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 149–153 CrossRef CAS.
- Y. Otsuki, I. Takebe, T. Ohno, M. Fukuda and Y. Okada, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 1913–1917 CrossRef CAS.
- C. Stussi, G. Lebeurier and L. Hirth, Virology, 1969, 38, 16–25 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr03897b |
| This journal is © The Royal Society of Chemistry 2016 |