
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A DFT study of endocyclic allenes: unprecedentedly superbasic hydrocarbons†
Davor
Margetić
* and
Ivana
Antol
Ruđer Bošković Institute, Bijenička cesta 54, HR-10002 Zagreb, Croatia. E-mail: margetid@irb.hr
First published on 23rd June 2016
Abstract
Density functional theory (DFT) calculations using the B3LYP, M05-2X and M06-2X functionals were employed in the study of basicity of allenes incorporated in seven-membered rings. These highly unstable species upon protonation give thermodynamically very stable tropylium cations, which are the origin of their extremely high basicity. An endocyclic allene → tropylium cation sequence was used for the design of novel hydrocarbon superbases, with calculated gas phase proton affinities exceeding well above 300 kcal mol−1.
Neutral organic superbases are of theoretical and synthetic interest, in particular their importance as aprotic and metal-free catalysts in organic reactions increases rapidly.1 Therefore, the design of novel superbasic molecules is an active research field.2,3 As a result, several functional groups with superbasic properties were identified: amidines, guanidines,4 biguanides,5 phosphazenes,6 and carbenes.7,8 Further increase in the basicity was achieved by imposing unfavourable interactions in the neutral form of the molecules, which are weakened upon protonation (proton sponges).9 Other effects that are employed in the design of superbases are the stabilization of protonated forms by resonance or intramolecular hydrogen bonding.10 For additional increase in basicity, various substituents were attached to delocalise the positive charge.11
We report here an in silico design of novel extremely basic hydrocarbon molecular systems. It is based on the cyclic allene → tropylium cation concept, where the incorporation of an allene moiety in a cycloheptadiene ring distorts the allene moiety from the ideal linear geometry to a less stable species. Upon protonation at the central carbon atom of the allene moiety the effective positive charge stabilization by the resonance of the tropylium cation and its aromatic character giving a highly stable system favors this process (Scheme 1). Cyclohepta-1,2,4,6-tetraene 1 is a known compound12 and it was shown that free 1 instantaneously yields 7-ethoxycyclohepta-1,3,5-triene if generated in ethanolic solution through the protonation of 1.13 Apparently, the basicity of 1 is high enough to cause its smooth conversion by HOEt and DOEt into (deutero)tropylium ethoxide. In addition to the estimation of basicities of 1 and its derivatives, investigation of their electronic structure is of interest, since the nature of binding in small ring endocyclic allenes (allene vs. carbene) was the subject of intensive discussion.14 These cyclic bent allenes are of importance in various organometallic reactions.15
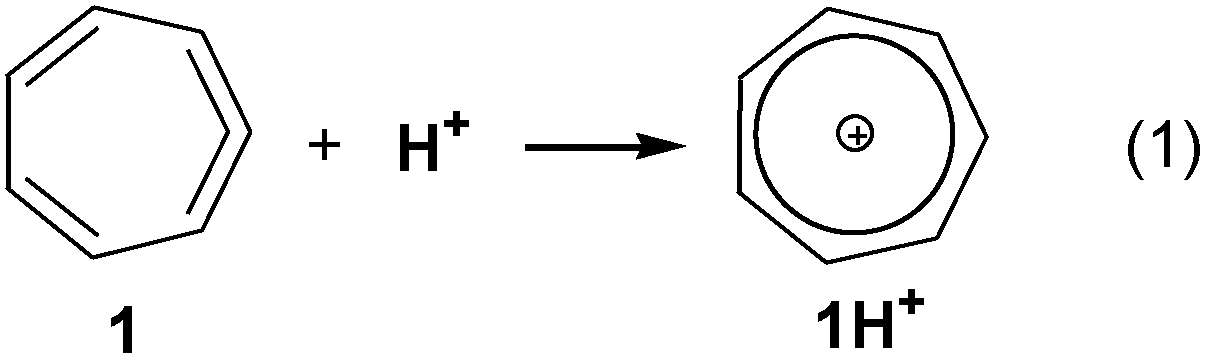 | ||
| Scheme 1 Formation of tropylium cation from allene 1. | ||
Structures of 1 and azuleno16 allenes 2–19 (Fig. 1) and their protonated forms were optimized by the B3LYP/6-31G(d), M052X/6-31G(d) and M062X/6-31G(d) methods and their gas phase basicities were calculated (for details see the ESI†). The results clearly favour cyclic allene-type structures over carbenes. These computationally studied exocyclic allenes possess extremely high basicity, as assessed by gas phase proton affinities (PAs) and basicities (GB) (Table 1).
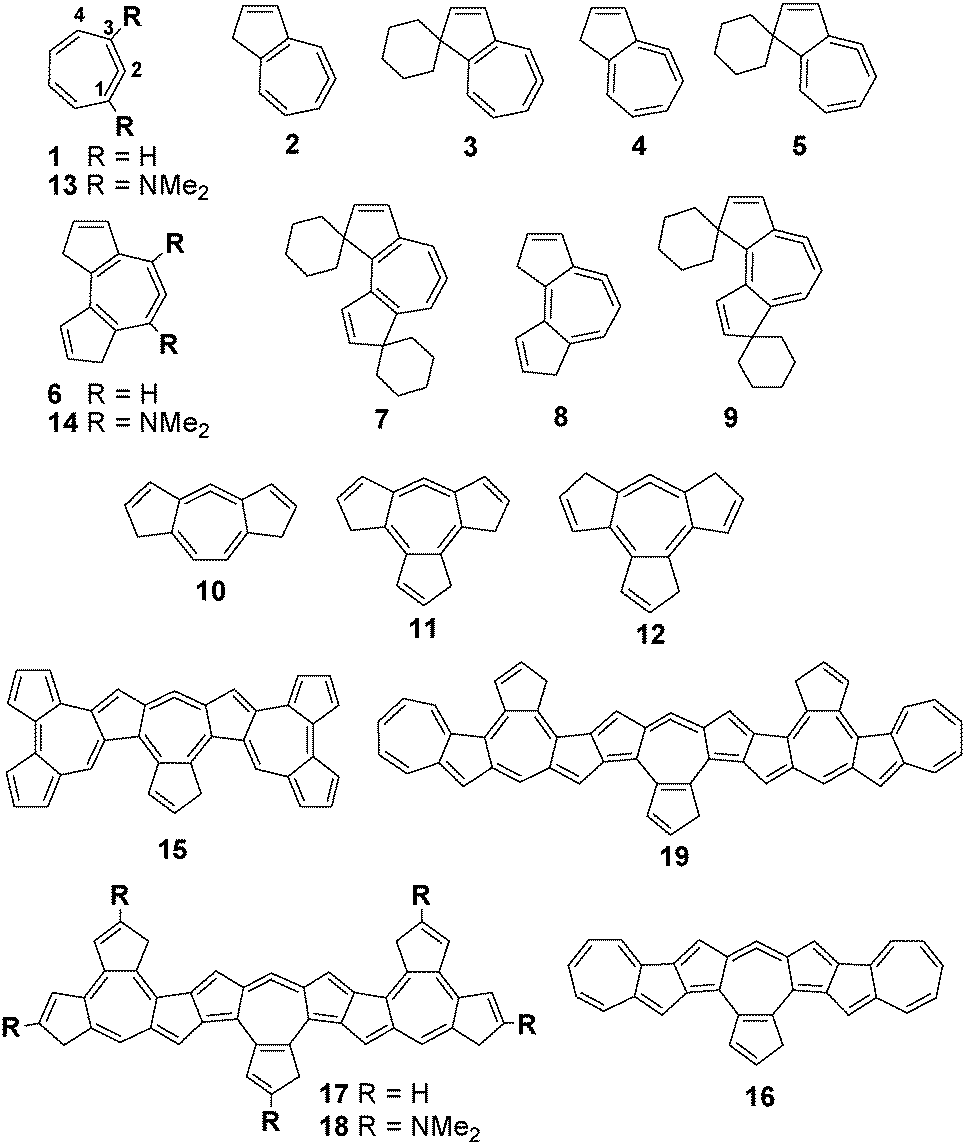 | ||
| Fig. 1 Superbases 1–19 studied in this work. | ||
| Base | PAB(2)a | GBBb | pKa(MeCN) | PAM05c | GBM05 | PAM06d | GBM06 |
|---|---|---|---|---|---|---|---|
| a PAB3LYP(2): B3LYP/6-311+G(d,p)//B3LYP/6-31G(d) + Evib(B3LYP/6-31G(d)).9 b B3LYP/6-31G(d). c PAM052X(2): M052X/6-311+G(d,p)//M052X/6-31G(d) + Evib(M052X/6-31G(d)). d PAM062X(2): M062X/6-311+G(d,p)//M062X/6-31G(d) + Evib(M062X/6-31G(d)). | |||||||
| 1 | 262.5 | 257.8 | 31.8 | 251.2 | 250.4 | 249.2 | 248.5 |
| 2 | 269.3 | 270.0 | 35.0 | 263.1 | 262.2 | 261.2 | 260.4 |
| 3 | 273.3 | 273.8 | 34.6 | 266.6 | 265.4 | 265.0 | 264.0 |
| 4 | 274.4 | 275.1 | 37.4 | 269.6 | 268.9 | 267.6 | 266.9 |
| 5 | 277.6 | 278.3 | 36.5 | 272.4 | 271.5 | 271.3 | 269.9 |
| 6 | 278.7 | 279.3 | 37.7 | 272.7 | 271.7 | 270.8 | 269.9 |
| 7 | 284.0 | 284.1 | 37.0 | 278.0 | 276.3 | 276.7 | 275.1 |
| 8 | 287.7 | 284.1 | 37.7 | 279.5 | 279.4 | 277.3 | 275.9 |
| 9 | 288.4 | 288.6 | 42.6 | 284.2 | 282.7 | 282.8 | 281.6 |
| 10 | 286.2 | 287.3 | 40.5 | 283.7 | 283.5 | 281.9 | 281.2 |
| 11 | 291.9 | 293.1 | 41.8 | 290.2 | 289.6 | 287.9 | 287.5 |
| 12 | 292.8 | 294.1 | 42.5 | 290.8 | 290.6 | 289.1 | 288.8 |
| 13 | 278.7 | 278.1 | 36.4 | 275.9 | 275.4 | 274.9 | 273.6 |
| 14 | 280.3 | 280.6 | 33.0 | 277.1 | 276.4 | 282.8 | 274.9 |
| 15 | 313.5 | 316.1 | 48.0 | 310.6 | 311.7 | 308.7 | 309.7 |
| 16 | 321.9 | 323.5 | 54.7 | 317.7 | 317.3 | 314.7 | 308.5 |
| 17 | 329.0 | 330.8 | 54.9 | 326.1 | 325.9 | 323.7 | 324.2 |
| 18 | 350.9 | 352.7 | 60.3 | 347.7 | 348.0 | 345.0 | 345.7 |
| 19 | 318.5 | 320.7 | 46.1 | 302.9 | 302.8 | 301.4 | 301.5 |
The proton affinities of the bases defined as a negative value of the enthalpy change for the protonation reaction (Scheme 1) of studied molecules vary from 262.5 to 350.9 kcal mol−1 at the B3LYP/6-311+G(d,p)//B3LYP/6-31G(d) level (Table 1). These values are significantly larger than 245 kcal mol−1 (PA of dimethylaminonaphthalene) thus designating allenes 1–14 as organic superbases in the gas phase. The borderline of 300 kcal mol−1 signifies the threshold for hyperbasicity,3 indicating that allenes 15–19 belong to this category. For comparison, the calculated (B3LYP/6-31+G(d,p)) and experimental enthalpies of protonation for the parent allene are 185.9 and 185.3 kcal mol−1, respectively,17 whereas the calculated PA of 1,4-dimethyl allene is 206.6 kcal mol−1 at the same computational level. PAs obtained in our study are well above previously calculated values for allenes.8,17,18 Analysis of results obtained by DFT calculations reveals that the fusion of five and seven-member rings leads to increased basicity by better delocalization of positive charge upon protonation. The enhanced basicity of 1,1-spiroalkylated azulenoallene 3 is attributed in the literature19 to the stabilization of both the inductive and σ–π conjugation effects of the exocyclic ring at the 1-position and the π–π conjugation of the double bond at the 2,3-positions in the respective 1H-azulenium cation. Presumably, the same effects stabilize the cations of 5, 7 and 9. Further increase in the basicity of endocyclic allenes could be achieved by the strategic attachment of substituents, which efficiently stabilizes positive charges such as the dimethylamino group (1/13, 6/14, and 17/18). In addition, the positioning of allene terminal carbon atom at the ring junction of five and seven-membered rings was found to further destabilize the endocyclic allene system (by 5 kcal mol−1, 2/4 and 6/8 pairs).
A similar relative basicity order was obtained for gas phase basicities (GB) reaching extremely high value (352.7 kcal mol−1) for 18 at the B3LYP/6-31G(d) level, while the lack of experimental values precludes the establishment of pKa correlation for allenes. However, an estimate of pKa values in acetonitrile using the correlation devised for guanidines20 still follows the trend of gas phase basicities, indicating that allenes listed in Table 1 have pKa values in acetonitrile (B3LYP) within the range of 32–60. The most basic is allene 18 with the pKa value certainly well above 50 pKa units.
Geometrical distortions imposed by the cyclic framework on the allene moiety and structural changes caused by protonation at the central carbon atom of the allene moiety are illustrated in Fig. 2 for azulenoallene 4.
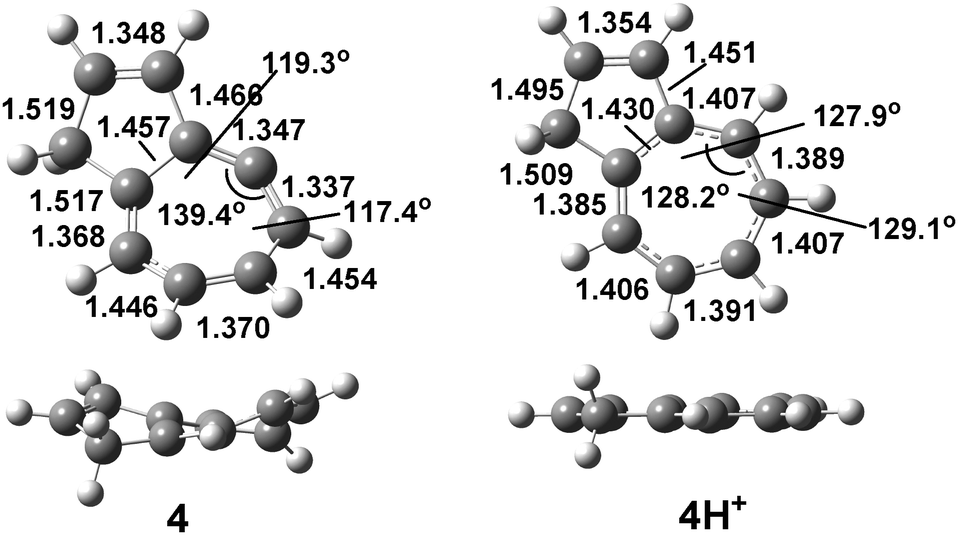 | ||
| Fig. 2 Neutral and protonated structures of 4 (B3LYP/6-31G(d)). | ||
The C1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2C3 angles in endocyclic allenes 1–19 vary from 131.2° to 146.4°, and the out-of-plane angles (C1C2C3–C4) are within a range of 17.8–33.9°, indicating a large deviation from the ideal allene geometry (180° and 45°, respectively). As a consequence of this distortion, the orbital overlap is not ideal and the double bonds are weakened; this is evidenced by the average allene C1C2/C2C3 distances of 1.330–1.375 Å, which are significantly elongated in comparison to the parent allene (1.309 Å). These geometrical features are reflected on the CC bond orders and hybridisation of the central allene carbon atom, which changes, for instance, from sp1 in parent 1,4-dimethylallene to sp1.29 in 15 (Table S1, ESI†). Upon protonation at the central allene carbon and its rehybridisation, effective cyclic conjugation is increased, which is demonstrated by the decreased bond alternation in the tropylium ring, such as for 4/4H+, as well as by the planarization of the cationic structure.
C2C3 angles in endocyclic allenes 1–19 vary from 131.2° to 146.4°, and the out-of-plane angles (C1C2C3–C4) are within a range of 17.8–33.9°, indicating a large deviation from the ideal allene geometry (180° and 45°, respectively). As a consequence of this distortion, the orbital overlap is not ideal and the double bonds are weakened; this is evidenced by the average allene C1C2/C2C3 distances of 1.330–1.375 Å, which are significantly elongated in comparison to the parent allene (1.309 Å). These geometrical features are reflected on the CC bond orders and hybridisation of the central allene carbon atom, which changes, for instance, from sp1 in parent 1,4-dimethylallene to sp1.29 in 15 (Table S1, ESI†). Upon protonation at the central allene carbon and its rehybridisation, effective cyclic conjugation is increased, which is demonstrated by the decreased bond alternation in the tropylium ring, such as for 4/4H+, as well as by the planarization of the cationic structure.
In conclusion, endocyclic allenes are recognized as potent neutral organic superbases and their unprecedented basicity was estimated by DFT calculations. Results suggest that they could effect in situ generation of anions and deprotonation of common organic solvents such as acetonitrile or DMSO.
Computational methodology
Calculations were performed using the Gaussian03 suite of programs. Geometry optimizations were carried out at the DFT level employing B3LYP, M052X and M062X hybrid functionals in conjunction with the 6-31G(d) basis set. Harmonic vibration frequencies were calculated to verify that the stationary structures are minima. Single point calculations were conducted, at the B3LYP/6-311+G(d,p) level, using B3LYP/6-31G(d) optimized structures. Gas phase basicity (GB) was calculated as the Gibbs free energy change of the protonation reaction. Proton affinities in the gas phase and pKa values in acetonitrile were calculated using equations given in the ESI.†Acknowledgements
We acknowledge the financial support of the Ministry of Science, Education and Sport of Croatia (Projects No. 098-0982933-2920 and 098-0982933-3218).Notes and references
- Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts, ed. T. Ishikawa, Wiley, Chichester, 2009 Search PubMed.
- D. Margetić, Physico-chemical properties of organosuperbases, in Ref. 1, 2009, pp. 9–48 Search PubMed.
- Z. B. Maksić, B. Kovačević and R. Vianello, Chem. Rev., 2012, 112, 5240–5270 CrossRef PubMed.
- Z. B. Maksić and B. Kovačević, J. Org. Chem., 2000, 65, 3303–3309 CrossRef.
- Z. Glasovac, P. Trošelj, I. Jušinski, D. Margetić and M. Eckert-Maksić, Synlett, 2013, 2540–2544 CAS.
- B. Kovačević, D. Barić and Z. B. Maksić, New J. Chem., 2004, 28, 284–288 RSC.
- R. Lo and B. Ganguly, Chem. Commun., 2011, 47, 7395–7397 RSC; M. Liu, I. Yang, B. Buckley and J. K. Lee, Org. Lett., 2010, 21, 4764–4767 CrossRef CAS PubMed.
- R. Tonner and G. Frenking, Angew. Chem., Int. Ed., 2007, 46, 8695–8698 CrossRef CAS PubMed.
- D. Margetić, T. Ishikawa and T. Kumamoto, Eur. J. Org. Chem., 2010, 6563–6572 CrossRef CAS; D. Margetić, P. Trošelj, T. Ishikawa and T. Kumamoto, Bull. Chem. Soc. Jpn., 2010, 83, 1055–1057 CrossRef; A. Singh and B. Ganguly, Eur. J. Org. Chem., 2007, 420–422 CrossRef; A. Singh and B. Ganguly, New J. Chem., 2009, 33, 583–587 RSC; V. Raab, K. Harms, J. Sundermeyer, B. Kovačević and Z. B. Maksić, J. Org. Chem., 2003, 68, 8790–8797 CrossRef PubMed.
- B. Kovačević, Z. Glasovac and Z. B. Maksić, J. Phys. Org. Chem., 2002, 15, 765–774 CrossRef.
- B. Kovačević and Z. B. Maksić, in NIC Symposium Proceedings, John von Neumann Institute for Computing, Jülich, NIC series 20, ed. D. Wolf, G. Münster and M. Kremer, 2003, pp. 71–80 Search PubMed.
- M. Christl, in Modern Allene Chemistry, ed. N. Krause and A. S. K. Hashmi, Wiley, Weinheim, 2004, ch. 6, vol. 1, pp. 243–357 Search PubMed.
- W. Kirmse, K. Loosen and H.-D. Sluma, J. Am. Chem. Soc., 1981, 103, 5935–5937 CrossRef CAS.
- I. Fernández, C. A. Dyker, A. DeHope, B. Donnadieu, G. Frenking and G. Bertrand, J. Am. Chem. Soc., 2009, 131, 11875–11881 CrossRef CAS PubMed; H. V. Huynh and G. Frison, J. Org. Chem., 2013, 78, 328–338 CrossRef PubMed; E. Soriano and I. Fernández, Chem. Soc. Rev., 2014, 43, 3041–3105 RSC , and references cited therein.
- M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed.
- T. Okazaki and K. K. Laali, Org. Biomol. Chem., 2003, 1, 3078–3093 Search PubMed.
- S. Gronert and J. R. Keefe, J. Org. Chem., 2007, 72, 6343–6352 CrossRef CAS PubMed.
- D. S. Patel and P. V. Bharatam, J. Org. Chem., 2011, 76, 2558–2567 CrossRef CAS PubMed.
- M. Oda, N. Nakajima, N. C. Thanh, K. Kitahara, R. Miyatake and S. Kuroda, Eur. J. Org. Chem., 2008, 5301–5307 CrossRef CAS.
- B. Kovačević and Z. B. Maksić, Org. Lett., 2001, 3, 1523–1526 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details and Cartesian coordinates of optimised structures. See DOI: 10.1039/c6nj00623j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |