
Extending the halogen-bonded supramolecular synthon concept to 1,3,4-oxadiazole derivatives†
Paulina I.
Hidalgo
 *a,
Sergio
Leal
a,
Claudio A.
Jiménez
*a,
Esteban
Vöhringer-Martinez
b,
Bárbara
Herrera
c,
Jorge
Pasán
de,
Catalina
Ruiz-Pérez
d and
Duncan W.
Bruce
f
*a,
Sergio
Leal
a,
Claudio A.
Jiménez
*a,
Esteban
Vöhringer-Martinez
b,
Bárbara
Herrera
c,
Jorge
Pasán
de,
Catalina
Ruiz-Pérez
d and
Duncan W.
Bruce
f
aDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad de Concepción, Casilla 160-C, Concepción, Chile. E-mail: pauhidal@udec.cl; cjimenez@udec.cl; Tel: +(56)(41)2204258
bDepartamento de Fisicoquímica, Facultad de Ciencias Químicas, Universidad de Concepción, Casilla 160-C, Concepción, Chile. E-mail: evohringer@udec.cl
cLaboratorio de Química Teórica Computacional (QTC), Departamento de Química-Física, Facultad de Química, Pontificia Universidad Católica de Chile, Av. Vicuña Mackenna 4860, Macul, Santiago, Chile. E-mail: bherrera@uc.cl
dLaboratorio de Rayos X y Materiales Moleculares, Departamento de Física, Facultad de Ciencias, Universidad de La Laguna, Av. Astrofísico Francisco Sánchez s/n, 38206 La Laguna (Tenerife), Spain. E-mail: jpasang@ull.es
eInstituto de Ciencia Molecular (ICMol), Universitat de València, C/Catedrático José Beltrán 2, 46980 Paterna, València, Spain
fDepartment of Chemistry, University of York, Heslington, York YO10 5DD, UK. E-mail: duncan.bruce@york.ac.uk
First published on 26th November 2015
Abstract
A series of five crystal structures of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 halogen-bonded complexes were obtained from 4-[5-(4-alkoxyphenyl)-1,3,4-oxadiazole-2-yl]pyridine and 1,3,5-trifluorotriiodobenzene. Electronic structure calculations show that the N(oxadiazole)⋯I interaction in the new synthon is as strong as the classic N(pyridine)⋯I interaction. Oxygen to sulfur atom subsitution on the oxadiazole ring results in a different supramolecular packing where the N(pyridine)⋯I interaction is favored, which could be rationalized by the changes in the molecular electrostatic potential predicted from the theoretical calculations.
:1 halogen-bonded complexes were obtained from 4-[5-(4-alkoxyphenyl)-1,3,4-oxadiazole-2-yl]pyridine and 1,3,5-trifluorotriiodobenzene. Electronic structure calculations show that the N(oxadiazole)⋯I interaction in the new synthon is as strong as the classic N(pyridine)⋯I interaction. Oxygen to sulfur atom subsitution on the oxadiazole ring results in a different supramolecular packing where the N(pyridine)⋯I interaction is favored, which could be rationalized by the changes in the molecular electrostatic potential predicted from the theoretical calculations.
Introduction
The overall goal of crystal engineering is the design and control of molecular packing in crystalline materials with desired properties.1 This approach has attracted attention in different areas such as biochemistry,2 medicinal chemistry,3 and materials chemistry4 among others. The precise understanding of intermolecular recognition between molecular building tectons has transformed this field into a multidisciplinary tool with impact in the development of organic devices such as organic conductors5 and magnets,6 chemical separation agents,7 NLO-active materials,8 liquid crystals9 and porous organic solids.10 The occurrence of particular properties in crystalline materials is in general the result of the combination of two features, i) the building block (molecular units), and ii) their arrangement in the crystal. The first is relatively simple to control, whereas the second is more difficult given that the crystal packing results from a delicate balance of all the intermolecular synthon interactions present in the crystal while normally adopting the densest accessible packing.4The concept of the supramolecular synthon was introduced into molecular crystal chemistry in 1995 (ref. 11) and involves the defined structural kernel of a crystal structure, which encapsulates the essence of a crystal in terms of molecular recognition.12 Robust supramolecular synthons, which involve strong intermolecular interactions, can be used successfully for crystal engineering purposes and there is a consensus that the final crystal structure can be analysed easily as a collection of robust synthons formed in the earlier stages of molecular association.13 In this context, the introduction of new synthons is relevant for the design of new solid materials with special properties. Because of the prevalence of their monovalent chemistry, halogen atoms are normally located at the periphery of the molecules. Thus, they are ideally positioned to be involved in intermolecular interactions and can play, therefore, a fundamental role in the aggregation of molecules into solids.4 In recent years, halogen bonding has grown from a scientific curiosity to one of the most interesting non-covalent interactions for constructing supramolecular assemblies.14
Halogen bonding (XB) interactions span over a very wide range, from the weak Cl⋯Cl interaction in chlorocarbons to the strong I−⋯I2 interaction in I3−;15 among them, those employing nitrogen-containing heterocycles are probably the most widely studied. Until now, almost exclusively six-membered rings such as pyridine,16 pyrazine,17 phenazine18 and bipyridine19 have been used as halogen bond acceptors. In the few cases reported where five-membered N-heterocycles are present in the tectons, they are not directly involved in the halogen bonding.20 In this regard, Aakeröy et al. recently reported the first case of an imidazole ring directly involved in halogen bonding.21 In this work, we have synthesised a series of 4-[5-(4-alkoxyphenyl)-1,3,4-oxadiazole-2-yl]pyridines, with hexyloxy- to decyloxy- alkyl chains, which have been co-crystallised with 1,3,5-trifluorotriiodobenzene (Scheme 1). The results reveal that the tri-iodo compound is able to form three halogen bonds simultaneously, with each iodine atom interacting with different nitrogen atoms, two of them with the oxadiazole ring and one with the pyridine ring. As such, this system represents the second example of 1,3,5-trifluorotriiodobenzene forming three N⋯I halogen bonds simultaneously and the second example also where a five-membered nitrogen-containing heterocycle is directly participating.
 | ||
| Scheme 1 A series of 4-[5-(4-alkoxyphenyl)-1,3,4-oxadiazole-2-yl]pyridine and 4-[5-(4-nonyloxyphenyl)-1,3,4-thiadiazole-2-yl]pyridine acceptors and the symmetric donor. | ||
Results and discussion
All complexes crystallised in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group and showed a 1:1 stoichiometry, despite being obtained from a 3:1 mixture of pyridine to tri-iodo compound. Using various pyridines as halogen bond acceptors (e.g. tris(1,3,5-{2-(pyridin-4-yl)vinyl}benzene), van der Boom and co-workers generated 2:1 complexes with 1,3,5-trifluorotriiodobenzene and hence argued that strong halogen bond acceptors and donors would be required simultaneously to form three halogen bonds with C6F3I3.22 It was shown later that C6F3I3 could form three halogen bonds simultaneously using chloride, bromide23 or iodide24 anions as acceptors giving a honeycomb-like structure in which the voids were filled with an appropriate bulky cation. Further work by Roper et al. showed that a 3:1 complex was possible with C6F3I3 using DMAP (4-(N,N-dimethylamino)pyridine) as acceptor.25 It seems, therefore, that a crucial aspect to form a 3:1 complex is their ability to fill the space effectively. This may be a major handicap to form 3:1 cocrystals with C6F3I3 since they tend to form star-like systems that do not pack efficiently. This is a reason for the small number of structures where C6F3I3 is engaged in three I⋯N halogen bond interactions. Remarkably, in cocrystals 1–5 reported here, the 1:1 stoichiometry shows a dense crystal packing and, at the same time, the 1,3,5-trifluorotriiodobenzene establishes three I⋯N interactions.
space group and showed a 1:1 stoichiometry, despite being obtained from a 3:1 mixture of pyridine to tri-iodo compound. Using various pyridines as halogen bond acceptors (e.g. tris(1,3,5-{2-(pyridin-4-yl)vinyl}benzene), van der Boom and co-workers generated 2:1 complexes with 1,3,5-trifluorotriiodobenzene and hence argued that strong halogen bond acceptors and donors would be required simultaneously to form three halogen bonds with C6F3I3.22 It was shown later that C6F3I3 could form three halogen bonds simultaneously using chloride, bromide23 or iodide24 anions as acceptors giving a honeycomb-like structure in which the voids were filled with an appropriate bulky cation. Further work by Roper et al. showed that a 3:1 complex was possible with C6F3I3 using DMAP (4-(N,N-dimethylamino)pyridine) as acceptor.25 It seems, therefore, that a crucial aspect to form a 3:1 complex is their ability to fill the space effectively. This may be a major handicap to form 3:1 cocrystals with C6F3I3 since they tend to form star-like systems that do not pack efficiently. This is a reason for the small number of structures where C6F3I3 is engaged in three I⋯N halogen bond interactions. Remarkably, in cocrystals 1–5 reported here, the 1:1 stoichiometry shows a dense crystal packing and, at the same time, the 1,3,5-trifluorotriiodobenzene establishes three I⋯N interactions.
The core motif for co-crystals 1 to 5 (Fig. 1) shows two C6F3I3 units connected through a type I F⋯F interaction, with a separation between fluorine atoms from 2.54 to 2.62 Å (Table S2 and Fig. S1†).26 The iodine para to this fluorine forms a halogen bond to the pyridine nitrogen atom of one oxadiazole molecule while the two ortho iodines bind to the oxadiazole nitrogen atoms of a different molecule. Typically, the donor⋯acceptor separation in one of the oxadiazole N⋯I halogen bonds [mean value of the N(3)⋯I bond distance is 3.019(8) Å; see Table 1] is close to what is observed for the pyridine N⋯I interaction [average value of 2.902(7) Å] while the other is a little longer [mean N(2)⋯I separation is 3.365(9) Å]. This situation represents one in which, in all the cases, the halogen bond distances observed are 12% shorter than the sum of van der Waals' radii.27 Since in the oxadiazole ring the Cipso does not exist, the analysis of the linearity of the halogen bond was made using the mid-point between the ring C–O bonds. This point corresponds to the best match of the line bisecting the C![[N with combining circumflex]](https://www.rsc.org/images/entities/char_004e_0302.gif) N angle, which represents the orientation of the lone pair of electrons in a Nsp2. In all cases, the CÎN angle and the ICipso are approximately linear, with values larger than 160° (see Table 1); also, the oxadiazole and the C6F3I3 rings are close to planarity, supporting the assertion that it is effectively a halogen bond (Table 1 and Fig. S2†). In the whole picture, each pair of 1,3,5-trifluorotriiodobenzene molecules is linked to four oxadiazole molecules, which in turn are linked to two pairs of C6F3I3 groups, building a ribbon-like structure (Fig. S3†). This leads to quite efficient packing and filling of space, and indeed the calculated density for co-crystals 1 to 5 is around 1.9 g dm−3.
N angle, which represents the orientation of the lone pair of electrons in a Nsp2. In all cases, the CÎN angle and the ICipso are approximately linear, with values larger than 160° (see Table 1); also, the oxadiazole and the C6F3I3 rings are close to planarity, supporting the assertion that it is effectively a halogen bond (Table 1 and Fig. S2†). In the whole picture, each pair of 1,3,5-trifluorotriiodobenzene molecules is linked to four oxadiazole molecules, which in turn are linked to two pairs of C6F3I3 groups, building a ribbon-like structure (Fig. S3†). This leads to quite efficient packing and filling of space, and indeed the calculated density for co-crystals 1 to 5 is around 1.9 g dm−3.
 | ||
| Fig. 1 1:1 Complex between 4-[5-(4-alkoxyphenyl)-1,3,4-oxadiazole-2-yl]pyridine and 1,3,5-trifluorotriiodobenzene. | ||
| Complex | d(N⋯I)a/Å | d(C–I)/Å | CÎN/° | ICipsob/° |
Anglec/° |
|---|---|---|---|---|---|
| a The first value corresponds to the N(pyridine)⋯I distance (N1⋯I) and the next two are the N(oxadiazole)⋯I distances, (N3⋯I) and (N2⋯I), respectively except for 6, where the two values correspond to N(pyridine)⋯I interactions (N1⋯I). b For oxadiazole rings the Cipso was substituted by the mid-point between C–O bonds. c Torsion angle between the plane of C6F3I3 and the pyridine or imidazole plane. | |||||
| 1 | 2.951(1) | 2.102(1) | 176.9(3) | 167.4(4) | 3.4(3) |
| 3.111(8) | 2.104(9) | 176.9(3) | 167.4(5) | 3.1(3) | |
| 3.258(9) | 2.101(9) | 169.7(3) | 164.7(6) | 3.1(3) | |
| 2 | 2.842(6)/2.913(6) | 2.105(6)/2.108(6) | 178.2(2)/177.2(2) | 170.9(3)/172.9(3) | 6.5(2)/1.7(2) |
| 3.029(6)/3.009(6) | 2.100(6)/2.010(6) | 167.6(4)/173.3(2) | 168.3(3)/161.7(3) | 5.3(2)/4.4(2) | |
| 3.457(6)/3.371(7) | 2.094(7)/2.102(7) | 160.0(2)/166.3(2) | 160.1(3)/172.8(3) | 5.2(2)/3.6(2) | |
| 3 | 2.863(5) | 2.096(3) | 177.0(1) | 174.2(2) | 4.0(1) |
| 2.956(3) | 2.086(3) | 173.7(1) | 165.0(2) | 2.2(1) | |
| 3.410(4) | 2.082(4) | 164.0(1) | 171.6(2) | 2.2(1) | |
| 4 | 2.877(7) | 2.092(6) | 177.3(2) | 173.6(3) | 2.1(2) |
| 2.954(6) | 2.078(6) | 174.5(2) | 166.3(3) | 0.9(2) | |
| 3.400(5) | 2.093(6) | 165.3(2) | 172.3(2) | 0.9(2) | |
| 5 | 2.965(2) | 2.087(1) | 177.3(4) | 168.7(6) | 5.6(4) |
| 3.049(1) | 2.090(1) | 176.3(4) | 167.8(6) | 3.1(3) | |
| 3.293(1) | 2.119(1) | 169.1(4) | 172.2(5) | 3.1(3) | |
| 6 | 2.959(4) | 2.092(4) | 169.7(13) | 159(2) | 88.5(8) |
| 2.962(4) | 2.097(4) | 168.9(13) | 165(2) | 83.0(8) | |
To rationalise the nature of the nitrogen–iodine interactions, electronic structure calculations were performed for 4-[5-(4-hexyloxyphenyl)-1,3,4-oxadiazole-2-yl]pyridine and 1,3,5-trifluorotriiodobenzene at the B3PW91/6-311G(d) level of theory with the Gaussian 03 package (for details see the ESI†).28 Since intermolecular halogen bonds have a large electrostatic contribution, the molecular electrostatic potential (MEP) mapped on the isodensity surface (isovalue = 0.001 e a0−3) was calculated for each molecule as shown in Fig. 2. Politzer et al.29 chose this surface, VS, due to its similarity to the van der Waals surface of molecules, which is representative of intermolecular interactions. 1,3,5-Trifluorotriiodobenzene displays the characteristic positive potential on the iodine atom along the extension of the carbon–iodine bond (σ-hole).29 This region of positive potential has been related to the formation of halogen bonds and the maximum value to the interaction energy. In 1,3,5-trifluorotriiodobenzene, the maximum value, Imax, on each iodine atom is +29.8 kcal mol−1, which agrees with previously reported values from Politzer et al.30 To form a halogen bond, the iodine atom has to interact with a nucleophilic site bearing a negative electrostatic potential on the molecule. Fig. 2 shows that the minimum value of the electrostatic potential in 4-[5-(4-hexyloxyphenyl)-1,3,4-oxadiazole-2-yl]pyridine is found on the nitrogen atoms in accordance with the interactions observed in the crystal. The most negative value of Imin, however, is on the N(3) nitrogen of the oxadiazole ring followed by the other ring nitrogen atom [N(2)] with values of −37.2 and −36.8 kcal mol−1, respectively.
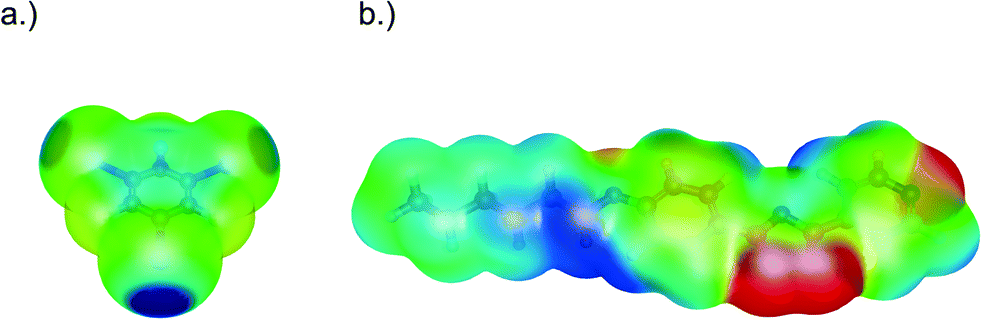 | ||
| Fig. 2 Molecular electrostatic potential mapped on the isodensity surface (isovalue = 0.001 e a0−3, blue corresponds to +18.8 kcal mol−1 and red to −18.8 kcal mol−1) for a) 1,3,5-trifluorotriiodobenzene and b) 4-[5-(4-hexyloxyphenyl)-1,3,4-oxadiazole-2-yl]pyridine. | ||
The pyridine nitrogen atom [N(1)] has the least negative value of the three nitrogen atoms with Imin = −36.5 kcal mol−1. These results suggest that if the interaction is only electrostatic in nature and no crystal packing effects are considered, the most favourable site for the interaction with the iodine atoms of 1,3,5-trifluorotriiodobenzene would be the N(3) nitrogen atom of the oxadiazole ring (the nitrogen closest to the phenyl ring). The N⋯I bond distances do not change considerably with alkyl chain length (Table 1), in agreement with the electronic structure calculations, which showed that changing the length of the alkyl chain does not alter the relative values of the MEP on the nitrogen atoms (Table S3†). To address the relevance of the N(2) atom as a possible second interaction site, Vs was recalculated once an interaction between the iodine atom and the most nucleophilic N(3) atom was formed in a ‘pair complex’. A ‘pair complex’ results from the separation of the periodic crystal structure into molecule pairs. These pairs are formed by two molecules that interact through the N(3)⋯I halogen bond. In the electronic structure calculation, their geometry is kept fixed representing the one in the crystal structure. We found that the most negative value of the ‘pair complex’ is now on the N(1) atom of the pyridine ring (Imin = −35.6 kcal mol−1) and not on the N(2) atom (Imin = −33.0 kcal mol−1) (see the ESI†). One can conclude that once the most favourable interaction with N(3) is formed, the ‘pair complex’ would favour the interaction over the pyridine nitrogen atom, which is the most nucleophilic site and additionally is sterically more accessible. The energy of this type of interaction was also estimated by calculating the dissociation energy of the ‘pair complexes’ formed by the two molecules interacting through either N(1), N(2) or N(3) atoms. These calculations were performed with the ORCA software package31 at the M06-2X/def-TZVP level of theory where the geometry was fixed. This level of theory was shown by Kozuch et al.32 to yield halogen bond interaction energies of model systems with errors below 1 kcal mol−1. It was found that the interaction energy is strongest for the N(1) nitrogen atom due to the short halogen bond distance, followed by the interaction energy involving the N(3) atom, which amounts to 5.7 kcal mol−1. This is in accordance with reported values of halogen bond interaction energies reported recently by Politzer et al.29 Therefore, it seems clear that N(3) and N(1) are the most favourable sites for the halogen bond interaction, however, the differences between the three N sites are small enough to consider that all the interactions can take place simultaneously and thus the driving force that leads to the crystal structure observed is Kitaigorodskii's rule of closest packing.
Considering the initial purpose to construct 3:1 star-like molecules where the 1,3,5-trifluorotriiodobenzene interacts with three oxadiazole molecules via the pyridine nitrogen atom, one can envisage several strategies to favour the negative electrostatic potential at the pyridine N(1) atom. The first approach involves the replacement of the alkyl chain by electron withdrawing substituents (such as –CF3); electronic structure calculations indicate that the order of Imin is reversed and N(1) becomes the preferred interaction site [Imin[N(3)] = −30.3 kcal mol−1, Imin[N(2)] = −30.5 kcal mol−1, Imin[N(1)] = −32.5 kcal mol−1]. The second strategy consists of introducing an electron-donating group onto the pyridyl ring. However, the available 3-position has many synthetic drawbacks that experimentally preclude the preparation of such a molecule. On the other hand, the substitution in the 2-position, even when it is synthetically feasible, faces an inherent steric problem in the formation of the co-crystals.
The third approximation is the substitution of the oxygen atom of the oxadiazole ring by sulfur. In this case, the Imin value at the N(1) atom is −37.5 kcal mol−1, whereas those of N(2) and N(3) are −33.6, and −33.9 kcal mol−1, respectively, showing a significant increase in the electron density of N(1), which should become the preferred interaction site.
To prove this concept, the thiadiazole derivative 4-[5-(4-nonyloxyphenyl)-1,3,4-thiadiazole-2-yl]pyridine (6) was synthesized and the structure was obtained from a crystal prepared from a 3:1 mixture of the thiadiazole with 1,3,5-trifluorotriiodobenzene. The resultant co-crystal (6) has a 2:1 stoichiometry and it crystallizes in the C2/c space group. In this case, halogen bonding linking both molecules occurs only at the N(1) nitrogen atom of the pyridine ring, with two thiadiazole molecules linked to one 1,3,5-trifluorotriiodobenzene. This situation is in agreement with the theoretical calculations regarding the preference of the halogen bond for the pyridine N atom (N⋯I geometrical parameters are listed in Table 1). The V-shaped adduct is depicted in Fig. 3, the two thiadiazole molecules forming an angle of 68.4(2)°, much less than the 120° expected from the configuration of the iodine atoms. These adducts are arranged in layers, and they are staggered with their neighbours, leading to a less efficient crystal packing with a calculated density of 1.64 g dm−3 (Fig. S4†).
 | ||
| Fig. 3 A view of the asymmetric unit of 6 a) along the c-axis and b) along the b-axis. | ||
The theoretical and experimental data reported here showed that when it comes to the fabrication of co-crystals based on halogen bonding, the archetypal iodine⋯pyridine interaction will not be necessarily the most favoured when acceptors such as oxadiazole are present. Theoretical calculations show that the oxadiazole nitrogen atoms [N(3) and N(2)], not the pyridine nitrogen atom [N(1)], have the highest electrostatic potential values. These results situate the three nitrogen atoms in equal conditions to interact, in agreement with the crystal structures of 1–5, where all the nitrogen atoms are involved in the formation of halogen bonds.
Moreover, there would be a certain degree of tunability and the energetically best acceptor can be changed in terms of the functionality of the substituents or the heteroatom in the diazole ring. The crystal structure of the thiadiazole represents the first step in this direction, where in the 1,3,5-trifluorotriiodobenzene adduct (6) only the pyridine nitrogen atoms participate in halogen bonds. Calculations show that in 4 (the equivalent oxadiazole adduct), the minimum value of the MEP is on the oxadiazole nitrogen N(3), whereas in 6, this value is located on the N(1) pyridine nitrogen atom with a clear difference from the values for N(2) or N(3). This property can be used for the construction of new halogen-bonded co-crystals based on the oxadiazole function as well as other five-membered N-heterocycles; research into this is now in progress.
Acknowledgements
This work has received financial support from FONDECYT N° 11100065, 11121179, 1120092, DIUC 212.23.49-1.0, NC 120082 and the Spanish Ministerio de Economía y Competitividad through projects MAT2013-43101-R, MAT2014-57465-R and CTQ2013-44844-P. J. P. also thanks the project CTQ2013-44844-P for a research contract.Notes and references
- (a) G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS; (b) G. R. Desiraju, The Crystal as a Supramolecular Entity, John Wiley & Sons, New York, 1996 Search PubMed; (c) J. W. Steed and J. J. Atwood, Supramolecular Chemistry, Wiley, Chinchester, 2000 Search PubMed; (d) C. B. Aakeröy, N. R. Champness and C. Janiak, CrystEngComm, 2010, 12, 22–43 RSC.
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS PubMed.
- A. R. Voth and P. S. Ho, Curr. Top. Med. Chem., 2007, 7, 1336–1348 CrossRef CAS PubMed.
- G. M. Espallargas, Ideas Chem. Mol. Sci.: Adv. Nanotechnol., Mater. Devices, 2010, 115–138 CAS.
- (a) M. Fourmigué and P. Batail, Chem. Rev., 2004, 104, 5379–5418 CrossRef PubMed; (b) M. Brezgunova, K. S. Shin, P. Auban-Senzier, O. Jeannin and M. Fourmigué, Chem. Commun., 2010, 3226–3228 Search PubMed; (c) H. M. Yamamoto, Y. Kosaka, R. Maeda, J. Yamaura, A. Nakao, T. Nakamura and R. Kato, ACS Nano, 2008, 2, 143–155 CrossRef CAS PubMed.
- G. R. Hanson, P. Jensen, J. McMurtrie, L. Rintoul and A. S. Micallef, Chem. – Eur. J., 2009, 15, 4156–4164 CrossRef CAS PubMed.
- P. Metrangolo, Y. Carcenac, M. Lahtinen, T. Pilati, K. Rissanen, A. Vij and G. Resnati, Science, 2009, 323, 1461–1464 CrossRef CAS PubMed.
- (a) S. George, A. Nangia, C.-K. Lam, T. C. W. Mak and J. F. Nicoud, Chem. Commun., 2004, 1202–1203 RSC; (b) E. Cariati, A. Forni, S. Biella, P. Metrangolo, F. Meyer, G. Resnati, S. Righetto, E. Tordin and R. Ugo, Chem. Commun., 2007, 2590–2592 RSC.
- (a) H. L. Nguyen, P. N. Horton, M. B. Hursthouse, A. C. Legon and D. W. Bruce, J. Am. Chem. Soc., 2004, 126, 16–17 CrossRef CAS PubMed; (b) C. Präsang, A. C. Whitwood and D. W. Bruce, Chem. Commun., 2008, 2137–2139 RSC.
- (a) K. Raatikainen, J. Huuskonen, M. Lahtinen, P. Metrangolo and K. Rissanen, Chem. Commun., 2009, 2160–2162 RSC; (b) P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Chem. Commun., 2008, 1635–1637 RSC; (c) B. K. Saha, R. K. R. Jetti, L. S. Reddy, S. Aitipamula and A. Nangia, Cryst. Growth Des., 2005, 5, 887–899 CrossRef CAS; (d) B. K. Saha and A. Nangia, CrystEngComm, 2006, 8, 440–4403 RSC.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS.
- K. Merz and V. Vasylyeva, CrystEngComm, 2010, 12, 3989–4002 RSC.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- P. Metrangolo and G. Resnati, Halogen Bonding: Fundamentals and Applications (Structure and Bonding), Springer, Heidelberg, 2010 Search PubMed.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed.
- D. W. Bruce, P. Metrangolo, F. Meyer, C. Präsang, G. Resnati, G. Terraneo and A. C. Whitwood, New J. Chem., 2008, 32, 477–482 RSC.
- C. A. Aakeröy, P. D. Chopade, C. Ganser and J. Desper, Chem. Commun., 2011, 47, 4688–4690 RSC.
- D. Cinčić, T. Friščić and W. Jones, CrystEngComm, 2011, 13, 3224–3231 RSC.
- R. Liantonio, P. Metrangolo, T. Pilati and G. Resnati, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, o575–o577 CAS.
- (a) J. I. Jay, C. W. Padgett, R. D. B. Walsh, T. W. Hanks and W. T. Pennington, Cryst. Growth Des., 2001, 1, 501–507 CrossRef CAS; (b) T. Mukai and K. Nishikawa, X-Ray Struct. Anal. Online, 2010, 26, 31–32 CrossRef CAS.
- C. B. Aakeröy, T. K. Wijenthunga and J. Desper, J. Mol. Struct., 2014, 1072, 20–27 CrossRef.
- (a) A. C. B. Lucassen, A. Karton, G. Leitus, L. J. W. Shimon, J. M. L. Martin and M. E. van der Boom, Cryst. Growth Des., 2007, 7, 386–392 CrossRef CAS; (b) M. Vartanian, A. C. B. Lucassen, L. J. W. Shimon and M. E. van der Boom, Cryst. Growth Des., 2008, 8, 786–790 CrossRef CAS.
- S. Triguero, R. Llusar, V. Polo and M. Fourmigué, Cryst. Growth Des., 2008, 8, 2241–2247 CAS.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Chem. Commun., 2008, 1635–1637 RSC.
- L. C. Roper, C. Präsang, V. N. Kozhevnikov, A. C. Whitwood, P. B. Karadakov and D. W. Bruce, Cryst. Growth Des., 2010, 10, 3710–3720 CAS.
- (a) K. Durka, S. Luliński, K. N. Jarzembska, J. Smętek, J. Serwatowskia and K. Woźniak, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 157–171 CAS; (b) R. B. Kanth Siram, D. P. Karothu, T. N. G. Row and S. Patil, Cryst. Growth Des., 2013, 13, 1045–1049 CrossRef; (c) V. R. Hathwar and T. N. G. Row, Cryst. Growth Des., 2011, 11, 1338–1346 CrossRef CAS; (d) R. Malavé Osuna, V. Hernández, J. T. López-Navarrete, E. D'Oria and J. J. Novoa, Theor. Chem. Acc., 2011, 128, 541–553 CrossRef.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 03, Revision E.01, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- F. A. Bulat, A. Toro-Labbe, T. Brinck, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 1679–1691 CrossRef CAS PubMed.
- F. Neese, WIREs Comput. Mol. Sci., 2011, 2, 73 CrossRef.
- S. Kozuch and J. Martin, J. Chem. Theory Comput., 2013, 135, 19282–19291 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1409978–1409983. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce02154e |
| This journal is © The Royal Society of Chemistry 2016 |