
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThioether bond formation by SPASM domain radical SAM enzymes: Cα H-atom abstraction in subtilosin A biosynthesis†
Alhosna
Benjdia
a,
Alain
Guillot
a,
Benjamin
Lefranc
bc,
Hubert
Vaudry
bc,
Jérôme
Leprince
bc and
Olivier
Berteau
*a
aMicalis Institute, ChemSyBio, INRA, AgroParisTech, Université Paris-Saclay, 78350 Jouy-en-Josas, France. E-mail: Olivier.Berteau@jouy.inra.fr
bINSERM U982, F-76821 Mont-Saint-Aignan, France
cInstitute for Research and Innovation in Biomedicine (IRIB), Regional Platform for Cell Imaging, PRIMACEN, University of Rouen, F-76821 Mont-Saint-Aignan, France
First published on 4th April 2016
Abstract
AlbA is a radical SAM enzyme catalyzing the formation of three unusual thioether bonds in the antibiotic subtilosin A. We demonstrate here that AlbA catalyzes direct Cα H-atom abstraction and likely contains three essential [4Fe–4S] centers. This leads us to propose novel mechanistic perspectives for thioether bond catalysis by radical SAM enzymes.
Subtilosin A is a bacteriocin produced by Bacillus subtilis targeting several pathogens such as Listeria monocytogenes.1 This bacteriocin is the founding member of sactipeptides, a diverse group of ribosomally synthesized and post-translationally modified peptides (RiPPs) characterized by the presence of unusual sulfur to α-carbon thioether bridges (Fig. 1a). Since the discovery of subtilosin A, which contains two Cys-to-Phe and one Cys-to-Thr linkages,2 several sactipeptides have been identified including the sporulation killing factor of Bacillus subtilis3 and two bacteriocins produced by Bacillus thuringiensis called thuricin CD4 and thurincin H,5 respectively. However, mechanistic details regarding the enzymes catalyzing these reactions are still lacking notably because of the high insolubility of their peptide substrates.2
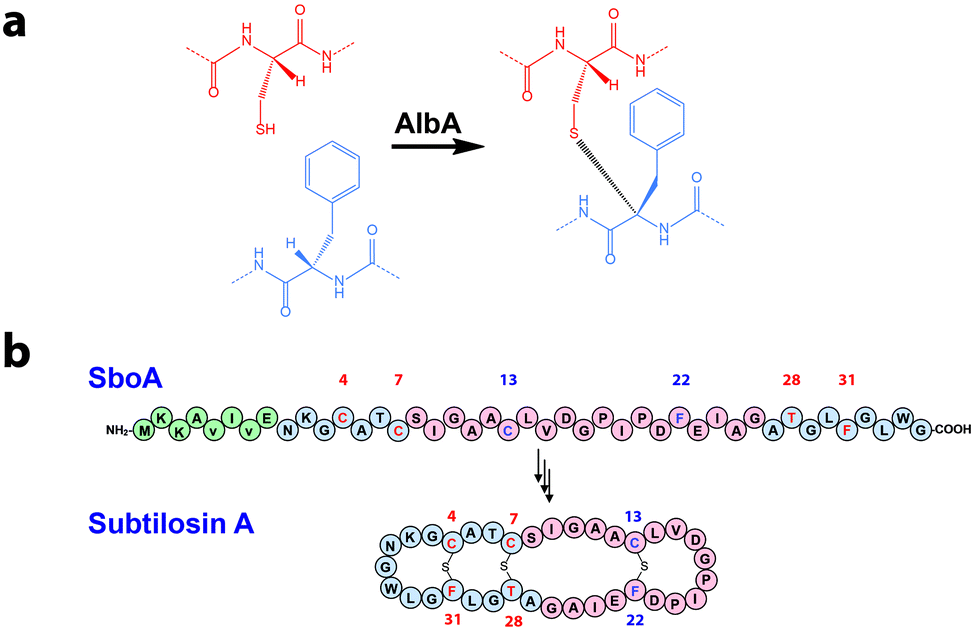 | ||
| Fig. 1 (a) Reaction catalyzed by AlbA. (b) Sequence of the pro-peptide SboA and structure of the mature subtilosin A. Numbers indicate the position of the residues involved in thioether bonds (i.e.4, 7 & 13: Cys; 22 & 31: Phe; 28: Thr). Amino acids involved in the L-configured thioether bond are labeled in blue (i.e.L-Phe 22) and those involved in D-configured bond are labeled in red (i.e.D-Phe 31 and D-Thr 28). Green circles indicate the leader peptide. Red circles correspond to the sequence of SboA8-27 and blue circles correspond to SboA27-6 (see Fig. 2). | ||
The biosynthesis of subtilosin A is dependent upon a 43-amino-acid peptide precursor (SboA) and the AlbA enzyme.1 Recently, it has been demonstrated that AlbA is a radical SAM (rSAM) enzyme catalyzing the formation of the three thioether bridges in subtilosin A providing the first biochemical information on the biosynthesis of sactipeptides.6 AlbA belongs to the SPASM-domain group of rSAM enzymes7 which catalyze peptide or protein post-translational modifications and are characterized by the presence of a specific protein domain containing [4Fe–4S] centers.6,8–10 However, to date, the function of the SPASM-domain is still unclear.7
To probe the mechanism of AlbA, we rationally designed synthetic peptides based on the sequence of the mature subtilosin A (Fig. 1b & Fig. S1, ESI†). These two peptides, SboA8-27 and SboA27-6, encompassed the full sequence of subtilosin A and were designed to allow the formation of only one thioether bond in order to dissect catalysis. The sequence of SboA8-27 corresponded to residues 8–27 while SboA27-6 contained the residues 27–35 covalently linked to residues 1–6 by an amide bond (Fig. 1b, 2a and b).
After anaerobic reconstitution (Fig. S2, ESI†), AlbA activity was assayed in the presence of SboA8-27 and S-adenosyl-L-methionine (SAM) under reducing conditions. As shown, AlbA efficiently cleaved SAM, although no substrate modification was evidenced, even after an extended incubation time (Fig. 2a, Fig. S3 & Table S1, ESI†). In contrast, with SboA27-6, a product (SboA27-6*) with a retention time of 42.15 min was formed along with 5′-deoxyadenosine (5′-dA) (Fig. 2b & Fig. S3, ESI†). LC-MS2 analysis showed that this new peptide had a mass shift of −2 Da (Fig. S4 and S5, ESI†) and contained a covalent bond between Cys4 and Phe31 as in authentic subtilosin A2 (Fig. 1b, 2c, d and Table S2, ESI†). This result was consistent with AlbA catalyzing the formation of a thioether bond on SboA27-6. Kinetic analysis showed that AlbA exhibited a specific activity of 1.59 ± 0.1 nmol min−1 mg−1 and 4.72 ± 0.02 nmol min−1 mg−1 for the production of the cyclic peptide and 5′-dA, respectively (Fig. 2e and f). In two hours ∼120 μM of a cyclic peptide was produced, a result similar to the one estimated using SboA as the substrate.6 Unexpectedly, the short peptide SboA27-6, despite being deprived of the leader peptide, proved to be an efficient substrate suitable to dissect enzyme catalysis.
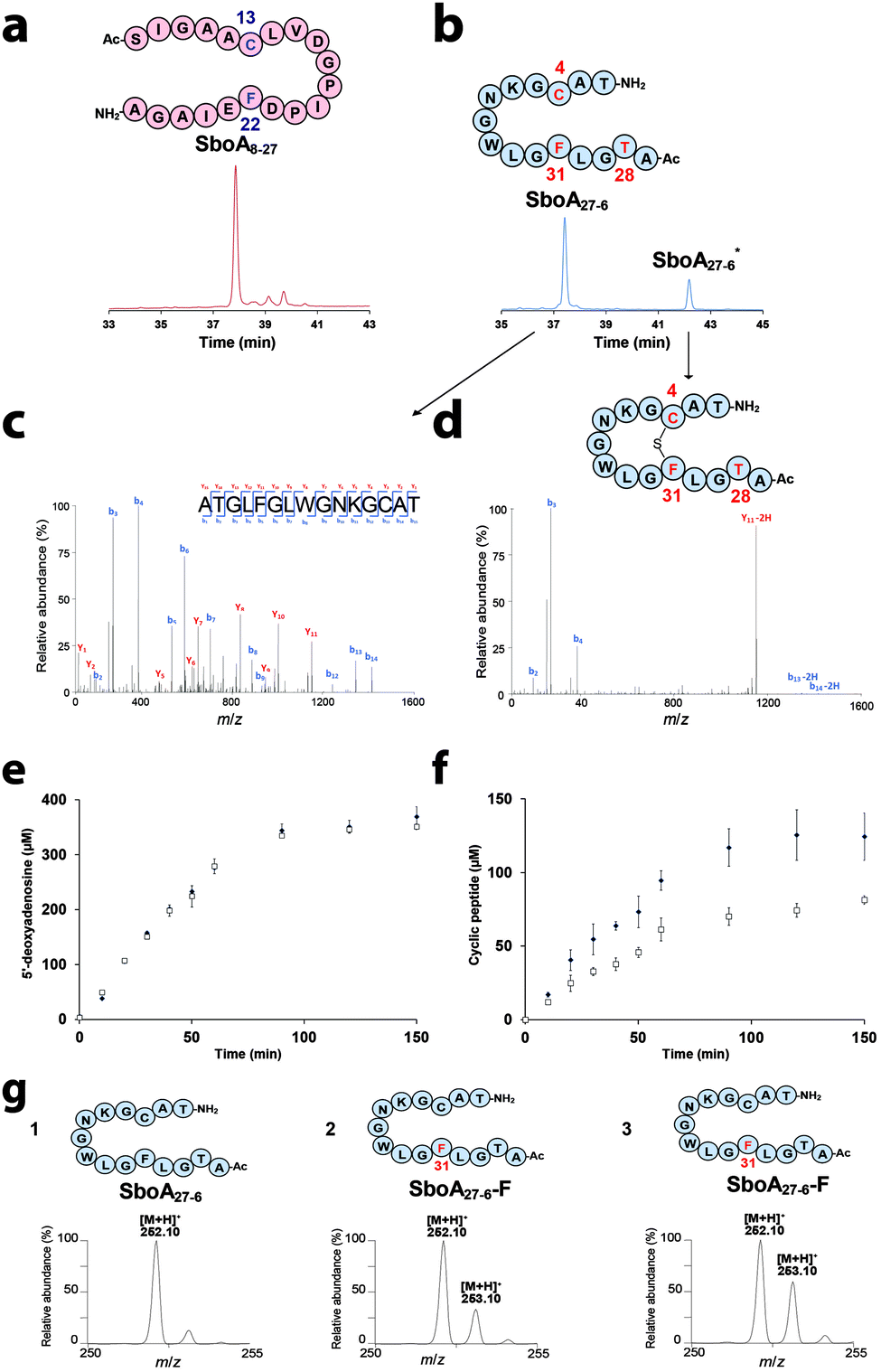 | ||
| Fig. 2 AlbA activity on synthetic peptides. HPLC analysis of AlbA reaction in the presence of (a) SboA8-27 or (b) SboA27-6 after 120 min (UV detection at 215 nm). LC-MS2 analysis of SboA27-6 (c) and the cyclic peptide SboA27-6* (d) produced by AlbA. Relevant fragments of the b and y series are indicated (see Fig. S5 and Table S2, ESI† for complete assignment). Time course for the production of 5′-dA (e) and cyclic peptide (f) by AlbA in the presence of SboA27-6 (■) or the deuterated substrate, SboA27-6-F (□). LC-MS analysis of 5′-dA produced by AlbA (g) in the presence of (1) SboA27-6, (2) SboA27-6-F using sodium dithionite (DT) or (3) SboA27-6-F using flavodoxin/flavodoxin reductase/NADPH (Fldx/Fpr) as a reducing system. AlbA (20 μM) was incubated in the presence of SAM (1 mM), peptide substrate (1 mM) and sodium dithionite (2 mM) or Fldx/Fpr. | ||
In order to demonstrate that AlbA catalyzes substrate H-atom abstraction and to identify the target of the 5′-dA˙ radical, we synthesized two SboA27-6 derivatives: SboA27-6-F and SboA27-6-C containing a per-deuterated phenylalanine or a cysteine (β,β-D2), respectively. With SboA27-6-F, we obtained a cyclic peptide with a mass shift of −3 Da corresponding to the loss of a H-atom and a D-atom. LC-MS2 confirmed the formation of a thioether bond between Cys4 and the deuterated Phe31 (Fig. S6, ESI†). In the presence of SboA27-6-F, AlbA exhibited a specific activity of 0.96 ± 0.01 nmol min−1 mg−1 for thioether bond formation and a SAM cleavage activity of 4.66 ± 0.07 nmol min−1 mg−1 (Fig. 2e and f).
Apparent from these kinetic experiments, a decoupling between SAM cleavage and product formation occurred with a ratio between 5′-dA and the cyclic peptide of ∼2.9. This ratio increased to 5 in the presence of the deuterated substrate. Comparing the reaction rate for cyclic peptide production using the unlabeled or the deuterated substrate, we determined a kinetic isotope effect of ∼1.7 (Fig. 2e and f). This behavior mirrored what has been reported for another SPASM-domain enzyme, the anaerobic Sulfatase-Maturating Enzyme (anSME)8–10 when the amino acid residue targeted by this enzyme was specifically deuterated.11
To definitively determine if Phe31 was the target of the 5′-dA˙ radical, we analyzed the 5′-dA produced in reaction using LC-MS (Fig. 2g). As shown, ∼20% deuterium was incorporated into the 5′-dA produced when SboA27-6-F was used as the substrate (Fig. 2g trace 2). This result was in-line with the 5-to-1 ratio measured between 5′-dA and cyclic peptide production. To improve the coupling between both reactions, we substituted sodium dithionite by the physiological reducing system: flavodoxin/flavodoxin reductase/NADPH.12,13 Under these conditions, deuterium incorporation increased up to 50% (Fig. 2g trace 3). In contrast, with SboA27-6-C (labeled on Cys4), no labeling was introduced into the 5′-dA produced, even when the physiological reducing system was used (Fig. S7, ESI†). This result excluded possible hydride shifts between residues during catalysis. Altogether, these labeling experiments strongly support that AlbA catalyzes direct Cα H-atom abstraction on Phe31 and it is the rate determining step of the reaction.
In addition of the rSAM center, AlbA was hypothesized to contain a second [4Fe–4S] center coordinated by three vicinal cysteine residues (i.e. Cys408, Cys414 and Cys417) with a critical role for substrate interaction.6 However, the crystal structure of anSME demonstrated that the [4Fe–4S] centers coordination in the SPASM-domain involves remote cysteine residues and that [4Fe–4S] centers do not coordinate the enzyme substrate.14 Furthermore, structural predictions positioned the SPASM [4Fe–4S] center of AlbA too far away from the enzyme active site to allow direct substrate interaction.7 Using a 3D structural model (see methods in the ESI†), we identified 7 cysteine residues in the AlbA SPASM-domain which perfectly superimposed and aligned with anSME (Fig. 3a and b & Fig. S8, ESI†). This model predicted that not only Cys408, Cys414 and Cys417 but also Cys344, Cys362, Cys403 and Cys433 are involved in [4Fe–4S] center coordination6. Hence, not one but two [4Fe–4S] centers were predicted in the SPASM-domain of AlbA. To probe this hypothesis, we generated the following single mutants: C344A, C362A, C403A and C433A. In addition, we also produced one mutant lacking these 4 cysteine residues (Mutant A4) and, as a control, a 6th mutant lacking the radical SAM binding motif CxxxCxxC (Mutant A3) (Fig. S9, ESI†).
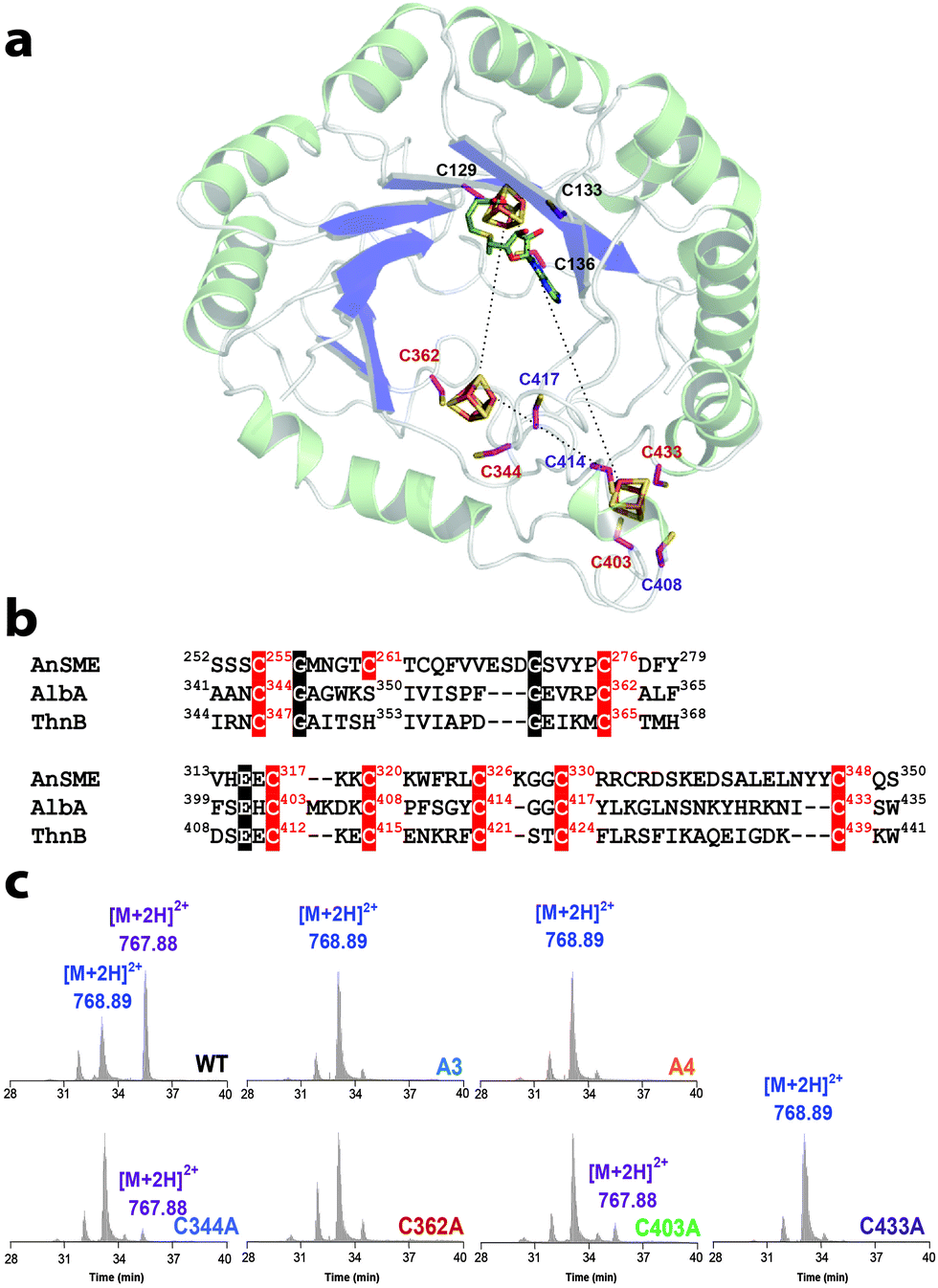 | ||
| Fig. 3 Role of the AlbA SPASM-domain. (a) Structural model of AlbA with three iron–sulfur centers. The cysteine residues involved in the coordination of [4Fe–4S] centers are depicted in pink. Numbers in purple indicate cysteine residues from the SPASM-domain previously mutated.6 (b) Sequence alignment of the rSAM enzymes: anSME, AlbA and ThnB. Highlighted in red, the anSME SPASM-domain cysteine residues involved in the coordination of [4Fe–4S] centers and the corresponding residues in AlbA and ThnB. In black are highlighted other strictly conserved residues. (c) Activity of AlbA wild-type (WT) and cysteine mutants: A3, A4, C344A, C362A, C403A and C433A measured using LC-MS. The SboA27-6 peptide has an m/z value of [M + 2H]2+ = 768.89 and the cyclic peptide SboA27-6* has an m/z value of [M + 2H]2+ = 767.88. Each protein (20 μM) was incubated under anaerobic and reducing conditions in the presence of SAM (1 mM), peptide substrate (1 mM) and sodium dithionite (2 mM). | ||
The UV-visible spectra of the reconstituted C344A, C362A, C403A and C433A mutants were identical to the wild-type enzyme while the A3 and A4 mutants exhibited a decrease in absorbance in the region between 320 and 420 nm, indicating partial iron–sulfur loading (Fig. S10, ESI†). Assayed in the presence of SAM and the SboA27-6 substrate, all mutants, except the A3, cleaved SAM into 5′-dA (Fig. S11, ESI†). However, they were all impaired for thioether bond formation with only the C344A and C403A mutants producing detectable levels of a cyclic peptide ([M + 2H]2+: 767.88) (Fig. 3c & Fig. S11, ESI†). The 7 conserved cysteine residues are thus critical for AlbA activity.
In addition to subtilosin A, recent studies on the sporulation killing factor,15 thuricin CD4 and thurincin H5 have shown that rSAM enzymes are responsible for the formation of sulfur to α-carbon thioether bridges. These rSAM enzymes belong either to the SPASM- (AlbA & ThnB) or twitch-domain (SkfB & TrnCD) groups.7 Interestingly, AlbA and ThnB despite having no significant homology (<25% identity) possess the 7 conserved cysteine residues (Fig. 3b). For all these enzymes, it has been speculated that they catalyze Cα H-atom abstraction on a diverse range of amino acid residues; however, no experimental proof has been provided thus far.
Our results support that AlbA, and likely all known rSAM enzymes involved in sactipeptide thioether bond formation, catalyzes Cα H-atom abstraction. Similarly to what was demonstrated for anSME,9 H-atom abstraction is the rate determining step of the reaction. Our study also supports that AlbA contains two SPASM [4Fe–4S] centers coordinated by 7 cysteine residues. While it remains to be determined whether they have partial or full ligation, sequence alignment (Fig. 3b) showed that Cys261 in anSME aligns with Ser350 in AlbA. Interestingly, a Ser residue has been recently reported to be involved in the coordination of the auxiliary [4Fe–4S] center in the rSAM enzyme LipA.16
Remarkably, the 3 thioether bonds catalyzed by AlbA have different (L/D) stereochemistries. In the case of lanthipeptides, which are by far the most well-studied thioether bond-containing peptides, it was elegantly demonstrated that substrate flexibility only governs thioether bond configuration.17 Furthermore, we showed here that even single mutations, which likely alter the redox properties of the [4Fe–4S] centers, are sufficient to dramatically alter enzyme activity. Collectively, these data support a role as an electron conduit for the auxiliary [4Fe–4S] centers rather than a role in substrate coordination which appears inconsistent with the formation of thioether bonds with both stereochemistry.10,17
In conclusion, we propose a novel mechanism for the catalysis of thioether bond formation by rSAM enzymes (Fig. 4). Following SAM cleavage, the 5′-dA radical abstracts a Cα H-atom on Phe (or Thr), a process thermodynamically favorable considering the CαH-atom bond dissociation energies of 348.9 kJ mol−1 and 345.1 kJ mol−1 for Phe and Thr respectively.18 Among rSAM enzymes, the pyruvate formate lyase activase (PFL-AE)19 and the ribonucleotide reductase activase (RNR-AE)20 have been shown to catalyze Cα H-atom abstraction of a glycine residue. However, because of steric interactions between the Phe side chain and the peptide backbone, AlbA generates a less stable tertiary Cα carbon-centered radical than the secondary glycyl radical produced by PFL-AE21 or RNA-AE.20 This carbon-centered radical intermediate will have a high propensity to abstract H-atoms and thus cannot react directly with cysteine to form a thioether bond. Hence, it has been proposed that the substrate interacts first through its cysteine residues with a SPASM [4Fe–4S] center,6 a process presumed to allow direct addition of the Cα carbon-centered radical to the S-atom. However, as detailed above, AlbA exhibits strong biochemical and structural analogies with anSME. This latter enzyme was shown to catalyze Cβ H-atom abstraction9 leading to the formation of a C–S bond between the Cβ-atom and the deprotonated thiol group of its cysteine substrate.22 Based on structural and biochemical data, it is established that the SPASM [4Fe–4S] centers of anSME fulfill a key redox function which remains to be fully elucidated.14,22 In the case of AlbA, our data support a similar mechanism. After H-atom abstraction and loss of the Cα-atom stereochemistry (of Phe or Thr), the radical intermediate likely oxidizes to an N-acyliminium ion (Fig. 4). This intermediate can be readily trapped by the nucleophilic thiolate group of a remote cysteine leading to the formation of the C–S bond. In support of this hypothesis, the chemical synthesis of cyclic peptides containing thioether bonds has been shown to involve N-acyliminium intermediates.23 Depending on the attack on the N-acyliminium intermediate, L- or D-configured bonds are formed, guided by substrate flexibility.17 In this mechanistic hypothesis, the SPASM [4Fe–4S] centers of AlbA are keys for the oxidation of the radical intermediate as proposed for anSME9,24 and for the formation of the N-acyliminium intermediate. This mechanism represents a novel redox alternative to the formation of thioether bonds in natural products.
 | ||
| Fig. 4 Proposed mechanism for AlbA. | ||
This work was supported in part by grants from the European Research Council (ERC) (Consolidator Grant 617053 to OB). We thank Dr Gaëlle Vassiliadis for her contribution to the early stage of this project and Ms Pauline Ruffié for technical assistance.
Notes and references
- G. Zheng, R. Hehn and P. Zuber, J. Bacteriol., 2000, 182, 3266–3273 CrossRef CAS PubMed.
- K. E. Kawulka, T. Sprules, C. M. Diaper, R. M. Whittal, R. T. McKay, P. Mercier, P. Zuber and J. C. Vederas, Biochemistry, 2004, 43, 3385–3395 CrossRef CAS PubMed.
- J. E. Gonzalez-Pastor, E. C. Hobbs and R. Losick, Science, 2003, 301, 510–513 CrossRef CAS PubMed.
- M. C. Rea, C. S. Sit, E. Clayton, P. M. O'Connor, R. M. Whittal, J. Zheng, J. C. Vederas, R. P. Ross and C. Hill, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9352–9357 CrossRef CAS PubMed.
- B. M. Wieckowski, J. D. Hegemann, A. Mielcarek, L. Boss, O. Burghaus and M. A. Marahiel, FEBS Lett., 2015, 589, 1802–1806 CrossRef CAS PubMed.
- L. Flühe, T. A. Knappe, M. J. Gattner, A. Schäfer, O. Burghaus, U. Linne and M. A. Marahiel, Nat. Chem. Biol., 2012, 8, 350–357 CrossRef PubMed.
- T. A. Grell, P. J. Goldman and C. L. Drennan, J. Biol. Chem., 2015, 290, 3964–3971 CrossRef CAS PubMed.
- A. Benjdia, J. Leprince, A. Guillot, H. Vaudry, S. Rabot and O. Berteau, J. Am. Chem. Soc., 2007, 129, 3462–3463 CrossRef CAS PubMed.
- A. Benjdia, J. Leprince, C. Sandstrom, H. Vaudry and O. Berteau, J. Am. Chem. Soc., 2009, 131, 8348–8349 CrossRef CAS PubMed.
- A. Benjdia, S. Subramanian, J. Leprince, H. Vaudry, M. K. Johnson and O. Berteau, J. Biol. Chem., 2008, 283, 17815–17826 CrossRef CAS PubMed.
- O. Berteau, A. Guillot, A. Benjdia and S. Rabot, J. Biol. Chem., 2006, 281, 22464–22470 CrossRef CAS PubMed.
- L. Decamps, B. Philmus, A. Benjdia, R. White, T. P. Begley and O. Berteau, J. Am. Chem. Soc., 2012, 134, 18173–18176 CrossRef CAS PubMed.
- B. Philmus, L. Decamps, O. Berteau and T. P. Begley, J. Am. Chem. Soc., 2015, 137, 5406–5413 CrossRef CAS PubMed.
- P. J. Goldman, T. L. Grove, L. A. Sites, M. I. McLaughlin, S. J. Booker and C. L. Drennan, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8519–8524 CrossRef CAS PubMed.
- L. Fluhe, O. Burghaus, B. M. Wieckowski, T. W. Giessen, U. Linne and M. A. Marahiel, J. Am. Chem. Soc., 2013, 135, 959–962 CrossRef PubMed.
- J. E. Harmer, M. J. Hiscox, P. C. Dinis, S. J. Fox, A. Iliopoulos, J. E. Hussey, J. Sandy, F. T. Van Beek, J. W. Essex and P. L. Roach, Biochem. J., 2014, 464, 123–133 CrossRef CAS PubMed.
- W. Tang, G. Jimenez-Oses, K. N. Houk and W. A. van der Donk, Nat. Chem., 2015, 7, 57–64 CrossRef CAS PubMed.
- B. N. Moore and R. R. Julian, Phys. Chem. Chem. Phys., 2012, 14, 3148–3154 RSC.
- J. Knappe, F. A. Neugebauer, H. P. Blaschkowski and M. Ganzler, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 1332–1335 CrossRef CAS.
- X. Sun, S. Ollagnier, P. P. Schmidt, M. Atta, E. Mulliez, L. Lepape, R. Eliasson, A. Graslund, M. Fontecave, P. Reichard and B. M. Sjoberg, J. Biol. Chem., 1996, 271, 6827–6831 CrossRef CAS PubMed.
- A. F. Wagner, M. Frey, F. A. Neugebauer, W. Schafer and J. Knappe, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 996–1000 CrossRef CAS.
- A. Benjdia, S. Subramanian, J. Leprince, H. Vaudry, M. K. Johnson and O. Berteau, FEBS J., 2010, 277, 1906–1920 CrossRef CAS PubMed.
- T. E. Nielsen, S. Le Quement and M. Meldal, Org. Lett., 2005, 7, 3601–3604 CrossRef CAS PubMed.
- A. Benjdia, G. Deho, S. Rabot and O. Berteau, FEBS Lett., 2007, 581, 1009–1014 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc01317a |
| This journal is © The Royal Society of Chemistry 2016 |