
Evaluation of CdTe/CdS/ZnS core/shell/shell quantum dot toxicity on three-dimensional spheroid cultures†
Mehriban
Ulusoy
a,
Antonina
Lavrentieva
a,
Johanna-Gabriela
Walter
a,
Franziska
Sambale
a,
Mark
Green
b,
Frank
Stahl
*a and
Thomas
Scheper
a
aGottfried Wilhelm Leibniz University of Hannover, Institute of Technical Chemistry, 30167 Hanover, Germany. E-mail: stahl@iftc.uni-hannover.de; Tel: +49 (0)511 762-2968
bKing's College London, Department of Physics, The Strand, WC2R LS London, UK. E-mail: mark.a.green@kcl.co.uk; Tel: +44 (0)2078 48212
First published on 7th August 2015
Abstract
In this work, three-dimensional (3D) spheroid cultures of human adipose-derived mesenchymal stem cells (hAD-MSCs), with tissue-mimetic morphology through well developed cell–cell and cell–matrix interactions and distinct diffusion/transport characteristics, were assessed for dose-dependent toxic effects of red-emitting CdTe/CdS/ZnS quantum dots (Qdots). Morphological investigations and time-resolved microscopy analysis in addition to cell metabolic activity studies revealed that 3D spheroid cultures are more resistant to Qdot-induced cytotoxicity in comparison to conventional 2D cultures. The obtained results suggest the presence of two distinct cell populations in 2D cultures with different sensitivity to Qdots, however that effect wasn't observed in 3D spheroids. Our investigations were aimed to improve the prediction of nanotoxicity of Qdot on tissue-level and provide the essential screening steps prior to any in vivo application. Moreover, penetration ability of highly fluorescent Qdots to densely-packed spheroids will fortify the biological application of developed Qdots in tissue-like structures.
Introduction
Conventional two-dimensional (2D) cell culture where a cell monolayer grows on flat plastic or glass surfaces does not reflect the essential physiology of real tissues. Since cells in the body grow in a three-dimensional (3D) environment, a 3D approach as an alternative to 2D culture can reduce the gap between cell culture and living tissue. Therefore, a 3D culture exhibits significant potential to improve the physiological relevance of cell-based assays.1–4 In many studies, 3D cell cultures were proven to display the induced extracellular matrix (ECM)-related biological functions such as intercellular signalling and interactions, cellular function and maintenance, molecular transport and tissue morphology.5–7 Cellular spheroids, being a simplified model of 3D cell cultures, take advantages of natural tendency of cells to aggregate.8 The cells produce ECM which in turn enhances intercellular adhesion. Thus, they don't require an external scaffold for the aggregation of the cells. For that reason, it is the most widely used model for high-throughput screening of straightforward cell function and toxicity analysis for biomedical applications.6,9,10 For nanoparticle (NP)-based cytotoxicity, labeling and delivery studies, 3D spheroid models have advantages over monolayer cell cultures.3 Monolayer cell cultures produce less dense ECM material on an apical side whereas cells grown in 3D extend ECM matrix production to all dimensions. Therefore, 2D models present a less significant barrier for transport and reduced cell binding compared to cells in 3D environment.11 For that reason, results obtained from NP-based research on 2D cell cultures do not reflect similar results as obtained from 3D cultures.3D models, however, possess some limitations in terms of translation compared to available 2D-based analysis methods. Some cell-based high-throughput screening methods for rapid analysis of drug or NP-based cellular responses (such as dose-dependent cell viability, cell migration and cell–cell/cell–matrix interaction) haven't been optimized for 3D culture models yet. Remaining limitations to overcome are scalability, reproducibility, sensitivity and compatibility of analytical methods with available screening systems. Despite the increasing number of publications for 3D-based cell culture studies, optimization of available analytical methods in order to address the nanoparticle and drug interactions remains a challenge.12
Nanocrystals of semiconductors, so-called quantum dots (Qdots), represent one of the most diverse NP class owing to a wide range of optical and structural properties which allow numerous application areas from molecular imaging to targeted drug delivery.13–15 Based on their size-tunable optical features, narrow emission and broad absorption bands in combination with broad-band excitations and long fluorescence life time, Qdots take a special position when compared with other nanoparticles and organic fluorophores.16,17 However, vast application of Qdots in cellular uptake studies come with some concerns, of which their toxicity comprises one of the major points of issue. Their fate in the organism as well as their toxicity which has a direct relation to their structure, size, surface chemistry and colloidal stability became an important research topic for their engineering and development. For that reason, investigation of any Qdot-related side effects on the cell level must be accomplished prior to their bioapplication. Until now, for almost all studies on Qdot toxicity, 2D cell cultures were used for the determination of dose-dependent adverse effects, and the results of the studies are difficult to translate to in vivo models. Regarding Qdot-related toxicity, to best of our knowledge, there is only one study by Lee and coworkers where effect of single dose CdTe Qdots on HepG2 spheroid cultures was reported.18 However, applied Qdot doses comprise one of the major parameters in toxicity studies; hence, dose-dependent adverse effects must be investigated in details in order to understand the Qdot interactions in a better way. Therefore, exploration of dose-dependent Qdot effects on 3D spheroid models should be considered as an essential step for the prediction of Qdot toxicity on micro-tissue structures.
In this report, we describe the evaluation of dose-dependent adverse effects of carboxyl-functionalized CdTe/CdS/ZnS core(small)/shell(thick)/shell(small) (CSS) Qdots on 3D spheroid cultures. One-pot aqueous synthesis of near-infrared emitting CdTe/CdS/ZnS core(small)/shell(thick)/shell(small) CSS Qdots with high quantum yields (∼64%) and impressive stability against photobleaching was recently reported in our previous study.19 The developed CSS Qdots hold a great potential for cellular imaging studies to owing near-infrared emission, which will give them an advantage to be monitored in thick and highly scattering 3D tissue samples.20 As a model system, we utilized human adipose-derived mesenchymal stem cells (hAD-MSCs) spheroid cultures as they are considered to be a promising candidate for cell therapy.21 Stem cells were also found to be more susceptible to Qdot toxicity (IC50 40 μg ml−1) in comparison to A549 lung adenocarcinoma cell lines (IC50 150 μg ml−1) according to our previous study.19 For that reason, hAD-MSCs were chosen as a suitable candidate to investigate Qdot-induced cytotoxicity. The adverse effects of Qdots were examined using two different approaches, including morphology and metabolic activity. Dose-dependent cell viability was determined with two different cell viability assays; CellTiter-Blue (CTB) and adenosine triphosphate (ATP) assays, and results were compared with monolayer 2D cultures. Morphological observations as well as metabolic activity studies were discussed in details. In the later step, the ability of Qdots to label spheroid structures was tested via fluorescence and confocal microscopy studies. Our results highlighted significant physiologically relevant data for Qdot-induced toxicity. We believe that outcomes of this study will provide important data to increase our understanding of Qdots-induced toxicity on tissue levels. They can also contribute to the establishment of analytical methods for nanoparticle-based cytotoxicity studies in tissue-like in vitro systems. Additionally, the penetration ability of Qdots to densely packed stem cell spheroids in combination with their high fluorescence yield with near-infrared emission feature will greatly aid future exploration of Qdots for the targeted imaging studies in 3D models, and potentially will broaden up their range of applications to the delivery of therapeutic agents to 3D compact tumors for diagnosis and disease treatment.
Experimental procedures
Materials
CdCl2·21/2H2O (cadmium chloride hemi(pentahydrate), >98%), Na2TeO3 (sodium tellurite, 99.5%), ZnCl2 (zinc chloride, anhydrous, 99.99%), 3-mercaptopropionic acid (MPA, 99%), NaBH4 (sodium borohydride, >98.0%) and trisodium citrate dehydrate were purchased from Sigma-Aldrich GmbH, Munich. CellTiter-Blue® and CellTiter-Glo® cell viability assay kits were purchased from Promega Corp., USA. For cell culture experiments, Minumum Essential Medium Alpha (MEM-α, Gibco Invitrogen, USA) supplemented with 10% human serum (HS, c.c.pro GmbH, Germany) and 0.5% gentamicin (Biochrom GmbH, Germany) was used as cell culture medium. For 2D cell cultures, 96-well flat bottom standard plates (Sarstedt AG&Co. Germany) and for 3D spheroid cultures, 96-well round bottom plates coated with ultra-low attachment surface (Corning Inc., USA) were used.Synthesis of CdTe/CdS/ZnS quantum dots
CdTe/CdS/ZnS Qdot were synthesized as described previously.19 First, CdTe/CdS Qdot having a core(small)/shell(thick) structure was synthesized as follows: 21 ml ddH2O, CdCl2 (125 mM, 1 ml), 18.5 μl 3-mercaptopropionic acid (MPA), trisodium citrate dehydrate (42.5 mM, 1 ml), sodium tellurite (5 mM, 1 ml) and sodium borohydride (125 mM, 1 ml) were added sequentially into a three-neck round bottom flask. The molar ratio of Cd/Te/MPA was set to be 1/0.04/1.7. Under mixing, the solution pH was adjusted to 11.4 with 1 M NaOH. The reaction solution was then refluxed with an air condenser (Findenser, Radleys, UK) at 90 °C. When the emission wavelength of CdTe/CdS Qdot reached to 655 ± 5 nm, the nanoparticle growth was stopped by simply cooling the reaction temperature to 4 °C. The as-prepared CdTe/CdS solution was directly used for the external synthesis of ZnS shell. 12 ml of the CdTe/CdS crude solution, 12 ml ddH2O and 1 ml ZnCl2 solution (25 mM) were added together giving a final volume of 25 ml. The final pH of the reaction solution was adjusted to 11.5 with 1 M NaOH. Then, the solution was heated slowly to 65 °C and the ZnS shell growth was proceeded for 3 h, yielding in CdTe/CdS/ZnS core(small)/shell(thick)/shell(small) Qdot (λemission = 670 ± 5 nm) nm, QYmax ∼ 64%). Later, samples were precipitated with 2-propanol and collected via centrifugation. The colloidal precipitate was weighed after drying under vacuum and re-dissolved in water. For storage, it was washed with ddH2O using 10 kDa MWCO PES membrane filters (Vivaspin, Sartorius Stedim Biotech GmbH, Germany) and kept at 4 °C. For cell culture applications, Qdots solutions after dilution with culture medium were filtrated with 0.2 μm pore-sized high-flow syringe filters (Sartorius Stedim Biotech GmbH, Germany) for sterilization.Cell culture and Qdot treatment
Human adipose-derived mesenchymal stem cells (hAD-MSCs) were isolated from subcutaneous adipose tissues of 3 different patients scheduled for abdominoplasty after obtaining informed written consent, as approved by the Institutional Review Board, project #2251-2014 on 15th May, 2014. The isolated populations have been extensively characterized as mesenchymal stem cells by surface marker analysis and functional properties (differentiation capacity). Cells were cultured with MEM-α medium supplemented with 10% HS and 0.5% gentamicin in standard T-175 culture flasks (Sarstedt AG&Co, Germany) until they reached desired sub-confluency (37 °C, 5% CO2). After washing with warm PBS, they were detached from the culture flask using accutase solution (Sigma-Aldrich). Following centrifugation at 300 g for 5 min, cell suspension was collected and the number of viable cells was determined by trypan blue exclusion. For 2D culture, 8000 cells in 100 μl of culture medium were seeded to 96-well flat bottom standard plates. After 24 h of adhesion at 37 °C, 5% CO2, medium was replaced with 100 μl of Qdot solutions diluted with culture medium. A control group was treated with fresh culture medium without Qdots. For 3D culture, two different treatment models were applied according to the Qdot exposure time. In the first model, Qdots were introduced to cells after 24 h when they completed spheroid formation (tQDOT = 24 h). The procedure was performed as follows: 3000 cells in 75 μl of culture medium were seeded to 96-well round bottom spheroid plates and incubated for 24 h for spheroid formation. Afterwards, 75 μl of Qdot solutions in culture medium were added to each well and incubated further for another 24 h. In the second model, cells were exposed to Qdots solution at the beginning of spheroid formation (tQDOT = 0 h). The procedure was performed as follows: 3000 cells in 75 μl of culture medium and 75 μl of Qdot solutions in culture medium were seeded together to spheroid well-plates and incubated for 24 h (37 °C, 5% CO2).For labeling experiments, hAD-MSCs after spheroid formation (tQDOT = 24 h) were treated with Qdot solution (250 μg ml−1) for 4 h at 37 °C, 5% CO2. After Qdot treatment, live cells in spheroids were stained with Calcein-AM (EMD Chemicals, Inc. CA) by adding 1 μl Calcein-AM (1 mg ml−1) solution to the culture medium. Spheroids were washed carefully with PBS prior to DAPI staining. After that, cell nuclei were stained with DAPI (Sigma-Aldrich GmbH, Munich) by adding 100 μl DAPI staining solution (1 μg ml−1). After 20 min of incubation, spheroids were washed gently with PBS solution and placed on μ-slide glass bottom microscopy chamber slides (ibidi GmbH, Germany) for fluorescence and confocal imaging studies.
Adenosine triphosphate (ATP) assay
ATP assay was performed by using CellTiter-Glo assay kit (Promega Corp., USA) according to manufacturer instructions. Intracellular ATP amount was measured via enzymatic reaction of luciferase in presence of ATP, Mg2+ and molecular oxygen. The luminescent signals produced from the reaction are proportional to the amount of ATP present in metabolically active cells. Assay kit enables cell lysis for the release of intracellular ATP for the production of luminescence signal. For 2D cell culture, after removing culture medium, 50 μl CellTiter-Glo stock solution and 50 μl supplement-free MEM-α medium was added to the wells and incubated for 10 minutes (37 °C, 5% CO2). For 3D spheroid cultures, 75 μl of culture medium was replaced carefully with 75 μl of CellTiter-Glo stock solution and incubated for 2 h under continuous shaking at 250 rpm (37 °C, 5% CO2) in order to allow spheroid lysis. The resulting luminescence signals were measured with Perkin-Elmer Wallac Victor 2v HTS Counter 1420.CellTiter-Blue (CTB) assay
The metabolic activity of viable cells in terms of their reduction capacity of resazurin was measured via CTB assay kit (Promega Corp., USA). For 2D cell culture, culture medium was removed gently and 100 μl of CTB reagent (diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 with supplement-free MEM-α medium) was added to each well and incubated for 2 h (37 °C, 5% CO2). For 3D spheroid cultures, 100 μl of medium was replaced with CTB reagent (diluted 1:4 with supplement-free MEM-α medium, giving a final dilution of 1:6). Spheroids incubated for 48 hours under continuous shaking at 250 rpm (37 °C, 5% CO2) for the release of fluorescence resorufin dye. The resulting fluorescence intensities (544Ex/590Em) were recorded with fluorometer (Fluoroskan Ascent, Thermo Fischer Scientific Inc. USA).
:6 with supplement-free MEM-α medium) was added to each well and incubated for 2 h (37 °C, 5% CO2). For 3D spheroid cultures, 100 μl of medium was replaced with CTB reagent (diluted 1:4 with supplement-free MEM-α medium, giving a final dilution of 1:6). Spheroids incubated for 48 hours under continuous shaking at 250 rpm (37 °C, 5% CO2) for the release of fluorescence resorufin dye. The resulting fluorescence intensities (544Ex/590Em) were recorded with fluorometer (Fluoroskan Ascent, Thermo Fischer Scientific Inc. USA).
Statistical analysis
Concentration-dependent normalized cell viability data obtained from ATP and CTB assays were fitted from 0 to 100 by using non-linear curve fitting/growth/sigmoidal/dose–response fitting functions (OriginPro 8.6.0 b70, OriginLab Corp. USA). Half-maximal inhibitory concentrations (IC50) were calculated from the fitted dose–response curves. The shown data are from at least two independent experiments, and all individual experiments were conducted with four replicates (n = 4). Levene's test was performed first to assess the homogeneity of variance of replicates at the level of 0.01 (α = 0.01). Then, one-way analysis of variance (ANOVA) and Tukey tests for the comparisons of mean values of independent groups were performed at the level of 0.01 (OriginPro 8.6.0 b70). A significant effect was reported at * p < α(0.01).Microscopy studies
Bright field images of spheroids were taken prior to viability assays with inverse microscope (Olympus IX50, Olympus Corp., Japan) using cellSens software (cellSens standard 1.7.1, Olympus). The mean diameter of spheroids was measured using areas of spheroids, assuming they are all spherical, with ImageJ 1.49b software. Time-lapse bright field microscope images of spheroids were captured using 4× objective with LumaScope 600 microscope (Etaluma Inc., USA). For fluorescence imaging experiments, a fluorescence microscope (Olympus Ix50) equipped with an Olympus camera (SC30, Japan) was used to capture the images of spheroids with cellSens Standard (Olympus Co. Japan) software (Excitation filter: BP530-550, barrier filter: BA590). Confocal microscope images were taken using Zeiss LSM-510 Meta (Zeiss, Germany).Results and discussion
Morphological investigations
Initially, single spheroids in the middle of each well were produced as liquid overlay spheroid cultures in commercially available low-attachment 96 well-plates. Spheroid growth, size and morphology were monitored by means of bright-field and time-lapse microscopy. Spheroid sizes between 100–500 μm have been commonly accepted to be representative of healthy spheroid structures owing to sufficient oxygen and nutrition transport.22–24 Alternatively, small spheroids (<100 μm) might fail to display the complexity of real tissue with little growth rates, whereas large spheroids (>500 μm) might have pronounced necrotic cores due to diffusion limitations for oxygen and nutrition.22 For that reason, we produced intermediate sized spheroids with a mean diameter of 312 μm ± 20 (CV = 6%, n = 30) to ensure optimum growth and diffusion rates. The low variation in spheroid size proved the reproducibility of spheroid formation by using low-attachment 96 well-plates which is one of the most important criteria in obtaining reproducible data.Following this, we investigated the morphological changes of hAD-MSCs spheroids when they were treated with different doses of Qdots, thereby providing initial data on any adverse effects. For this, two different treatment models were used according to Qdot exposure time. In the first model, cells were exposed to Qdots 24 h after formation of the spheroids (tQDOT = 24 h) in order to investigate the effect of Qdots to spheroids. In the second model, cells were treated with Qdots at the beginning of spheroid formation (tQDOT = 0 h) to observe the Qdots effect on the spheroid formation. Firstly, different Qdots concentrations were administered to hAD-MSCs spheroids for 24 h after cells were incubated for 24 h for spheroidal formation (tQDOT = 24 h). Microscope images of spheroids revealed that Qdots induced significant effects to the morphology of hAD-MSCs spheroids with a concentration-dependent manner (Fig. 1). Low Qdot concentrations (<300 μg ml−1) did not result in any observable alterations to spheroids whereas higher Qdot doses caused a rough surface formation due to protruding of granular shaped cells. As the Qdot concentration was increased to 1200 μg ml−1, an observable increase in the surface roughness was pronounced and formation of round cells became obvious, corresponding to a significant increase in cell death. The round cells, however, did not detach from the spheroid, preserving the overall spherical structure, but they increased the diameter of the spheroid by 48%. hAD-MSCs form very tightly packed spheroids due to tight cell junctions preventing cells from losing their interactions and allowing them to remain in the spheroid.18 Time-lapse microscope images of hAD-MSCs spheroid treated with 1200 μg ml−1 Qdots are given in Fig. S1 (see also ESI, Video S1†). After 3 h of Qdot exposure, cells started to slowly morph into granular shapes and to swell outward from the spheroid forming a rugged surface, resulting in a concomitant increase in spheroid diameter. Interestingly, the increase in spheroid diameter showed a linear relationship for exposure times in the range of 2 h–8 h (Adj. R2 = 0.977). After 8 h of exposure, the increase in diameter slowed down and reached a plateau until 20 h (see ESI, Fig. S2†).
 | ||
| Fig. 1 Bright-field microscope images of hAD-MSCs spheroids treated with different Qdot concentrations for 24 h. The Qdot solutions were introduced to the spheroids after cells were incubated for 24 h for spheroid formation (tQDOT = 24 h). Scale bar is 200 μm. | ||
Stem cell spheroids with compact structures might create stronger diffusion barrier for nanoparticle transport, thus mitigate nanoparticle interactions. Despite the fact that NPs in vivo face very similar and potentially severe diffusion limitations, we also aimed to explore the effect of Qdots on spheroid formation by seeding the hAD-MSCs directly with Qdots (tQDOT = 0 h), eliminating diffusion related issues, and ensuring that all cells made contact with the Qdots. Following that, the morphologies of spheroids were imaged after 24 h of incubation (Fig. 2). Herein, the Qdots induced the cells to undergo more significant morphological alterations in comparison to previous model (Fig. 1). At concentrations of 300 μg ml−1 and lower, the Qdots didn't result in any observable effect, whereas higher amounts elicited distinct changes in the cellular aggregation behaviour of hAD-MSCs. Cell–cell/cell–ECM interactions were weakened significantly resulting in the formation of several smaller spheroids instead of one large spheroid. When cells were exposed to 1200 μg ml−1 of Qdot, the effect became more significant and cells lost their ability to form aggregates completely. Furthermore, in respect to their granular structure, it can be assumed that they also lost their viability. Consequently, we can confirm that high Qdot doses impaired cell–cell/cell–ECM interactions, thus preventing cells to form spheroids. This drastic phenomenon wasn't observed in the previous 3D model (tQDOT = 24 h). We suggest that, in the later 3D model (tQDOT = 0 h), the cells were not protected by well-developed ECM when they were subjected to toxic Qdot doses. Due to the absence of ECM, their contact with neighboring cells was reduced to a minimum level. Time-lapse microscope images of hAD-MSCs, which were treated with 600 μg ml−1 Qdots before spheroid formation (tQDOT = 0 h) are presented in Fig. 3 (see also ESI, Video S2†). After 6 h of incubation in the presence of high Qdots concentration, hAD-MSCs started to feature distinct cell aggregation behaviour. Afterwards uneven sized spheroids formation was observed. One explanation for this effect may be the nonspecific interaction of Qdots with transmembrane integrin proteins which may impair the receptor–ligand interactions for cell adhesion. Integrins regulate cell–cell/cell–ECM interactions and therefore play critical roles in cell proliferation, migration, differentiation, adhesion and apoptosis.25 They were also identified on MSCs cell surfaces and various studies showed their importance for the attachment and survival of MSCs.26 Shinto et al. proposed a nonspecific carboxyl (COOH)–integrin interaction mechanism which may cause blocking of actual integrin–ligand interactions.27 Since Qdots used in this study utilized surface COOH groups for their aqueous stabilization, such nonspecific COOH/integrin interactions might have occurred. Additionally, unmodified Qdots tend to bind non-specifically to cell membranes in order to reduce their high surface energy.28 These two phenomena both separately or in combination can result in nonspecific binding of Qdots to cell surface integrin receptors, and in turn might impair cell–cell/cell–ECM interactions. Furthermore, it was found that when the integrin–ligand binding was blocked, the cells rounded up and elevated expression of matrix metalloproteinases (MMPs) which are responsible for partial degradation of surrounding ECM in order to facilitate cell motility.29,30 Fujita et al. supported these finding by discovering titanium dioxide (TiO2) nanoparticles induced overexpression of MMPs in 2D cell culture of HaCaT cells.31 Recently, Liu and co-workers have also found out that gold nanoparticles after being internalized by lung cancer cells modulated cell invasion by causing overexpression of MMPs.32 Thereby, we can presume that high Qdot doses might exhibit similar inflammatory effects which can be later associated with diminishing cell–cell/cell–ECM interactions in hAD-MSCs. These hypotheses, however, need further examinations.
 | ||
| Fig. 2 Effect of Qdot nanoparticles to the formation of hAD-MSCs spheroids. hAD-MSCs were exposed to different Qdot concentrations at the beginning of spheroid formation (tQDOT = 0 h). Bright-field microscope images were taken after 24 h of Qdot treatment. Scale bar is 200 μm. | ||
 | ||
| Fig. 3 Time-lapse bright-field microscope images of hAD-MSCs exposed to 600 μg ml−1 Qdots solution at the beginning of spheroid formation (tQDOT = 0 h). The effect of Qdots to the spheroid formation was monitored for 24 h. | ||
Cell viability studies
Following the morphology investigations, cell viability assays were performed in order to give more detailed insight into Qdots-based cellular toxicity with respect to hAD-MSCs metabolic activities. Nanoparticles featuring broad absorption spectra such as Qdots, carbon nanotubes and silver nanoparticles might restrict the application of various conventional viability assays based on colorimetric measurements, such as acid phosphatase (APH) and methyl tetrazolium (MTT) assays,33–35 although they were successfully utilized for determination of cell viability in untreated 3D cell cultures.18,35,36 Moreover, high Qdot concentrations can interfere with the signals, thus causing significant false positive results as well as distorted data in viability results.37 For that reason, we performed adenosine triphosphate (ATP) luminescence cell viability assay and CellTiter-Blue (CTB) fluorescence viability assay as an alternative. The ATP assay is based on the enzymatic activity of luciferase enzyme to produce luminescent oxyluciferin in the presence of ATP, molecular oxygen and Mg2+. The amount of luminescence signal produced from the enzymatic reaction is proportional to the ATP content present in viable cells. ATP assays also require cell lysis for the release of ATP via disrupting the cell membrane. Alternatively, the CTB viability assay is based on intracellular reduction of resazurin by metabolically active cells which yield a highly fluorescent resorufin compound. The disruption of cells is not particularly necessary due to the diffusion of reduced resazurin outside of the cells. Therefore, cells maintain their activity during the assay for given incubation periods. Both assays were tested for their cross-reactivity with highest Qdots dose (1200 μg ml−1) in cell culture medium in absence of cells and there wasn't any signals detected higher than the background signals.Concentration-dependent cell viability results including dose–response fit curves obtained from ATP assay are shown in Fig. 4. Monolayer 2D cell culture exhibited distinct dose–response pattern featuring biphasic dose–response (non-monotonic) unlike 3D spheroid cultures which exhibited monophasic (monotonic) dose–response pattern. Biphasic behaviour of hAD-MSCs in 2D culture might be explained by presence of two distinct cell populations with different sensitivity to Qdots. Initially, there occurred a sharp decrease in viability to ∼50% at concentrations approaching to 20 μg ml−1. Beyond this concentration point, the viability of cells was decreased slightly when the dose was increased to 150 μg ml−1. This was followed by a moderate decrease at the highest Qdot concentration (1200 μg ml−1) at which all cells were dead. Meanwhile, 3D spheroid cultures demonstrated higher resistance to Qdot induced cellular toxicity than 2D culture with a mean IC50 value of 190 μg ml−1 for 3D (tQDOT = 24 h) and 187 μg ml−1 for 3D (tQDOT = 0 h). Accordingly, the exposure of cells to Qdots didn't result in pronounced adverse alterations of the viability of the 3D (tQDOT = 0 h) model despite cells being exposed to Qdots at the beginning of spheroid formation. Consequently, cells sustained their viability at the similar levels in both 3D models. The dose-dependent viability responses acquired from CTB assay for 3D cultures demonstrated an increase in viability at low Qdot doses in comparison to control samples (see ESI Fig. S3†). One may attribute this result to hormesis, which is a low-dose stimulatory or inhibitory effect in dose–responses above or below the toxicological thresholds.38 NP-induced hormesis was attributed to increasing ROS levels with low-doses, giving rise to cell activities to suppress the imbalance, thus causing an increase in metabolic activities.39 So far, various studies reported for several nanoparticles (CdTe, TiO2, aluminium NPs, single-walled carbon nanotubes) inducing hormesis.40,41 Though, CTB assay for 2D culture and ATP assay for 3D cultures didn't demonstrate similar patterns. Hence, we can neglect the possibility of hormesis. Meanwhile, O'Brien et al. described that cell lines with high metabolic rates can reduce resazurin further to colourless and nonfluorescent hydroresorufin.42 In such a case, untreated cells with high metabolic activity can further reduce resazurin to nonfluorescent hydroresorufin resulting in low fluorescent signals at the end-point in comparison to treated cells.42–44 This can lead to unsubstantial results causing misinterpretation of data and underestimation of cell activity. Moreover, in this study, the resazurin incubation time was prolonged to 48 h because of the slow diffusion of reduced resazurin from highly compact structured hAD-MSCs spheroids. Fluorescence signals only reached detectable levels after 48 h. For that reason, we can presume that the prolonged incubation time for resazurin (48 h) may have enabled over-reduction of resazurin by untreated cells followed by a decrease in their fluorescence signals. In the meantime, cells treated with Qdots sustained their reduction capacity and respectively produced higher signals than control samples. We should also note that, resazurin dye, also known as Alamar Blue, was proved to be toxic to the cells for long incubation periods (>24 h).42,45 Finally, signal qualities of ATP and CTB assays were compared by determining signal-to-noise (S/N) ratios for all culture types. For 2D cultures, S/N ratios were calculated to be 172 ± 19 for ATP assay and 11 ± 4 for CTB assay. For 3D cultures, S/N ratios were estimated to be 27 ± 4 for ATP assay and 4 ± 1 for CTB assay. Consequently, ATP assay signal qualities were found to be 16-fold greater in 2D culture and 7-fold in 3D culture than CTB assay. We therefore concluded that the CTB assay is not a reliable method for the determination of Qdot toxicity in the case of highly active and densely-packed hAD-MSCs spheroids. However, other cell lines particularly those forming loose cell aggregates and have relatively low metabolic activities, such as A549 lung carcinoma, HepG2 hepatocellular carcinoma and medulloblastoma tumour cells, may be suitable for CTB assay.22,46
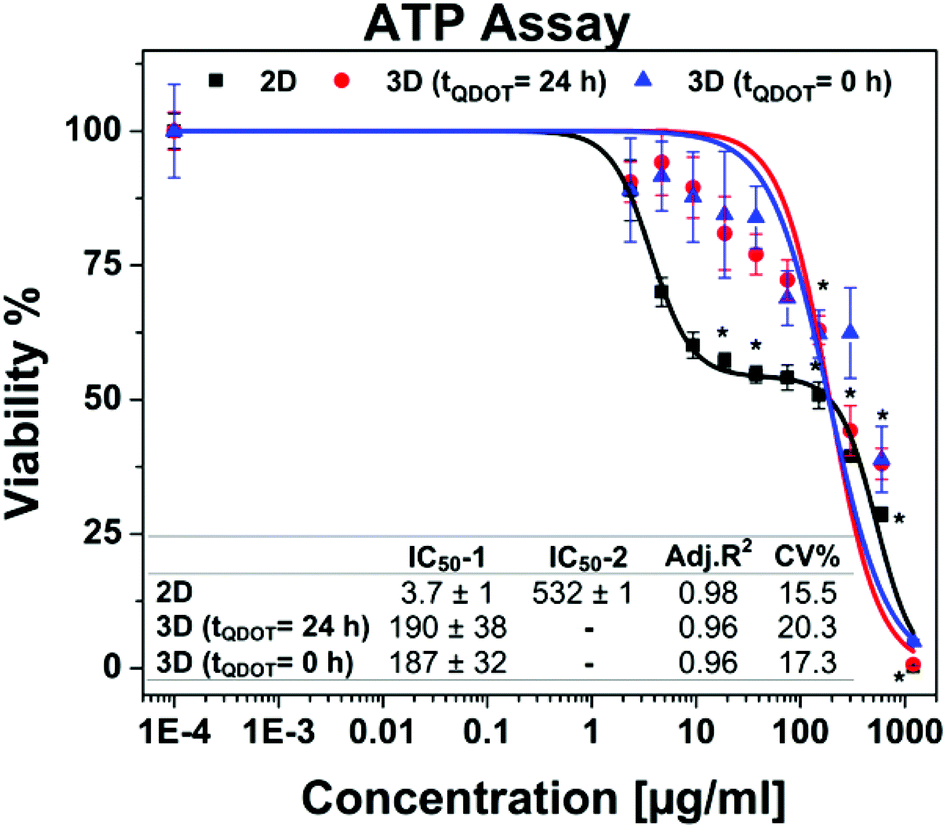 | ||
| Fig. 4 Cell viability data obtained from ATP assay. Data points are means ± standard error of 3 ± 1 independent experiments which each were conducted in 4 replicates. * indicates significant difference at 0.01 level. p < α = 0.01 (ANOVA one-way). Inset table shows half-maximal inhibitory concentrations (IC50) in μg ml−1. 2D cell culture having a biphasic dose–response curve possesses two IC50 values (IC50-1 and IC50-2) whereas each 3D spheroid cultures have one IC50 value owing to monotonic dose–response pattern. Acceptance criteria is CV (coefficient of variance) < 20%. | ||
Similar to our findings, conventional monolayer cell cultures being more sensitive against NPs or drug molecules in comparison to various 3D cell culture systems have been also reported by other groups.5,47–50 According to the Lee and co-workers report, where they assessed single-dose toxicity of CdTe Qdots on HepG2 spheroid cultures, 10 μg ml−1 of CdTe reduced the cell viability to 56% in 2D culture and 31% in 3D culture after 12 h of Qdot exposure.18 The results of the study cannot be directly compared to our findings since different cell types as well as different Qdot compositions were tested. Nevertheless, we can state that the same dose (10 μg ml−1) for core/shell/shell CdTe/CdS/ZnS Qdots did not cause any damage to hAD-MSCs spheroids. This may be attributed to the presence of protective outer shell layers acting as a strong barrier against Cd2+ leakage.19
Regarding in vivo Qdot-induced toxicity analysis; Hauck et al. reported that CdSe/ZnS core/shell Qdot did not show any significant toxicity in healthy Sprague–Dawley rats.51 Later, Chou et al. confirmed that Cd, Se and Zn containing Qdot did not cause toxicity in rats in 90 days.52 Another study reported by Nurunnabi et al. pointing out that carboxylated graphene Qdots did not induce apparent toxicity in rats after 22 days of exposure.53 The discrepancy in results in the context of Qdot toxicity between monolayer cell cultures and animal models was attributed to the presence of complex physiological processes in the body that NPs inevitable undergo after their uptake. Their circulation in blood stream, accumulation by specific organs and renal excretion play a deterministic role for their fate in the body.35 Yet, structural diversities in Qdot properties, such as size, shape, charge, material composition, and surface chemistry etc., necessitate the investigation of every Qdot structure-based effects individually. We also note that petri-dish cultures lack all of these physiological processes, being isolated on plastic substrates and therefore all cells facilitate NP interactions easily without facing any transport limitations. On the other hand, complex and diverse in vivo uptake mechanisms reduce the interaction of Qdots with the target organ and therefore they cooperatively act to minimize the adverse effects of Qdots. Nevertheless, developments for in vitro 3D cultures fortify their potential to fill the gap between petri-dish cultures and animal models with the hope that they will also decrease the use of laboratory animals.
Labeling spheroids with Qdots
Carboxyl-functionalized Qdot were tested for their applicability in 3D spheroid imaging. For that, hAD-MSC spheroids treated with Qdots were monitored with fluorescence and confocal microscopes, and the obtained images are presented in Fig. 5. Fluorescence microscope images with a standard (x, y) view illustrated surface fluorescence of labelled spheroid (Fig. 5a). On the other hand, z-stacked confocal images provided a better insight to the penetration and the uptake of Qdots in spheroids.54 According to the fluorescence confocal images, after 4 h Qdots appeared to be uptaken mostly by peripheral cells in the spheroid structure. As it can be seen from Fig. 5b, strong red fluorescence signals appeared only at the outer region, while signals decreased towards the inner region of the spheroid. In Fig. 5c, z-stacked images taken at individual z-planes at 3 μm step size from bottom surface towards centre of spheroid showed that highest Qdot labelling occurred at peripheral regions. Z-stack mean intensity plot (Fig. 5d-right) reveals that fluorescence intensity of Qdots decreases by 50% within 36 μm focal distance. These observations might suggest presence of diffusion limitations for the transport of Qdots into spheroid inner region.55 Fluorescence images taken from the bottom edge of the spheroid (Fig. 5d) show that Qdots were internalized by the cells rather than being non-specifically captured by extracellular matrix material. Live cell staining with Calcein-AM (Fig. 5d) shows that cells at the bottom surface of the spheroid retained their viability during Qdot treatment; thus, uptake of Qdots did not induce cell death. As a result, within the given incubation time (4 h) Qdots enabled staining into intact stem cell spheroids due to their inherent small size and surface structure. We can conclude that penetration of Qdots into tissue-like intact spheroid structures can make them a suitable tool for targeted imaging and drug delivery studies. | ||
| Fig. 5 Fluorescence imaging of hAD-MSCs spheroids treated with CdTe/CdS/ZnS Qdots for 4 h. (A) Fluorescence images captured from fluorescence microscopy from left-to-right: bright-field, Qdot (red), Calcein-AM (green) and DAPI (blue). (B) Single plane confocal images collected with 10× objective, from left-to-right: bright-field, DAPI (blue), Qdot (red), merge image of Qdot + DAPI and magnified spheroid. (C) Z-stacked confocal images of spheroid at individual z-planes. Z-stacks were acquired with 3 μm step size. The inserted graphic shows z-stack plot profile for normalized Qdot fluorescence mean intensity versus z-stack height (μm). (D) Single-plane confocal fluorescence images of spheroid at the bottom surface, from left-to-right: Qdot (red), Calcein-AM (green), DAPI (blue), merged image of Qdot + Calcein-AM + DAPI, and merge image of bright-field + Qdot + Calcein-AM + DAPI. | ||
Conclusions
In summary, we described for the first time the dose-dependent adverse effects of CdTe/CdS/ZnS core(small)/shell(thick)/shell(small) Qdots on the 3D spheroid culture of primary human mesenchymal stem cells. Morphological alterations in spheroid models (tQDOT = 0 h, tQDOT = 24 h) caused by toxic levels of Qdots revealed distinct characteristics. Either end-point microscope images or time-lapse studies demonstrated that Qdots expressed more drastic effect on spheroid formation, where cells were incubated in presence of Qdots during cell aggregation. Additionally, we detected a correlation in the spheroid diameters with increasing Qdot concentrations when hAD-MSCs were exposed to Qdots after spheroid formation (tQDOT = 24 h). Further, ATP and CTB cell viability tests were assayed to address the impact of dose-dependent Qdot toxicity to cell metabolic activities. According to ATP assays, significant differences were observed between 2D and 3D cell cultures. Interestingly, dose–response curves for 2D culture featured a reproducible biphasic curve model in contrast to spheroid cultures with monotonic dose–response patterns. As a result, monolayer cell cultures, on one side attaching to plastic substrate and on the other side being exposed to medium and having only periphery cell contacts, were found to be more susceptible to Qdot toxicity. Alternatively, in 3D spheroids, fully formed cell–cell and cell–ECM interactions in all three dimensions played a protective role and resulted in greater resistance to Qdot-induced toxicity. Meanwhile, CTB assays were proved to be an unreliable viability assay for densely packed stem cell spheroids due to diffusion limitations for resazurin/resorufin and therefore produced aberrant results. Overall, well-defined 3D spheroid cultures with the presence of microtissue-like cell clusters and ECM layer as well as mass transport characteristics constitute an initial testing platform to address physiologically relevant toxicity issues of nanoparticles for automated cell based high-throughput screening methods. Lastly, penetration ability of highly-fluorescent CdTe/CdS/ZnS Qdots demonstrates versatility of Qdots in 3D spheroid imaging. Their optical and structural features can be utilized in the future for targeted imaging and delivery studies using solid tumor spheroids.Acknowledgements
This work was supported by BioFabrication for NIFE (initiative, which is financially supported by the Lower Saxony ministry of Science and Culture and the Volkswagen Foundation). We also would like to thank Dr Patrick Lindner for his support for statistical analysis, Prof. Dr-Ing. Birgit Glasmacher and Dr Lutz Dreyer (Institute for Multiphase Processes, Leibniz University of Hanover) for providing us access to confocal microscope and for their additional support with the analysis.References
- F. Pampaloni, E. G. Reynaud and E. H. K. Stelzer, Nat. Rev. Mol. Cell Biol., 2007, 8, 839–845 CrossRef CAS PubMed.
- M. M. Stevens, Nat. Nanotechnol., 2009, 4, 342–343 CrossRef CAS PubMed.
- E. L. da Rocha, L. M. Porto and C. R. Rambo, Mater. Sci. Eng., C, 2014, 34, 270–279 CrossRef CAS PubMed.
- L. G. Griffith and M. A. Swartz, Nat. Rev. Mol. Cell Biol., 2006, 7, 211–224 CrossRef CAS PubMed.
- E. Kim, W. B. Jeon, S. Kim and S. K. Lee, J. Nanosci. Nanotechnol., 2014, 14, 3356–3365 CrossRef CAS PubMed.
- E. Fennema, N. Rivron, J. Rouwkema, C. van Blitterswijk and J. de Boer, Trends Biotechnol., 2013, 31, 108–115 CrossRef CAS PubMed.
- A. C. Luca, S. Mersch, R. Deenen, S. Schmidt, I. Messner, K. L. Schafer, S. E. Baldus, W. Huckenbeck, R. P. Piekorz, W. T. Knoefel, A. Krieg and N. H. Stoecklein, PLoS One, 2013, 8, e59689 CAS.
- N. T. Elliott and F. Yuan, J. Pharm. Sci., 2011, 100, 59–74 CrossRef CAS PubMed.
- A. Ivascu and M. Kubbies, J. Biomol. Screening, 2006, 11, 922–932 CrossRef CAS PubMed.
- X. L. Zhang, W. Wang, W. T. Yu, Y. B. Xie, X. H. Zhang, Y. Zhang and X. J. Ma, Biotechnol. Prog., 2005, 21, 1289–1296 CrossRef CAS PubMed.
- T. T. Goodman, C. P. Ng and S. H. Pun, Bioconjugate Chem., 2008, 19, 1951–1959 CrossRef CAS PubMed.
- Z. H. Li and Z. F. Cui, Biotechnol. Adv., 2014, 32, 243–254 CrossRef CAS PubMed.
- M. Akin, R. Bongartz, J. G. Walter, D. O. Demirkol, F. Stahl, S. Timur and T. Scheper, J. Mater. Chem., 2012, 22, 11529–11536 RSC.
- R. Tang, J. Xue, B. Xu, D. Shen, G. P. Sudlow and S. Achilefu, ACS Nano, 2015, 9, 220–230 CrossRef CAS PubMed.
- C. E. Probst, P. Zrazhevskiy, V. Bagalkot and X. Gao, Adv. Drug Delivery Rev., 2013, 65, 703–718 CrossRef CAS PubMed.
- D. Ag, R. Bongartz, L. E. Dogan, M. Seleci, J. G. Walter, D. O. Demirkol, F. Stahl, S. Ozcelik, S. Timur and T. Scheper, Colloids Surf., B, 2014, 114, 96–103 CrossRef CAS PubMed.
- M. Bottrill and M. Green, Chem. Commun., 2011, 47, 7039–7050 RSC.
- J. Lee, G. D. Lilly, R. C. Doty, P. Podsiadlo and N. A. Kotov, Small, 2009, 5, 1213–1221 CAS.
- M. Ulusoy, J. G. Walter, A. Lavrentieva, I. Kretschmer, L. Sandiford, A. Le Marois, R. Bongartz, P. Aliuos, K. Suhling, F. Stahl, M. Green and T. Scheper, RSC Adv., 2015, 5, 7485–7494 RSC.
- A. L. Rogach and M. Ogris, Curr. Opin. Mol. Ther., 2010, 12, 331–339 CAS.
- P. Chang, Y. Qu, Y. Liu, S. Cui, D. Zhu, H. Wang and X. Jin, Cell Death Dis., 2013, 4, e99175 Search PubMed.
- D. P. Ivanov, T. L. Parker, D. A. Walker, C. Alexander, M. B. Ashford, P. R. Gellert and M. C. Garnett, PLoS One, 2014, 9 Search PubMed.
- A. Asthana and W. S. Kisaalita, Drug Discovery Today, 2012, 17, 810–817 CrossRef PubMed.
- Y. Gao, M. G. Li, B. Chen, Z. C. Shen, P. Guo, M. GuillaumeWientjes and J. L. S. Au, AAPS J., 2013, 15, 816–831 CrossRef CAS PubMed.
- O. Lieleg, M. Lopez-Garcia, C. Semmrich, J. Auernheimer, H. Kessler and A. R. Bausch, Small, 2007, 3, 1560–1565 CrossRef CAS PubMed.
- D. Docheva, C. Popov, W. Mutschler and M. Schieker, J. Cell. Mol. Med., 2007, 11, 21–38 CrossRef CAS PubMed.
- H. Shinto, Y. Aso, T. Fukasawa and K. Higashitani, Colloids Surf., B, 2012, 91, 114–121 CrossRef CAS PubMed.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Aberg, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS PubMed.
- C. Brakebusch, D. Bouvard, F. Stanchi, T. Saki and R. Fassler, J. Clin. Invest., 2002, 109, 999–1006 CrossRef CAS PubMed.
- L. Heckmann, J. Fiedler, T. Mattes and R. E. Brenner, Cells Tissues Organs, 2006, 182, 143–154 CrossRef CAS PubMed.
- K. Fujita, M. Horie, H. Kato, S. Endoh, M. Suzuki, A. Nakamura, A. Miyauchi, K. Yamamoto, S. Kinugasa, K. Nishio, Y. Yoshida, H. Iwahashi and J. Nakanishi, Toxicol. Lett., 2009, 191, 109–117 CrossRef CAS PubMed.
- Z. X. Liu, Y. C. Wu, Z. R. Guo, Y. Liu, Y. J. Shen, P. Zhou and X. Lu, PLoS One, 2014, 9, e99175 Search PubMed.
- P. V. AshaRani, G. L. K. Mun, M. P. Hande and S. Valiyaveettil, ACS Nano, 2009, 3, 279–290 CrossRef CAS PubMed.
- J. M. Worle-Knirsch, K. Pulskamp and H. F. Krug, Nano Lett., 2006, 6, 1261–1268 CrossRef CAS PubMed.
- K. M. Tsoi, Q. Dai, B. A. Alman and W. C. W. Chan, Acc. Chem. Res., 2013, 46, 662–671 CrossRef CAS PubMed.
- J. Friedrich, W. Eder, J. Castaneda, M. Doss, E. Huber, R. Ebner and L. A. Kunz-Schughart, J. Biomol. Screening, 2007, 12, 925–937 CrossRef CAS PubMed.
- N. A. Monteiro-Riviere, A. O. Inman and L. W. Zhang, Toxicol. Appl. Pharmacol., 2009, 234, 222–235 CrossRef CAS PubMed.
- M. A. Nascarella and E. J. Calabrese, Dose-Response, 2012, 10, 344–354 CrossRef CAS PubMed.
- Y. Y. Guo, J. Zhang, Y. F. Zheng, J. Yang and X. Q. Zhu, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2011, 721, 184–191 CrossRef CAS PubMed.
- E. Jan, S. J. Byrne, M. Cuddihy, A. M. Davies, Y. Volkov, Y. K. Gun'ko and N. A. Kotov, ACS Nano, 2008, 2, 928–938 CrossRef CAS PubMed.
- I. Iavicoli, E. J. Calabrese and M. A. Nascarella, Dose-Response, 2010, 8, 501–517 CrossRef CAS PubMed.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Eur. J. Biochem., 2000, 267, 5421–5426 CrossRef.
- A. Romoser, D. Ritter, R. Majitha, K. E. Meissner, M. McShane and C. M. Sayes, PLoS One, 2011, 6, e22079 CAS.
- D. Breznan, D. Das, C. MacKinnon-Roy, B. Simard, P. Kumarathasan and R. Vincent, Toxicol. in Vitro, 2015, 29, 142–147 CrossRef CAS PubMed.
- H. Gloeckner, T. Jonuleit and H. D. Lemke, J. Immunol. Methods, 2001, 252, 131–138 CrossRef CAS PubMed.
- F. Sambale, A. Lavrentieva, F. Stahl, C. Blume, M. Stiesch, C. Kasper, D. Bahnemann and T. Scheper, J. Biotechnol., 2015, 205, 120–129 CrossRef CAS PubMed.
- D. R. Albrecht, G. H. Underhill, T. B. Wassermann, R. L. Sah and S. N. Bhatia, Nat. Methods, 2006, 3, 369–375 CrossRef CAS PubMed.
- M. Bokhari, R. J. Carnachan, N. R. Cameron and S. A. Przyborski, J. Anat, 2007, 211, 567–576 CAS.
- M. Oishi, Y. Nagasaki, N. Nishiyama, K. Itaka, M. Takagi, A. Shimamoto, Y. Furuichi and K. Kataoka, ChemMedChem, 2007, 2, 1290–1297 CrossRef CAS PubMed.
- D. Mueller, L. Kramer, E. Hoffmann, S. Klein and F. Noor, Toxicol. in Vitro, 2014, 28, 104–112 CrossRef CAS PubMed.
- T. S. Hauck, R. E. Anderson, H. C. Fischer, S. Newbigging and W. C. W. Chan, Small, 2010, 6, 138–144 CrossRef CAS PubMed.
- L. Y. T. Chou and W. C. W. Chan, Nat. Nanotechnol., 2012, 7, 416–417 CrossRef CAS PubMed.
- M. Nurunnabi, Z. Khatun, K. M. Huh, S. Y. Park, D. Y. Lee, K. J. Cho and Y. K. Lee, ACS Nano, 2013, 7, 6858–6867 CrossRef CAS PubMed.
- V. Charwat, K. Schutze, W. Holnthoner, A. Lavrentieva, R. Gangnus, P. Hofbauer, C. Hoffmann, B. Angres and C. Kasper, J. Biotechnol., 2015, 205, 70–81 CrossRef CAS PubMed.
- A. I. Astashkina, C. F. Jones, G. Thiagarajan, K. Kurtzeborn, H. Ghandehari, B. D. Brooks and D. W. Grainger, Biomaterials, 2014, 35, 6323–6331 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures and videos. See DOI: 10.1039/c5tx00236b |
| This journal is © The Royal Society of Chemistry 2016 |