DOI:
10.1039/C4TX00070F
(Paper)
Toxicol. Res., 2015,
4, 121-131
Kinetic and density functional theory (DFT) studies of in vitro reactions of acrylamide with the thiols: captopril, L-cysteine, and glutathione†
Received
8th July 2014
, Accepted 24th September 2014
First published on 25th September 2014
Abstract
The kinetics for the in vitro reactions of acrylamide (AA), a potentially toxic food contaminant formed during high-temperature food preparation, with captopril (CapSH), L-cysteine (CySH) and glutathione (GSH) were determined under basic conditions and constant ionic strength (pH 7.10–9.10; I = 0.2 mol dm−3, NaCl), and pseudo first-order conditions with respect to AA. The second-order rate constants, k, and activation parameters (ΔH‡, ΔS‡, and ΔG‡) were determined over the ranges of 293 ≤ θ ≤ 303 K for CySH and 303 ≤ θ ≤ 315 K for GSH and CapSH. Comparison of experimental second-order rate constants at 303 K for CapSH, CySH, and GSH were: 0.13 ± 0.01, 0.34 ± 0.02, and 0.18 ± 0.02 dm3 mol−1 s−1, respectively and DFT calculations show evidence of diminished intra-molecular hydrogen abstraction reaction in aqueous solution specific only to GSH. An isokinetic plot of ΔH‡versus ΔS‡ yields an isokinetic temperature of 260 ± 24 K and an intercept, ΔG‡, of 71 ± 4 kJ mol−1 indicating that AA reacts with all three thiols via a similar reaction mechanism. Theoretically determined ΔG‡ values in aqueous solution (DFT-BVP86/Ahlrichs-TZVP/COSMO-RS) for CapSH, CySH, and GSH, including their zero point vibrational energy, are 212.2, 217.8, and 253.4 kJ mol−1, respectively at 298 K.
Introduction
Acrylamide (AA), a known neurotoxin and potential human carcinogen1–4 was recently discovered in foods.5 Research is currently underway to quantify the levels of AA in various foods.6,7 Limited data is available on AA's interactions within biological systems and as such studies (in vivo and in vitro) are being conducted to assess the potential of AA toxicity and carcinogenicity.8 α,β-Unsaturated carbonyl compounds such as AA react readily with thiolate anions (RS−) by Michael addition and have been widely used to selectively modify thiol groups of structural and functional proteins.9 Friedman10 has reported on the use of AA to alkylate the thiol groups of various enzymes. However, the relationship of such reactions to AA's toxicity and potential carcinogenicity is unknown. Kinetically, the Michael additional reaction scheme of thiols with AA is shown below:| | 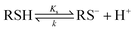 | (1) |
| | RS− + H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCONH2 + H2O → RS–CH2CH2CONH2 + OH− CHCONH2 + H2O → RS–CH2CH2CONH2 + OH− | (2) |
Tong et al.9 reported on the reactions of AA with glutathione (GSH) and human serum albumin (HSA), two of the most abundant thiols within the human body. The rate constants, with phosphate buffer, were found to be 0.021 dm3 mol−1 s−1 and 0.0054 dm3 mol−1 s−1, respectively under physiological conditions. The reaction was conducted under pseudo-first order conditions with AA in large excess. The amount of unreacted thiol was determined by withdrawing portions of the reaction mixture at various times and reacting with Ellman's reagent, 5,5′-dithiobis(2-nitrobenzoic acid), commonly symbolized as ESSE. The residual Ellman's thiolate anion (ES−) was monitored spectrophotometrically following the Ellman's scheme.11
L-Cysteine (CySH) and GSH are important thiols synthesised within the body. They form part of the body's natural defence mechanism12,13 by scavenging free radicals and helping to maintain the secondary structure of certain proteins.14,15 Captopril (CapSH) is also important as a vasodilator in the treatment of congestive heart failure where it protects NO from oxidation by scavenging free oxygen radicals.16–19
In this report, the reactions of AA with CapSH, CySH and GSH were investigated with a Tris/HCl buffer system by monitoring the rate of disappearance of ES− at 409 nm. We present a comprehensive study on the reaction rates of CapSH with AA, including theoretical investigation of the minimum energy pathway for the reaction by density functional theory (DFT). How AA reacts with these thiols, thus reducing or enhancing their ability to carry out their function within the body, is of great importance in understanding the mechanism and metabolism of AA.
Materials and methods
Chemicals
Acrylamide (electrophoresis grade), AR grade captopril, L-cysteine, glutathione, Ellman's reagent (ESSE), sodium chloride, Tris·HCl, potassium hydrogen phthalate, potassium dihydrogen phosphate, L-ascorbic acid (H2ASc), and HPLC grade methanol were purchased from Aldrich (Fairlawn, NJ). Deionised water was prepared in house using a Labconco Water Pro PS deionizer (Labconco Water Corporation, Kansas City, MO). All stock and working solutions were prepared in deionised water. HCl (Analar grade) was obtained from BDH Laboratories (Poole, England).
Spectrophotometric measurements
The acrylamide–thiolate (AA–SR) complexes were monitored spectrophotometrically on a HP 8453 Diode Array Spectrophotometer with 1 cm path length quartz cuvettes. The wavelengths monitored were 327 nm for ESSE and 409 nm for the Ellman's thiolate (ES−) anion. The diode array spectrophotometer was operated in the kinetic mode for determination of reaction parameters and in the standard mode for quantifying the rate constants associated with each AA–thiol reaction. Temperature was maintained by a Lauda-Brinkmann RM6 B Ciculator/Thermostated Water Bath (Lauda Dr R. Wobser, GMBH and Co., Lauda-Königshofen, Germany).
In the kinetic mode, all reactions were conducted at 310 K. In the standard mode with varied AA concentrations, the reactions were conducted at 310 K. For varied thiol concentrations, the reactions were conducted at 293, 298, 303 K for CySH and 303, 310, and 315 K for CapSH and GSH. Mixtures containing CapSH and GSH were measured at 1 minute intervals while CySH mixtures were measured at 30 s intervals. The pH of the final solutions were recorded using an Orion Research Expandable ion Analyzer EA 920 (Orion Research Inc., Boston, MA, USA) calibrated with buffers: 4.01 (0.0496 mol dm−3 K2HC8H4O4) and 6.87 (0.025 mol dm−3 KH2PO4).
LC/DAD/MS analysis
The AA–SR complexes were analyzed on an Agilent 1100 Series LC coupled to an Agilent 1100 Series MS Detector (Agilent, Technologies, Palo Alto, CA, USA). Separation of the complexes was achieved using a Zorbax Eclipse XDB–C18 column (150 mm × 4.6 mm ID × 5 μm particle size; Agilent Technologies, Palo Alto, CA, USA) with a Zorbax XDB–C18 guard column (12.5 mm × 4.6 mm i.d.) at a flow rate of 0.2 mL min−1; Operating parameters were: positive electrospray ionization (+ESI) in the SIM and scan modes; SIM ions: 308, 379, 380, 613; scan range: 300–400 amu (for the AA–SG complex); Mobile phase: methanol/water (5 + 95 v/v)%; run time 8.00 min; MS: Drying gas temperature: 350 °C; drying gas flow: 12 L min−1; nebulizer gas pressure: 35 psig; capillary voltage: 3000 V; Fragmentor voltage: 80 V (scan), 70 V (SIM); DAD: 195 ± 10 nm with a reference of 360 ± 100 nm.
Preparation of thiol solutions
Preliminary studies.
Stability of thiols.
Solutions of CapSH, CySH, and GSH, were all prepared at concentrations of 6.5 × 10−4 mol dm−3 in deionised water. 1.00 mL of each thiol solution was transferred to 10.00 mL volumetric flasks containing Tris/HCl buffer solutions (pH 7.4; ionic strength (I) = 0.2 mol dm−3 (NaCl)) or HCl (pH 4.5; I = 0.2 mol dm−3 (NaCl)) and made up to the mark with deionised water for a final thiol concentration of 6.5 × 10−5 mol dm−3.
A 1.0 × 10−4 mol dm−3 stock solution of ESSE was prepared in deionised water. 1.00 mL of each of the buffered thiol solutions and 1.00 mL of ESSE were mixed in 1 cm quartz cuvettes. The reactions were monitored spectrophotometrically in the kinetic mode at 310 K at a cycle time of 10 s for 9000 s.
Variation of acrylamide concentration.
1.00 mL of each thiol solution was transferred to 10.00 mL volumetric flasks containing Tris/HCl buffer solutions (pH 7.4; I = 0.2 mol dm−3 (NaCl)). A 1.0 mol dm−3 AA solution was prepared in deionised water and various amounts added to the buffer solutions for final AA concentrations ranging from 9.963 × 10−3 mol dm−3 to 9.963 × 10−2 mol dm−3i.e. pseudo-first order conditions with respect to AA. 1.00 mL of each of the thiol buffer solutions and 1.00 mL of 1.0 × 10−4 mol dm−3 ESSE were added to quartz cuvettes at various times to measure the rate of thiol decomposition.
Variation of thiol concentration.
Varying volumes of each thiol stock solution were added to 10.00 mL volumetric flasks containing Tris/HCl buffer solutions (pH 7.4; I = 0.2 mol dm−3 (NaCl)) to give final thiol concentrations within the range of 1.0 × 10−5 mol dm−3 to 1.0 × 10−4 mol dm−3. The AA concentration was kept constant for each thiol. The AA concentration used for CapSH was 4.986 × 10−2 mol dm−3 and 9.963 × 10−3 mol dm−3 for both CySH and GSH. 1.00 mL of each of the thiol solutions and 1.00 mL of ESSE solution were added to quartz cuvettes at 30 s and 1 min intervals as described in Spectrophotometric Measurements. The reactions were monitored in the standard mode at a temperature at 310 K and the pseudo-first order rate constants, kobs, for the thiol decomposition were calculated.
Variable temperature studies
Values of kobs, for each thiol were determined at three temperatures while varying the pH of the mixtures. The pH range used was 7.10–9.10 (Tris/HCl buffer). The ionic strength was kept constant at 0.2 mol dm−3 (NaCl). Thiol concentration was kept at 6.5 × 10−5 mol dm−3, and the AA concentration was also kept constant for each thiol.
The AA concentration used for CapSH, CySH, and GSH were: 4.986 × 10−2 mol dm−3, 9.963 × 10−3 mol dm−3, and 1.993 × 10−2 mol dm−3, respectively. GSH and CapSH solutions were monitored at 303, 310, and 315 K while CySH solutions were monitored at 293, 298, and 303 K.
Statistical analysis of data
The pseudo-first order rate constants, kobs, for the thiol decomposition were calculated from absorbance-time data using the StatGraphics Centurion XV.I Software (SGS). kobs values at each temperature were used to calculate the specific rate constants, k, and the pKs for each thiol. Activation parameters, ΔH‡, ΔS‡, ΔG‡ for each thiol were calculated from Eyring plots. Parameters generated from the Eyring plots were used to construct an isokinetic plot.
Computational studies
Gas phase (1 atm) geometries and energies were optimized using the BVP86 DFT functional20 and Ahlrichs’ TZVP basis set21 as available in the PQS programme.22 Transition state geometries were optimized until a single imaginary vibrational frequency corresponding to displacement along the intrinsic reaction coordinate was found. The energy differences between reactants, transition states and products were computed relative to that of the combined energy of the isolated reactants in each case. Energy partition function analysis was used to determine the thermal corrections to the energy of each molecule (enthalpic and entropic) at different temperatures from translational, rotational and vibrational modes. With water as solvent, energies and optimized geometries were computed using the COSMO23 electrostatic screening model and the output transferred to the ADF24 programme for COSMO-RS25 calculations at infinite dilution and computing the atomic charges with the Hirshfeld Atom In Molecule (AIM) method.26,27
Results and discussion
The rate of disappearance of the Ellman's thiolate (ES−) species at 409 nm is equivalent to the rate of reaction between AA and RS−. As time progressed, more RS− reacted with AA to form the AA–SR complex, so less RS− was available to react with ESSE (species at 327 nm) to generate ES−. Eventually all the RS− was consumed and no more ES− was formed.
Preliminary work in this study involved analysis of the reaction with AA and RSH under acidic conditions (pH 4.2 (HCl); I = 0.2 mol dm−3 (NaCl)), where no visible reaction was observed, supporting the reaction steps shown in eqn (1) and (2); RS− is the reactive species that reacts with AA. At 310 K, variation of AA and thiol concentrations gives a linear increase in rate at constant thiol concentration; i.e. the rate remained unchanged when the AA concentration is unchanged. This indicates that the reaction is first order with respect to AA. The pseudo-first order rate constant, kobs, for each reaction is calculated according to eqn (3);
| | | At = A∞ + (Ao − A∞)e−kt | (3) |
where:
Ao is the initial absorbance;
At is the absorbance at time
t;
k is the pseudo-first order rate constant.
Based on observations of increased kobs with increasing pH, (Table 1), and the proposed reaction mechanism depicted in eqn (1) and (2), the rate law (eqn (4)) was derived:
| | 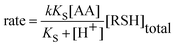 | (4) |
which can be simplified to
eqn (5):
| | | rate = kobs [RSH]total, | (5) |
where
kobs is given by:
| | 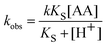 | (6) |
Table 1 Pseudo-first order rate constants for the reaction between AA and CapSH, CySH, and GSH at 303 K: pH dependence. [RSH] = 6.5 × 10−5 mol dm−3, I = 0.2 mol dm−3 (NaCl), λ = 409 nm. [H2Asc] = 1 mM for CySH
| CapSH |
CySH |
GSH |
| [AA] = 4.986 × 10−2 mol dm−3 |
[AA] = 9.963 × 10−3 mol dm−3 |
[AA] = 1.993 × 10−2 mol dm−3 |
| pH |
k
obs (10−3 s−1) |
pH |
k
obs (10−3 s−1) |
pH |
k
obs (10−3 s−1) |
| 7.07 |
0.123 |
7.33 |
1.44 |
7.33 |
0.329 |
| 7.33 |
0.182 |
7.42 |
2.13 |
7.57 |
0.394 |
| 7.78 |
0.373 |
7.80 |
2.40 |
7.89 |
0.672 |
| 7.91 |
0.463 |
7.96 |
2.72 |
8.14 |
0.964 |
| 8.13 |
0.693 |
8.19 |
3.06 |
8.50 |
1.73 |
| 8.48 |
1.32 |
8.55 |
3.11 |
8.78 |
2.29 |
| 9.11 |
3.46 |
9.09 |
3.25 |
9.09 |
3.58 |
Values for k and Ks are obtained from eqn (6) using non-linear regression analysis. The pKs for each thiol was determined from the analysis of the kinetic data. The activation parameters: ΔH‡ (enthalpy of activation) and ΔS‡ (entropy of activation), for the reaction between AA and the various thiols (RSH) were obtained from the values of the specific rate constants obtained at different temperatures using the Eyring equation:
| | 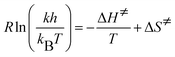 | (7) |
where
T is temperature in Kelvin,
kB is Boltzman's constant,
h is Planck's constant, and
R is the standard molar gas constant.
A plot of 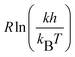 versus 1/T yields a slope of −ΔH‡ and an intercept of ΔS‡. The Gibbs free energy of activation, ΔG‡, (at 298 K) is calculated from eqn (8):
versus 1/T yields a slope of −ΔH‡ and an intercept of ΔS‡. The Gibbs free energy of activation, ΔG‡, (at 298 K) is calculated from eqn (8):
Reaction of acrylamide and glutathione
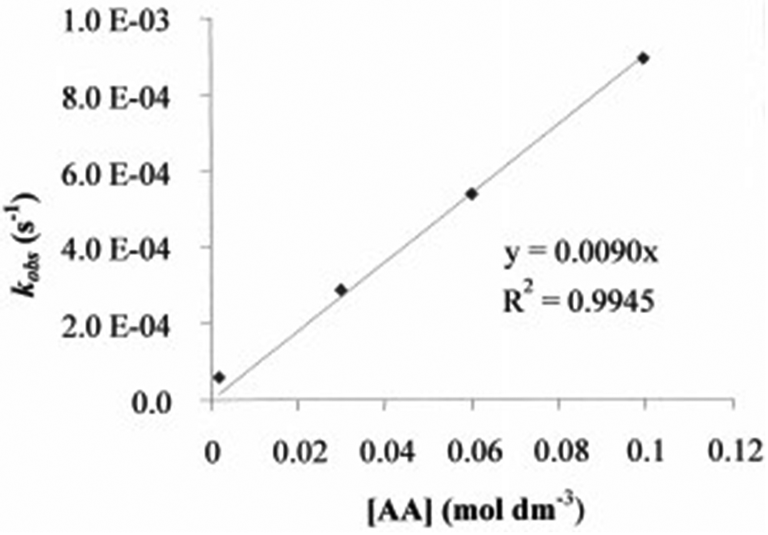
The repetitive-scan spectra and time trace (inset) for the reaction between AA and GSH is shown in Fig. 1. Data for the reaction between GSH and AA with varying [GSH] are shown in Table 2. The plot of kobsvs. [AA] shows a zero intercept (Fig. 2). Table 2 and Fig. 2 show that kobs remains unchanged when the [GSH] was varied and increased when the AA concentration increased, indicating that the reaction was pseudo-first order with respect to AA.
 |
| | Fig. 1 Repetitive-scan spectra of the reaction between AA and GSH (Inset: Absorbance-time trace showing the rate of disappearance of the ES− species). | |
 |
| | Fig. 2 Pseudo-first order rate constants for the reaction of the thiolate group of GSH with varying AA at 310 K; pH 7.4; [GSH] = 6.5 × 10−5 mol dm−3. | |
Table 2 Independence of kobs with varying [GSH] in excess AA at 310 K; pH 7.4; [AA] = 9.963 × 10−2 mol dm−3
| [GSH] (10−5 mol dm−3) |
k
obs (10−3 s−1) |
| 0.863 |
1.1 |
| 1.29 |
1.1 |
| 3.47 |
1.3 |
| 6.93 |
1.0 |
| Average and std. dev. |
1.1 ± 0.1 |
The calculated second order rate constant for the reaction of AA with GSH at 310 K is 0.25 ± 0.01 dm3 mol−1 s−1, which is 10 times faster than that reported by Tong et al.9 This could be explained by potential interaction of the phosphate buffer system that was used by Tong et al. Similar buffer interactions were absent in the Tris/HCl buffer reaction system. Hence Tris/HCl buffer was used instead of phosphate buffer system in this work. A zero intercept for the plot of kobsvs. [AA] (Fig. 2) implies that the reaction of AA with the thiol group of GSH far exceeds the rate of oxidation of GSH by oxygen in air to form the disulphide, GS–SG. This was confirmed by LC/DAD/MS with +ESI operated in the SIM and scan modes (Fig. 3). The chromatogram shows two peaks at 2.808 min and 3.170 min which correspond to GSH (unreacted) and the AA–SG adduct, respectively. The ion fragment, 308 is due to the cleavage of the AA–SG bond and subsequent reformation of GSH which picks up an additional hydrogen to become (GSH + H)+. The ion fragment, 379 confirms the formation of the AA–SG species. The absence of a peak and ion fragments corresponding to the disulphide, GS–SG support our findings that the Michael addition reaction between AA and GSH occurs much faster than the oxidation of GSH by oxygen in air which results in the formation of the disulphide, GS–SG.
 |
| | Fig. 3 LC/MS Chromatogram, SIM, and scan spectra of a solution containing AA and GSH AA–SG complex (3.170 min); unreacted GSH (2.808 min). A and B are the scan and SIM mass spectra, respectively of AA–SG. The highlighted ion represents the protonated molecular ion, [AA–SG + H]+. | |
Variation of pH and temperature
A plot of kobsvs. [H+] showing pH dependency for GSH at 310 K is shown in Fig. 4. The model (line) shows excellent correlation to the experimental data indicating that the model fits the proposed mechanism depicted in eqn (1) and (2). The second-order rate constant (k) and pKs are calculated from this plot.
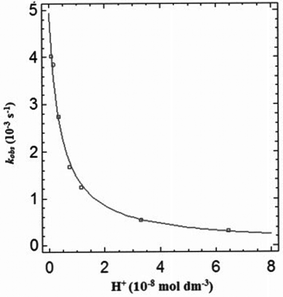 |
| | Fig. 4 Plot of kobsversus [H+] for GSH; Temperature 310 K; [AA] = 1.993 × 10−2 mol dm−3. | |
Reactions of L-cysteine
Reaction of L-cysteine and Ellman's reagent in Tris/HCl buffer.
The repetitive-scan spectra and time traces (inset) for the reaction between CySH and ESSE are shown in Fig. 5. The absorbance vs. wavelength plot shows a clearly defined isosbestic point at 360 nm. The time trace shows that maximum absorbance of ES− occurred after 30 seconds. This was maintained up to 600 seconds and gradually decreased thereafter; the reverse was true for the ESSE species. It was observed that the yellow solution (denoting the presence of ES−) became colourless with time. Like GSH and other thiols, CySH forms the thiolate anion, CyS− in basic media. The CyS− ion is very reactive. It was suspected that CyS− was being quickly oxidized in the presence of oxygen in air to form the disulphide CyS–SCy, cystine, and ES− was reverting to ESSE.
 |
| | Fig. 5 Repetitive-scan spectra of the reaction between ESSE and CySH (Inset: Absorbance-time trace showing the rates of appearance and disappearance of the ES− and ESSE species. (blue arrows: initial reaction; green arrows: final reaction). | |
Reaction of L-cysteine and acrylamide.
Like GSH, the reaction of CySH with a constant large excess of AA with varying [CySH] gives unchanged pseudo-first order rate constants, and increased linearly with an increase in [AA]T, i.e. the reaction is pseudo-first order with respect to AA. Unlike GSH however, the plot does not pass through the origin; suggesting the occurrence of a reaction that is independent of the reaction between CySH and AA (Fig. 6). A modified rate constant, kobs, accounts for the non-zero intercept in eqn (6) to give eqn (9):| | 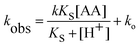 | (9) |
 |
| | Fig. 6 Pseudo-first order rate constants for the reaction of the thiol group of CySH with varying AA at 310 K; pH 7.4; [CySH] = 6.5 × 10−5 mol dm−3. | |
It is proposed that this independent reaction was the competitive oxidation of CySH by oxygen in air to form the disulphide, CyS–SCy, where ko is the calculated rate constant for the oxidation of CySH by oxygen in air at 310 K. This independent reaction was investigated by adding a ten times excess of the known anti-oxidant, ascorbic acid (H2Asc), to a reaction flask containing CySH. This inhibited the oxidation of CySH to the disulphide by reacting with the oxygen in air thus allowing CySH to react with AA.
This reaction mixture containing CySH and H2Asc was reacted with ESSE as described in Preliminary Studies. It was observed that the reaction was stabilized by the presence of H2Asc (evident by the yellow colour of the solution at the end of the reaction time of 9000 s, indicative of the presence of ES−). This indicated that H2Asc prevented the one electron oxidation of CySH as reported in a previous study conducted by Ohno et al.28 As a result, 1.0 × 10−3 mol dm−3 H2Asc was subsequently added to all CySH stock solutions prior to reaction with AA.
Eqn (10) shows the reduction of cystine (CyS–SCy), the disulphide, to two molecules of cysteine. The oxidation of the other thiols by oxygen is slow in comparison to their reaction with AA and as a result no H2Asc was added to those solutions.
| | | CyS–SCy + 2e− + 2H+ ⇌ 2CySH | (10) |
Summary of kinetic results
Calculated second order rate constants, pKs, and activation parameters for the reaction between AA with CySH, GSH, and CapSH are shown in Table 3.
Table 3 Summary of experimental results: calculated second-order rate constants (k), activation parameters (ΔH‡ ΔG‡ ΔS‡), and pKs for the reactions between AA and RSH
| Parameter |
CapSH |
CySH |
GSH |
|
Ref. 29.
Ref. 16.
|
|
k (dm3 mol−1 s−1) (293 K) |
— |
0.21 ± 0.01 |
— |
|
k (dm3 mol−1 s−1) (298 K) |
— |
0.27 ± 0.02 |
— |
|
k (dm3 mol−1 s−1) (303 K) |
0.13 ± 0.01 |
0.34 ± 0.02 |
0.18 ± 0.02 |
|
k (dm3 mol−1 s−1) (310 K) |
0.163 ± 0.003 |
— |
0.25 ± 0.01 |
|
k (dm3 mol−1 s−1) (315 K) |
0.19 ± 0.01 |
— |
0.31 ± 0.04 |
| Average pKs |
9.01 ± 0.05 |
7.4 ± 0.2 |
8.5 ± 0.2 |
| Literature pKs (298 K) |
9.8b |
8.15a |
8.56a |
| ΔH‡ (kJ mol−1) |
22 ± 7 |
34 ± 5 |
33 ± 5 |
| ΔS‡(J K−1 mol−1) |
−190 ± 2 |
−143 ± 2 |
−149 ± 2 |
| ΔG‡ (kJ mol−1) (298 K) |
78 ± 3 |
76 ± 1 |
78 ± 1 |
The pseudo-first order rate constants of the reaction between AA and the thiols: CapSH, CySH and GSH are greatly affected by pH in a manner that depends on the speciation of each thiol in solution. As pH increases, the thiol group becomes progressively more ionized, resulting in a higher concentration of the thiolate anion. The increase in kobs values with increasing pH, for all three thiols, indicates that the thiolate anion is more reactive than the thiol.
The smaller rate constant obtained for CapSH's reaction with AA (compared to CySH and GSH) may be due to differences in minimized geometry (vide infra), acidity, hydrogen bonding, and solvent interactions at any stage of the reaction. The reported pKs for CySH is 8.15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29 while the experimental value is 7.4 ± 0.2 which is at the lower range of the buffer system that was used. The pKs for GSH and CapSH are progressively larger (8.5629 and 9.816 respectively), while our experimentally calculated values are: 8.5 ± 0.2 and 9.01 ± 0.05 for GSH and CapSH, respectively. The pKs represents the pH at which there are equal amounts of RSH and RS−. This implies that at the buffer range used (7.10–9.10 for Tris/HCl), a larger amount of CyS− is present in solution in comparison to GS− or CapS−. This may explain why the reaction between AA and the three thiols has apparent order CySH > GSH > CapSH as indicated by kobs at 303 K (Table 1). Although pKs is temperature dependent, the measured values in our studies were unaffected by small variations in temperature. For all thiols, the experimentally derived entropies of activation (ΔS‡) were negative, as may be expected for an associative mechanism in the transition state of the Michael-type addition mechanism. The experimental ΔS‡ for the addition reactions was in the order: CapSH < GSH ∼ CySH which can be attributed to significant intramolecular H-bonding in GSH stabilising the AA–SG transition state. Given that values of k for GSH are smaller than for CySH, a small steric effect on the Michael addition reaction is also possible. Furthermore, the possibility of intra-molecular H-bonding would make the S− less nucleophilic in GSH than CySH but not as significant as CapSH. We have investigated these possibilities with ab initio methods as presented in the next section.
29 while the experimental value is 7.4 ± 0.2 which is at the lower range of the buffer system that was used. The pKs for GSH and CapSH are progressively larger (8.5629 and 9.816 respectively), while our experimentally calculated values are: 8.5 ± 0.2 and 9.01 ± 0.05 for GSH and CapSH, respectively. The pKs represents the pH at which there are equal amounts of RSH and RS−. This implies that at the buffer range used (7.10–9.10 for Tris/HCl), a larger amount of CyS− is present in solution in comparison to GS− or CapS−. This may explain why the reaction between AA and the three thiols has apparent order CySH > GSH > CapSH as indicated by kobs at 303 K (Table 1). Although pKs is temperature dependent, the measured values in our studies were unaffected by small variations in temperature. For all thiols, the experimentally derived entropies of activation (ΔS‡) were negative, as may be expected for an associative mechanism in the transition state of the Michael-type addition mechanism. The experimental ΔS‡ for the addition reactions was in the order: CapSH < GSH ∼ CySH which can be attributed to significant intramolecular H-bonding in GSH stabilising the AA–SG transition state. Given that values of k for GSH are smaller than for CySH, a small steric effect on the Michael addition reaction is also possible. Furthermore, the possibility of intra-molecular H-bonding would make the S− less nucleophilic in GSH than CySH but not as significant as CapSH. We have investigated these possibilities with ab initio methods as presented in the next section.
Calculated transition state geometries and reaction path energies
Fig. 7 shows the calculated lowest energy conformations in the transition states (TS) that occur for CapSH, CySH and GSH in the gas phase. Similar structures are obtained when the optimization includes the effect of solvent water molecules (COSMO), except for CySH, where O6–H13 (intra-molecular) and H13–O24 (inter-molecular) distances (Table 4; h, i respectively) exceed that of typical medium strength H-bonds.30 This likely arises from rotation of the terminal carboxyl group to maximize electrostatic interactions with the polar solvent. Otherwise, the number of significant O⋯HX contacts is unchanged by solvent, so that only water-phase predicted geometries (within the polarized continuum model) are discussed further below.
 |
| | Fig. 7 Perspective views of computed transition state structures for (a) CySH, (b) CapSH and (c) GSH reaction with acrylamide (non-essential hydrogens omitted). | |
Table 4 Measurements (Å/°) in calculated TS structures for CapSH, CySH and GSH
| Measure |
Label |
CapSH–AA |
CySH–AA |
GSH–AA |
| Gas |
Water |
Gas |
Water |
Gas |
Water |
| S(11)–C(9) |
a |
2.355 |
2.515 |
2.299 |
2.499 |
1.995 |
1.938 |
| S(11)–H(12) |
b |
1.650 |
1.730 |
1.616 |
1.728 |
1.490 |
1.495 |
| H(12)–C(7) |
c |
1.482 |
1.389 |
1.539 |
1.402 |
1.822 |
1.812 |
| C(9)–C(7) |
d |
1.427 |
1.417 |
1.433 |
1.417 |
1.468 |
1.486 |
| C(7)–C(5) |
e |
1.477 |
1.487 |
1.469 |
1.485 |
1.432 |
1.422 |
| C(5)–O(6) |
f |
1.240 |
1.248 |
1.248 |
1.251 |
1.281 |
1.286 |
| C(5)–N(1) |
g |
1.383 |
1.362 |
1.381 |
1.361 |
1.370 |
1.385 |
| O⋯HX |
h |
2.527 |
2.785 |
2.201 |
2.470 |
2.034 |
2.123 |
| i |
4.021 |
4.056 |
2.412 |
2.883 |
2.048 |
2.066 |
| j |
|
|
2.473 |
2.664 |
1.755 |
1.788 |
| k |
|
|
3.048 |
2.657 |
1.837 |
1.880 |
| l |
|
|
|
|
1.712 |
1.627 |
| ∠ (S12–C9–H10) |
m |
97.67 |
93.88 |
95.78 |
92.65 |
104.61 |
105.63 |
The gas phase energy barrier calculated for TS formation with AA and GSH (Fig. 8, left) is lowest for the thiols studied, consistent with the intra-molecular H-bond contacts that are present in GSH but absent from the others. However, the net relative stabilization of the TS is only ∼3.2 kcal mol−1 compared to the most energetic activation path shown by CapSH, suggesting that the positive entropy change which accompanies the formation of the TS is large enough to oppose the stabilizing enthalpic effect of the many intra-molecular H-bonds typical for many systems that have been studied in literature.30,31 The role of entropic contributions to the free energy changes is investigated in more detail below.
 |
| | Fig. 8 Theoretical minimum energy barriers for the reaction of CySH, CapSH and GSH with acrylamide in the gas phase and aqueous solution (DFT BVP86/TZVP/COSMO). | |
The solution phase energetics (Fig. 8, right) are obtained from the gas phase data using the Hess's analysis shown in Fig. 9 and summarized in eqn (11) and (12);
| | 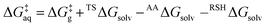 | (11) |
| | 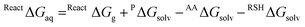 | (12) |
where AA = acrylamide, RSH = thiol, TS = transition state, P = product and Δ
G‡ is the activation free energy change in gas phase or aqueous medium.
 |
| | Fig. 9 Thermodynamic scheme for free energy changes in gas phase and solution. | |
Following gas phase ab-initio calculations, the free energy of solvation at infinite dilution, H-bond interaction energy, and the activity coefficient for each component in water were calculated with the COSMO-RS module of the ADF software. By assuming that the heat capacity of each molecule is constant over a small temperature range,32 the slope of the free energy of solvation with temperature affords an estimate of the enthalpic and entropic contributions to the total free energy change of solvation.33 We use a ±30 K margin at 6 K intervals around 298.15 and 303.15 K to determine the relevant entropic terms in each case. Gas phase free energies are also corrected by degrees of freedom component analysis at each specified temperature. Table 5 presents the combined thermodynamic results.
Table 5 DFT-computed thermodynamic parameters for the reaction of CapSH, CySH, and GSH with acrylamide at 298.15 and 303.15 K
| |
ΔG‡ (kcal mol−1) |
ΔH‡ (kcal mol−1) |
ΔS‡ (cal K −1 mol−1) |
ΔGReact (kcal mol−1) |
Equilibrium constant |
| Temp (K) |
298.15 |
303.15 |
298.15 |
303.15 |
298.15 |
303.15 |
298.15 |
303.15 |
298.15 |
303.15 |
| CapSH(g) |
51.14 |
51.38 |
36.77 |
36.76 |
−48.20 |
−48.22 |
−1.27 |
−1.03 |
8.52 |
5.54 |
| CapSH(aq) |
50.73 |
50.90 |
38.42 |
38.35 |
−41.27 |
−41.40 |
−2.77 |
−2.61 |
107.82 |
76.56 |
| CySH(g) |
49.72 |
49.94 |
36.33 |
36.34 |
−44.89 |
−44.88 |
−1.82 |
−1.62 |
21.75 |
14.70 |
| CySH(aq) |
52.05 |
52.17 |
41.86 |
41.77 |
−34.18 |
−34.31 |
−1.95 |
−1.72 |
27.05 |
17.39 |
| GSH(g) |
47.90 |
48.18 |
31.27 |
31.27 |
−55.77 |
−55.78 |
−1.22 |
−1.07 |
7.90 |
5.88 |
| GSH(aq) |
60.57 |
60.56 |
52.58 |
52.29 |
−26.81 |
−27.28 |
−1.15 |
−0.90 |
6.93 |
4.45 |
| |
ΔGsolv (kcal mol−1) |
ΔHsolv (kcal mol−1) |
ΔSsolv (cal K−1 mol−1) |
H-bonding (kcal mol−1) |
Activity coefficient |
| Acrylamide |
−12.80 |
−12.57 |
−19.49 |
−19.14 |
−22.45 |
−22.03 |
−10.09 |
−9.76 |
2.45 |
2.49 |
| CapSH(Prod) |
−31.50 |
−31.08 |
−43.99 |
−43.26 |
−41.89 |
−40.86 |
−18.78 |
−18.16 |
6.89 |
7.51 |
| CapSH(React) |
−17.20 |
−16.93 |
−25.38 |
−24.91 |
−27.45 |
−26.76 |
−9.29 |
−8.98 |
61.44 |
64.46 |
| CapSH(TS) |
−30.41 |
−29.98 |
−43.22 |
−42.50 |
−42.98 |
−41.98 |
−18.92 |
−18.29 |
13.32 |
14.20 |
| CySH(Prod) |
−25.94 |
−25.50 |
−39.28 |
−38.58 |
−44.72 |
−43.86 |
−21.53 |
−20.81 |
4.86 |
4.76 |
| CySH(React) |
−13.02 |
−12.83 |
−18.87 |
−18.53 |
−19.64 |
−19.14 |
−8.01 |
−7.71 |
8.11 |
8.11 |
| CySH(TS) |
−23.48 |
−23.17 |
−32.84 |
−32.29 |
−31.38 |
−30.60 |
−16.64 |
−16.06 |
15.24 |
14.77 |
| GSH(Prod) |
−45.24 |
−44.47 |
−68.29 |
−67.09 |
−77.32 |
−75.85 |
−34.59 |
−33.44 |
1.85 |
1.97 |
| GSH(React) |
−32.52 |
−32.07 |
−46.22 |
−45.42 |
−45.94 |
−44.79 |
−21.98 |
−21.24 |
9.19 |
9.47 |
| GSH(TS) |
−32.64 |
−32.25 |
−44.40 |
−43.68 |
−39.44 |
−38.32 |
−16.13 |
−15.56 |
25.55 |
27.31 |
The differences in activation free energy for CapSH, CySH and GSH at 298 K when the effects of solvation are included in the computation (Fig. 8, right), are −0.41, +1.33 and +12.7 kcal mol−1, respectively. The small decrease for CapSH and slight increase for CySH are unlikely to be significant given the intrinsic limitations of each step in the computation, but the TS of GSH is predicted to be significantly less accessible in water. If dissociation of the thiol is a necessary first step in the reaction mechanism, then a polar solvent such as water will stabilize the charged reactant species and hence increase the energy of activation as observed in Fig. 8.
According to the experimental acid dissociation constants (Table 3), GSH is more easily dissociated than CapSH, so its stabilization by water should be larger. For theoretical comparison, we compute the Hirshfeld charges on the reactive functionality, C-SH, as an estimate of nucleophilicity in the minimum energy conformations obtained in gas and aqueous phase (Table 6).
Table 6 Atomic Hirshfeld charges in gas phase and aqueous solutiona
| |
C |
S |
H |
| (g) |
(aq) |
(g) |
(aq) |
(g) |
(aq) |
|
†Transition state.
|
| CapSH |
−0.069 |
−0.069 |
−0.035 |
−0.037 |
0.044 |
0.044 |
| CapSH† |
−0.070 |
−0.075 |
0.026 |
−0.036 |
0.048 |
0.045 |
| CySH |
−0.076 |
−0.075 |
−0.056 |
−0.058 |
0.032 |
0.032 |
| CySH† |
−0.074 |
−0.079 |
0.036 |
−0.050 |
0.050 |
0.048 |
| GSH |
−0.073 |
−0.078 |
−0.017 |
−0.018 |
0.045 |
0.040 |
| GSH† |
−0.056 |
−0.054 |
0.206 |
0.221 |
0.060 |
0.058 |
In both gas and solution, the computed sequence of estimated nucleophilicity of the sulphur atom in the thiol is CySH > CapSH > GSH, where CapSH and GSH are reordered compared to experiment. Coupled with the reordering in reaction rates, this finding suggests that the extent of formation of the thiolate anion is variable and is not only dependent on pKs and that solvent and hydrogen bonding interactions may be important. In particular, inter- and intra-molecular hydrogen bonding that stabilize a gaseous transition state are likely to be disrupted by water molecules in solution, adding to the relative increase in activation energy. On inspecting the derived enthalpy (ΔH‡) and entropy (ΔS‡) contributions to ΔG‡ for GSH (Table 5), we note the reaction in aqueous solution gives the smallest change in ΔS‡ and the largest change in ΔH‡ of the thiols studied. This pattern is a complete reversal of the findings for the gas phase reaction, supporting the effect of H-bond interactions on the activation energy in solution; the highest free energy change with solvation (ΔGsolv) for the AA product, reactant and transition states all occur in the reaction of GSH. We may also assign the reversal of activation energies for CySH and CapSH in water as being due to the combination of limited H-bonding capability of CapSH and its large pKs (9.0 experimental), but the computed difference is small at 1.32 kcal mol−1.
The computed free energy of reaction (ΔGReact) for all three thiols with acrylamide are also small, the largest at −1.82 kcal mol−1 for CapSH. However, the small differences between thiols are expected to have significant impact on the thermodynamic equilibrium constants computed in each case according to:
| | 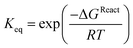 | (13) |
We obtain values of 108, 27 and 7 mol L−1 for CapSH, CySH, and GSH, respectively at 298 K (Table 5). There are several transition state models which seek to relate, with varying degrees of success, theoretical activation parameters to experimentally derived rate constants.34–36 Our computed kinetic values of rate constants, ks, activation free energy, ΔG‡s and Arrhenius pre-factor As (s for in silico) are shown in Table 7.
Table 7 Experimental and theoretical estimates of rate constants, k, Arrhenius pre-factor, A and activation parameters (ΔG‡, ΔH‡, and ΔS‡) for the reaction of AA with CapSH, CySH and GSHa
| |
ΔG°‡ |
ΔH°‡ |
ΔS°‡ |
Rate constant (dm3 mol−1 s−1) |
Arrhenius |
| (kcal mol−1) |
(kcal mol−1) |
(cal mol−1 K−1) |
k
|
k
s
|
lnA |
lnAs |
|
s for in silico.
|
| CapSH |
18.64 |
5.26 |
−45.41 |
0.13 |
1.28 × 10−24 |
7.93 |
9.64 |
| CySH |
18.16 |
8.13 |
−34.18 |
0.34 |
1.55 × 10−25 |
13.04 |
13.21 |
| GSH |
18.64 |
7.89 |
−35.61 |
0.18 |
1.38 × 10−31 |
12.58 |
16.74 |
There is marked disparity between the experimental and theoretical kinetic parameters in Table 7, but relatively good agreement is obtained for the standard entropy change of activation with all three thiols (−41.3, −34.2, −26.8 cal mol−1 K−1) for CapSH, CySH and GSH from theory, Table 5. Closely matching the kinetic variables with experimental data for these reactants in aqueous solution will likely require higher level DFT computational methods with the Moller–Plessett Perturbation Theory37 (MP2), larger basis sets, Variational Transition-State Theory32 (VTST) and isotopic labelling experiments since the transition state involves a proton migration. However, given that AA may react ∼500 times faster with –SH than with –NH2 that are both present in biologically active thiols10,38 our results have established that dominant reaction between AA and thiols is probably the Michael addition between the more nucleophilic –S− groups and AA. In particular, a linear isokinetic plot of ΔH‡versus ΔS‡ for the reactions described above (a correlation coefficient >0.99) gave an isokinetic temperature of 260 ± 24 K and an intercept value for ΔG‡ of 71 ± 4 kJ mol−1 (17 ± 1 kcal mol−1, Fig. 10). Although there is controversy in literature about the nature of enthalpy–entropy compensation (whether it is real39–41 or artefact42,43) most workers support its existence. The isokinetic temperature represents the temperature at which the rates of reaction between AA and all three thiols are equal. The presence of a linear isokinetic plot for RSHs confirms that the reactions between AA and all three thiols follow a similar general reaction mechanism i.e. the Michael addition.
 |
| | Fig. 10 Isokinetic plot for the reactions between AA and the thiols – CapSH, CySH and GSH. | |
Conclusions
Thiols are known to react with acrylamide, a potential carcinogen, formed in carbohydrate food preparations above 100 °C. In this paper, we report the kinetics of the reaction of acrylamide with three common thiols, CapSH, CySH and GSH according to a Michael addition reaction scheme. Assuming that the nucleophilicity of all three thiols is approximately the same, the rates of the reaction with AA were expected to follow: CySH > CapSH > GSH, reflecting the increasing molecular size of the thiols. However this was not observed. Rather, the experimental reaction rate follows the order CySH > GSH > CapSH. DFT calculations support the experimental results through marked effects of solvation and hydrogen bonding on the activation energies, where the GSH–AA activated complex is computed at 8–10 kcal mol−1 higher than CapSH and CySH. This is consistent with diminished intra-molecular hydrogen bonding in GSH–AA due to more inter-molecular interactions with the polar water solvent molecules. We obtained an experimental linear entropic-enthalpy compensation plot from ΔG‡, ΔH‡, ΔS‡, activation parameters which support a single pathway for all three thiols. Recent reports32 suggest that the calculation of rate constants could be bettered using Variational Transition State Theory with MP2 correlation, or with explicit treatment of the role of water molecules (ex Solvent Assisted Proton Exchange or SAPE models44,45) which we expect to form part of our future work.
Acknowledgements
This work was supported by funding provided by the Office of Graduate Studies and Research, The University of the West Indies, Mona, Jamaica, West Indies and the Departments of Chemistry, The University of the West Indies, Mona, Jamaica and St. Augustine, Trinidad and Tobago.
References
- I.A.R.C., IARC Monogr. Eval. Carcinog. Risks Hum., 1994, 60, 389–433 Search PubMed.
- I.A.R.C., IARC Monogr. Eval. Carcinog. Risks Hum., 1986, 39, 403 Search PubMed.
- I.A.R.C., IARC Monogr. Eval. Carcinog. Risks Hum., 1987,(Suppl. 7), 56 Search PubMed.
- Office of Environmental Health Hazard Assessment (O.E.H.H.A.) California Environmental Protection Agency, Meeting of the Carcinogen Identification Committee (CIC). Safe drinking water and toxic enforcement act of 1986 (proposition 65), http://www.oehha.ca.gov/prop65/docs_state/pdf/acrylbrief.pdf, Access Date: July 12, 2014.
- E. Tareke, P. Rydberg, P. Karlsson, M. Törnqvist and S. Eriksson, Chem. Res. Toxicol., 2000, 13, 517–522 CrossRef CAS PubMed.
- G.-A. Bent, P. Maragh and T. Dasgupta, Food Chem., 2012, 133, 451–457 CrossRef CAS PubMed.
- E. Tareke, P. Rydberg, P. Karlsson and M. Törnqvist, J. Agric. Food Chem., 2002, 50, 4998–5006 CrossRef CAS PubMed.
- K. Galesa, U. Bren, A. Kranjc and J. Mavri, J. Agric. Food Chem., 2008, 56, 8720–8727 CrossRef CAS PubMed.
- G. Tong, W. Cornwell and G. Means, Toxicol. Lett., 2004, 147, 127–131 CrossRef CAS PubMed.
- M. Friedman, J. Agric. Food Chem., 2003, 51, 4504–4526 CrossRef CAS PubMed.
- J. Wilson, D. Wu, R. Motiu-Degrood and D. A. Hupe, J. Am. Chem. Soc., 1980, 102, 359–363 CrossRef CAS.
- K. Kolšek, M. Sollner Dolenc and J. Mavri, Chem. Res. Toxicol., 2013, 26, 106–111 CrossRef PubMed.
- J. Mavri, Toxicol. In Vitro, 2013, 27, 479–485 CrossRef CAS PubMed.
- A. Pastore, F. Piemonte, M. Locatelli, A. L. Lo Russo, L. M. Gaeta, G. Tozzi and G. Federici, Clin. Chem., 2003, 8, 1467–1469 Search PubMed.
- J. Castell, M. Gomez-Lechon, X. Ponsoda and R. Bort, Arch. Toxicol., 1997, 313–321 CAS.
- P. Ribeiro, A. Santini, H. Pezza and L. Pezza, Ecletica Quim., 2010, 35, 179–188 CrossRef PubMed.
- O. Pecháñova, Phys. Res., 2007, 56, S41–S48 Search PubMed.
- C. Napoli, V. Sica, F. De Nigris, O. Pignalosa, M. Condorelli, L. Ignarro and A. Liguori, Am. Heart J., 2004, 148, e5 CrossRef CAS PubMed.
- M. Harrison and M. Baldwin, Org. Mass Spectrosc., 1989, 24, 689–693 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef PubMed.
- J. Baker, T. Janowski, K. Wolinski and P. Pulay, WIREs Comput. Mol. Sci., 2012, 2, 63–72 CrossRef CAS.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- Vrije Universiteit, Amsterdam, The Netherlands, ADF2013 COSMO-RS, SCM, Theoretical Chemistry, http://www.scm.com, Last Accessed: July 12, 2014.
- C. C. Pye, T. Ziegler, E. van Lenthe and J. N. Louwen, Can. J. Chem., 2009, 87, 790–797 CrossRef CAS.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129 CrossRef CAS.
- K. B. Wiberg and P. R. Rablen, J. Comput. Chem., 1993, 14, 1504 CrossRef CAS.
- Y. Ohno, K. Ormstad, D. Ross and S. Orrenius, Toxicol. Appl. Pharmacol., 1985, 78, 169–179 CrossRef CAS.
- M. Friedman, J. Cavins and J. Wall, J. Am. Chem. Soc., 1965, 87, 3672–3682 CrossRef CAS.
- G. A. Jeffrey and S. Takagi, Acc. Chem. Res., 1978, 11, 264–270 CrossRef CAS.
- C. L. Perrin and J. B. Nielson, Annu. Rev. Phys. Chem., 1997, 48, 511–544 CrossRef CAS PubMed.
- D. E. Smith and A. D. J. Haymet, J. Chem. Phys., 1993, 98, 6445–6454 CrossRef CAS PubMed.
- N. R. Syme, C. Dennis, A. Bronowska, G. C. Paesen and S. W. Homans, J. Am. Chem. Soc., 2010, 132, 8682–8689 CrossRef CAS PubMed.
-
B. C. Garrett and D. G. Truhlar, Variational Transition State Theory, in Theory and Applications of Computational Chemistry: The First Forty Years, Elsevier B.V., 2005 Search PubMed.
- T. Bartsch, J. M. Moix, R. Hernandez, S. Kawai and T. Uzer, Adv. Chem. Phys., 2008, 140, 191–238 CrossRef CAS.
-
C. J. Cramer, Essentials of Computational Chemistry: Theories and Models, John Wiley and Sons Ltd, 2004 Search PubMed.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- J. M. Rice, Mutat. Res., 2005, 580, 3–20 CAS.
- L. Liu and Q.-X. Guo, Chem. Rev., 2001, 101, 673–695 CrossRef CAS PubMed.
- K. F. Freed, J. Phys. Chem. B, 2001, 115, 1689–1692 CrossRef PubMed.
- J. F. Douglas, J. Dudowicz and K. F. Freed, Phys. Rev. Lett., 2009, 103, 135701–135704 CrossRef.
- A. Cornish-Bowden, J. Biosci., 2002, 27, 121–126 CrossRef.
- A. Cooper, C. M. Johnson, J. H. Lackey and M. Nollmann, Biophys. Chem., 2001, 93, 215–230 CrossRef CAS.
- C. A. Bayse, J. Phys. Chem. A, 2007, 111, 9070–9075 CrossRef CAS PubMed.
- C. A. Bayse, Org. Biomol. Chem., 2011, 9, 4748 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tx00070f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 




 versus 1/T yields a slope of −ΔH‡ and an intercept of ΔS‡. The Gibbs free energy of activation, ΔG‡, (at 298 K) is calculated from eqn (8):
versus 1/T yields a slope of −ΔH‡ and an intercept of ΔS‡. The Gibbs free energy of activation, ΔG‡, (at 298 K) is calculated from eqn (8):