
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phthalimide-based π-conjugated small molecules with tailored electronic energy levels for use as acceptors in organic solar cells†
Arthur D.
Hendsbee
ab,
Seth M.
McAfee
ab,
Jon-Paul
Sun
c,
Theresa M.
McCormick
d,
Ian G.
Hill
c and
Gregory C.
Welch
*ab
aDepartment of Chemistry, Dalhousie University, 6274 Coburg Road, Halifax, Nova Scotia, Canada B3H 4R2
bDepartment of Chemistry, University of Calgary, 2500 University Drive NW, Calgary, Alberta, Canada T2N 1N4. E-mail: Gregory.welch@ucalgary.ca
cDepartment of Physics, Dalhousie University, 6274 Coburg Road, Halifax, Nova Scotia, Canada B3H 4R2
dDepartment of Chemistry, Portland State University, Portland, OR 97207-0751, USA
First published on 29th July 2015
Abstract
The design, synthesis, and characterization of seven phthalimide-based organic π-conjugated small molecules are reported. The new materials are based on a phthalimide–thiophene–CORE–thiophene–phthalimide architecture. The CORE units utilized were phthalimide (M2), diketopyrrolopyrrole (M3), isoindigo (M4), naphthalene diimide (M5), perylene diimide (M6), and difluorobenzothiadiazole (M7); they were specifically selected to progressively increase the electron affinity of the resulting compound. A small molecule with no core (M1) was synthesized for comparison. Each material was synthesized through optimized direct heteroarylation cross-coupling procedures using bench top solvents in air. Combinations of UV-visible spectroscopy (UV-vis), cyclic voltammetry (CV), differential scanning calorimetry (DSC), ultraviolet photoelectron spectroscopy (UPS) and density functional theory (DFT) were used to characterize each material. The use of various core acceptor building blocks with differing electron affinities resulted in the series M1–M7 having a range of energetically deep LUMO levels and a range of HOMO–LUMO gap energies. Meanwhile, the melting and crystallization temperatures of the molecules M1–M7 were also found to vary according to the change in central acceptor unit. Compounds M1–M7 were employed as acceptors in combination with either the polymeric donor P3HT or small molecule donor DTS(FBTTh2)2 to understand how the LUMO levels of each acceptor influences the open circuit voltage (Voc). It was found that, in general, Voc was only weakly related to the offset between the HOMO energy level of the donor and LUMO level of the acceptor used, with a Voc of up to 1.2 V being achieved for M1.
Introduction
Donor–acceptor π-conjugated materials have been extensively used as the active components in sensing devices, thin film transistors, photoswitches, photovoltaics and a variety of other useful applications.1–3 With respect to solution processed photovoltaics, the development of both donor–acceptor (D–A) conjugated polymers and small molecules has led to the realization of fullerene-based heterojunction (BHJ) organic solar cells (OSCs) with power conversion efficiencies (PCE) reaching beyond 9%.4–7 More recently, D–A organic π-conjugated compounds have found utility as electron-accepting components (vide infra) in solution processed BHJ OSCs, a position that is dominated by fullerene derivatives. Key advantages of D–A compounds compared to fullerene include the fact that electronic energy levels, absorption profiles, solubility parameters, and self-assembly tendencies can be precisely controlled through both selection and functionalization of the D and A building blocks.8–10 In addition, the majority of D–A compounds are easily synthesized though standard organic chemistry techniques using widely available starting materials. These advantages of D–A compounds can potentially allow for specifically matched electron-donor electron-acceptor pairings within the BHJ architecture where photon harvesting by the donor (channel I process) and acceptor (channel II process) are equally efficient, maximizing the photon to electron conversion efficiency.11–13 Donor materials making use of the D–A design strategy are well studied and thousands of materials with different physical properties have been presented in the literature, allowing for versatility when selecting the donor component for use in OSCs. On the other hand, acceptor materials based upon the D–A design strategy have traditionally been far less studied.Within the past year there has been a surge of publications reporting the use of D–A conjugated polymers and small molecules as an alternative to fullerene in BHJ OSCs with PCEs surpassing 6% for both polymer and small molecule acceptors.14–16 In particular, the use of small molecule based D–A compounds as electron-acceptors has been met with great success. For instance, Sellinger et al. have used benzothiadiazaole (BT)–phthalimide based acceptors to achieve PCEs up to 3.7% when paired with thiophene based donor polymers.17,18 Fluorene19–22 and fused fluorene–thiophene14,23 building blocks have also been used in several high performance OSC systems. Jenekhe and co-workers have also explored imide based acceptors,24 recently reporting on an electron deficient imide based framework to create OSCs with PCEs reaching 5%.25 Perylene diimides are well known for their electron deficient character and have been used to produce some of the highest performing small molecule acceptor based solar cells.26–30 There exist many additional examples that utilize the D–A strategy of combining building blocks; however, for additional details the reader is directed to a selection of recent reviews discussing this subject.31–35
In the design of new electron accepting materials one of the key criteria sought after is relatively deep highest occupied molecular orbital (HOMO) energy levels and low lying lowest unoccupied molecular orbital (LUMO) energy levels to ensure efficient channel I and channel II processes. The synthetic diversity of D–A type compounds allows for precise control over electronic energy levels thereby allowing for optimization of energy offsets.9 Specifically, by increasing the offset between the donor HOMO and acceptor LUMO in a donor–acceptor solar cell, one expects a progressive increase in open circuit voltages.
Previously in our lab group we have investigated the use of a phthalimide–thiophene–thiophene–phthalimide (Phth–Th–Th–Phth) architecture with respect to its utility as a charge transporter and found it to be a good electron transporting material with mobilities of ∼0.2 cm2 V−1 s−1.36 This phthalimide–thiophene based small molecule has also been used in our group to produce OSCs with high Voc, ∼1.0 V, when paired as an acceptor with P3HT.37 Moving forward, we incorporated the diketopyrrolopyrrole (DPP) chromophore as a central building bock in this design and demonstrated that DPP and phthalimide can be used together to make low band gap electron transporting materials.38 In another study we have investigated the use of the isoindigo (IS) chromophore as a central building block and demonstrated that it can also be used to produce low band gap materials, that when used as the electron-accepting component in OSCs, gave devices with a Voc of ∼1.0 V.39
We have also performed a theoretical investigation on a series of electron deficient building blocks with acceptor–donor–acceptor–donor–acceptor (A1DA2DA1) architecture (where A1 and A2 have different electron affinities). This has demonstrated that electronic energy levels and band gaps can be systematically tuned by varying the terminal and/or core electron acceptor units (A1 and A2, respectively).40
To expand on the Phth–Th–CORE–Th–Phth architecture we have set out to incorporate naphthalene diimide (NDI), perylene diimide (PDI), difluorobenzothiadiazole (F2BT) and phthalimide (Phth), in addition to the previously utilized IS and DPP chromophores, as the central acceptor units (Fig. 1). Each of the selected building blocks has a different electron affinity due to the nature of the functional groups on their respective aromatic cores. This change in electron-accepting character in the central acceptor unit of our design was predicted to produce a series of molecules with a range of LUMO energy levels. Herein, we evaluate the effect that each electron deficient building block has on the optical, thermal, and electronic properties of these molecular systems and discuss the impact that the inclusion of each acceptor core has on the Phth–Th–CORE–Th–Phth structure.
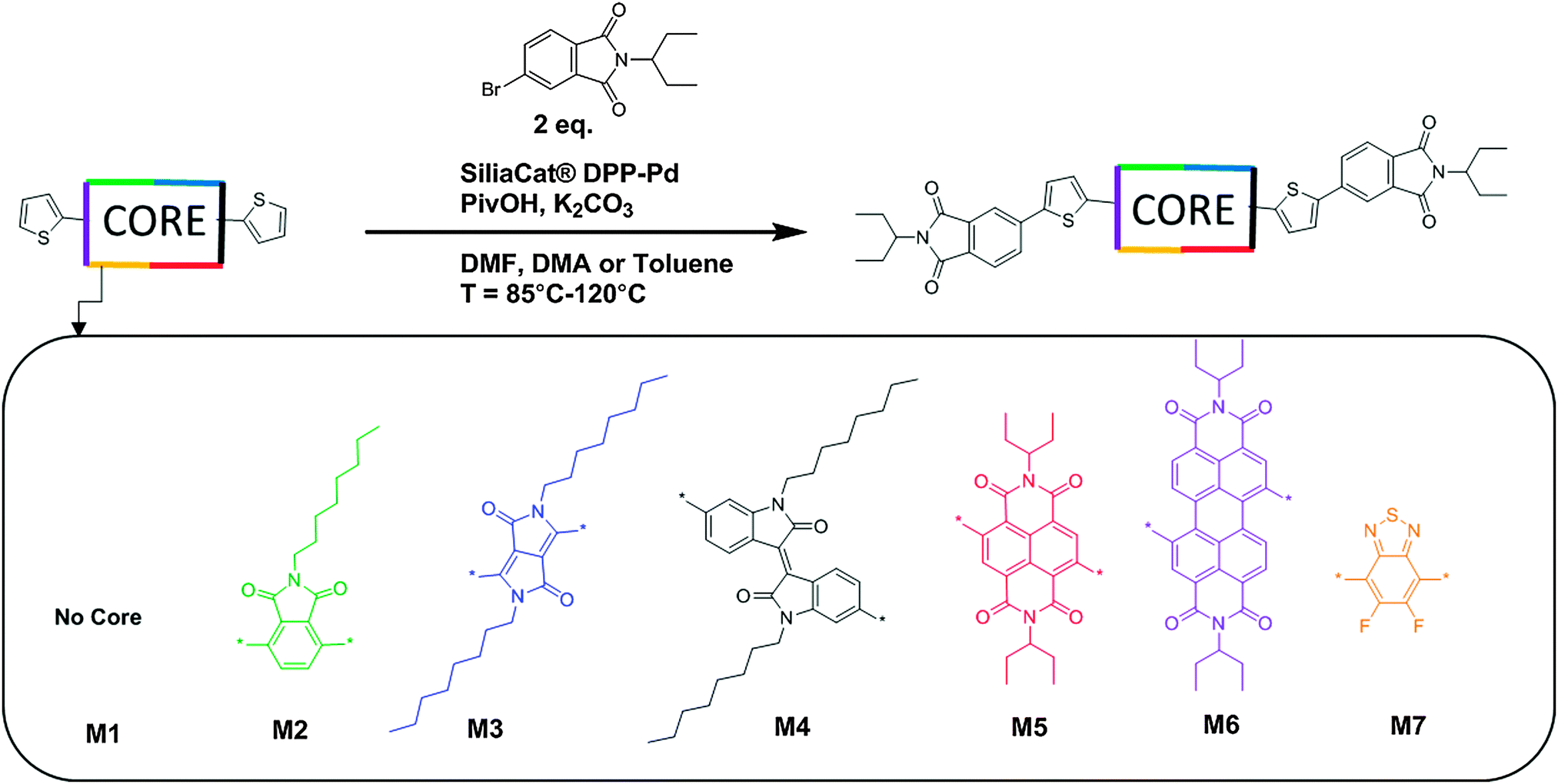 | ||
| Fig. 1 Synthetic routes towards molecules M1–M7 using direct heteroarylation conditions. 5 mol% Pd catalyst, 20 mol% pivalic acid, 2.5 eq. potassium carbonate. Solvents used in this work include DMA, DMF and toluene. Reaction temperatures used in this work range between 85–120 °C, full synthetic details for small molecules M1–M7 and relevant precursor materials are given in the ESI,† Section S1. | ||
Materials synthesis
The synthesis of small molecules M1–M7 was completed via direct heteroarylation (DHA) using the heterogeneous catalyst SiliaCat® DPP-Pd, following our previously reported methods.27,41 All precursor materials were synthesized using literature or modified literature procedures with full details in the ESI.† For each DHA reaction, catalyst loadings of 5 mol% Pd with 20 mol% pivalic acid and potassium carbonate as the base were used. For the phthalimide end capping units, 1-ethylpropyl alkyl chains were added via condensation of the anhydride with 1-ethylpropylamine. Branched alkyl chains such as this can provide increased solubility compared to their linear counterparts while minimizing the amount of non-conjugated moieties in the molecule.42 For both of the CORE components NDI and PDI the same 1-ethylpropyl chains were used which are well known to promote the solubility of these chromophores.42,43 For the phthalimide, IS, and DPP CORE fragments, alkylation with linear C8 alkyl chains, known to promote solubility of these chromophores was accomplished using previously reported methods.38,39Synthesis proceeded in a similar manner for compounds M1–M7, and synthetic results are summarized in Table S1 (ESI†). Notably, small molecule M7 required the use of toluene as a solvent for the reaction due to a lack of product formation when the reaction was attempted in polar aprotic solvents such as DMA or DMF, even over a range of temperatures (see ESI†). The effective use of toluene as a solvent for promoting direct heteroarylation reactions involving the BT building block has been noted recently in the literature.44 All products were purified in the same manner, by first separating the heterogeneous catalyst from the reaction mixture using a filtration apparatus, followed by column chromatography on silica-gel with a pentane–CH2Cl2 gradient to elute the products. Each of the new compounds M1–M7 was found to be soluble in common organic solvents used for solution processing.
Optical characterization of small molecules M1–M7
To examine the optical properties of M1–M7, the UV-vis spectra of M1–M7 were obtained in CHCl3, spectra are shown in Fig. 2 and data is tabulated in Table 1. Due to the differing electronic properties of the core units used in the design architecture, small molecules M1–M7 were each a noticeably different colour. Compound M1 appears yellow in CHCl3 solution and the UV-vis profile of M1 is broad with λmax at 424 nm and λonset at 496 nm. Compound M2, bearing a phthalimide core exhibits a very similar absorption profile to that of M1, and also appears yellow in solution having λmax at 395 nm and λonset at 453 nm. Interestingly the absorbance maxima do not change significantly from M1 to M2, despite the relative extension of conjugation via an additional electron withdrawing phthalimide unit. Compound M3 appears dark blue in CHCl3 solution and has λmax at 611 nm and λonset at 666 nm. This strong low energy absorbance maxima is commonly seen from DPP small molecules and polymers and is attributed to intramolecular charge transfer (ICT).38,45–47 The small molecule M4 bearing an IS core absorbs light across the entire visible spectrum and appears black in solution. The λmax is 440 nm and λonset is 678 nm for compound M4. This type of absorption profile is typical for IS based D–A systems, which frequently produce strongly absorbing materials with narrow band-gaps.39,48–51 Small molecule M5, bearing a NDI core appears dark red in solution and has a broad low energy absorbance band extending beyond 600 nm. For compound M5 the peak absorbance occurs at 364 nm and the onset of absorbance at 587 nm. Compound M6, which contains a PDI core appears purple in solution and displays a similar absorbance profile to M5 having a strong high-energy absorbance maxima (λmax = 374 nm) and a broad absorbance band at lower energies (λonset = 704 nm). The absorbance profiles of both molecules M5 and M6 display a strong high energy absorption band and a relatively weaker low energy absorption band, which are features often observed for both NDI52–54 and PDI55–57 based chromophores. Small molecule M7 containing a F2BT core appears red-orange in solution and exhibits a strong absorbance band (λmax = 464 nm, λonset = 567 nm).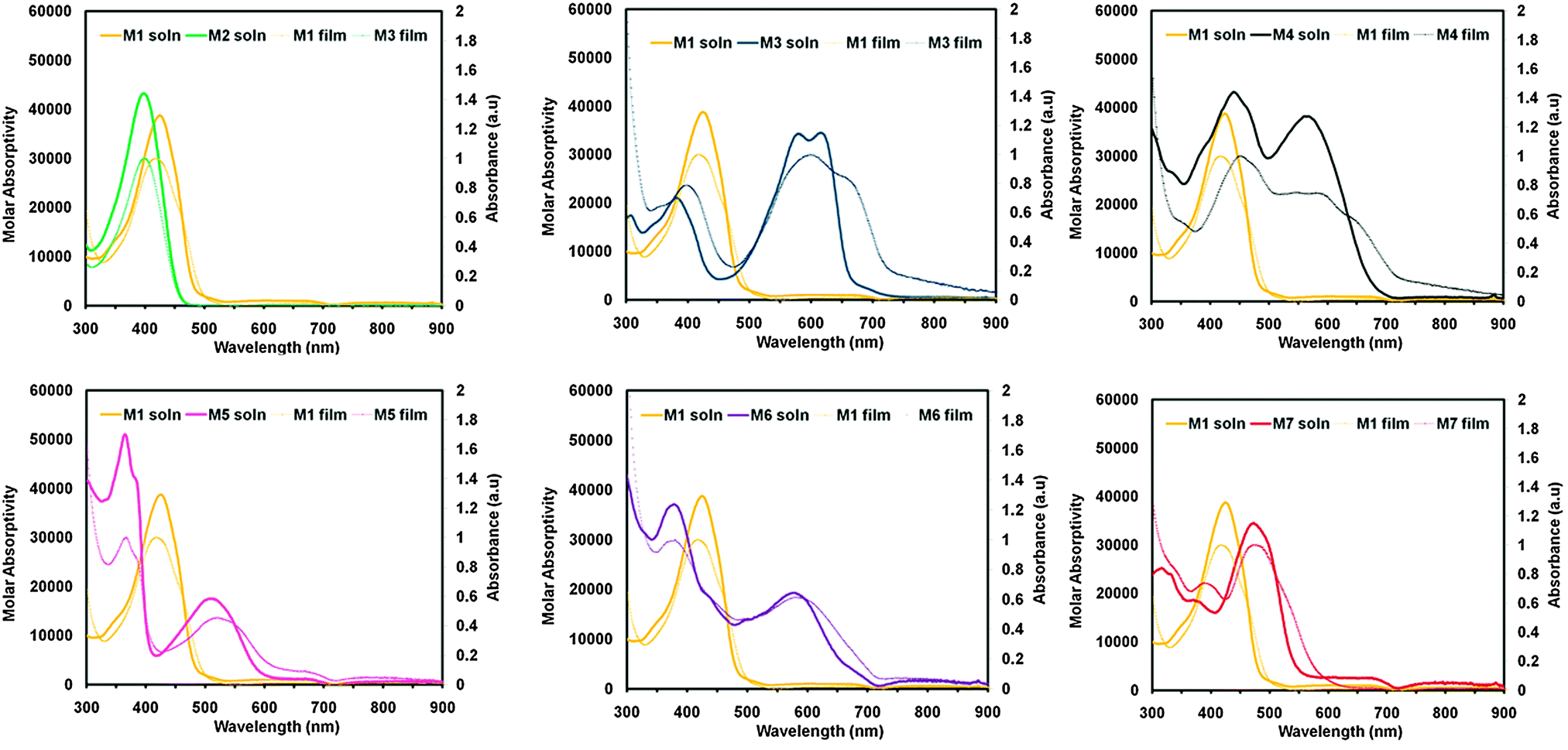 | ||
| Fig. 2 UV-visible absorption spectra of compounds M1–M7 in CHCl3 solution (solid lines), and thin films cast from 1% wt/vol CHCl3 (dashed lines). The spectra of M1 is plotted against each of M2–M7 for reference. | ||
| λ max,sol'n (nm) | λ onset,sol'n (nm) | λ max,film (nm) | λ onset,film (nm) | EACV (eV) | IECV (eV) | IEUPS (eV) | T melt (°C) | T cryst (°C) | |
|---|---|---|---|---|---|---|---|---|---|
| M1 | 424 | 496 | 413 | 499 | 3.03 | 5.60 | 6.0 | 277 | 181 |
| M2 | 395 | 453 | 404 | 483 | 2.98 | 5.92 | 6.1 | — | — |
| M3 | 611 | 666 | 592 | 727 | 3.44 | 5.23 | 5.4 | 144, 254 | 126, 209 |
| M4 | 440 | 678 | 450 | 738 | 3.64 | 5.47 | 5.7 | 259 | 209 |
| M5 | 364 | 587 | 364 | 611 | 3.82 | 5.92 | 6.1 | 279 | — |
| M6 | 374 | 704 | 373 | 709 | 3.90 | 5.69 | 6.0 | — | — |
| M7 | 464 | 567 | 468 | 577 | 3.29 | 5.66 | 5.9 | — | — |
The UV-visible absorbance profiles of thin films on glass substrates were obtained and plots of absorbance vs. wavelength for compounds M1–M7 are shown in Fig. 2, with the data summarized in Table 1. Each of the cores used in M1–M7 can have a different influence on the supramolecular structure of the resultant materials when transitioning from solution into the solid state. M1, for example has an absorbance maxima in the solid state that is blue shifted from the solution (λmax = 413 nm). Compound M2 bearing a phthalimide core has absorbance maxima that is only slightly red shifted compared to solution (λmax = 404 nm). The absorbance maximum of M3 shifts towards higher energies in the solid state (λmax = 592 nm) and the absorbance profile broadens with a low energy shoulder observable around 680 nm (λonset = 727 nm). Compound M4, which has a central IS unit undergoes a substantial change in its absorbance profile upon thin film formation. The absorbance maximum for M4 shifts towards higher energies (λmax = 450 nm), and the absorbance profile is slightly broadened with a red shifted onset of absorption (λonset = 738 nm). In contrast, compound M5, which has a NDI core shows very little change in its UV-visible absorption profile upon transitioning from solution to film with λmax = 364 nm and λonset = 611 nm. M6, which contains a PDI core, displayed minimal change in the absorption profile upon transitioning from solution to the solid state (λmax = 373 nm and λonset = 709 nm). Compound M7, bearing a F2BT core also underwent minimal change in its absorption profile upon transitioning from the solution to the solid state (λmax = 468 nm λonset = 577 nm). It is clear from the solution and thin film absorption experiments that each core included in the Phth–Th–CORE–Th–Phth architecture has a definite effect on the optical properties of the resultant molecule. In addition the different observations made for M1–M7 upon transitioning from solution to film indicated that the inclusion of the core acceptor unit was having a definite effect on the self-assembly of the molecules from solution.
Thermal characterization of small molecules M1–M7
In order to gain more insight into the solid phase properties, small molecules M1–M7 were evaluated using differential scanning calorimetry (DSC) to probe their melting and crystallization behaviour. Samples were heated from 50 °C to 300 °C for 3 cycles under air. Compound M1 displayed a sharp melting transition at 277 °C and a crystallization transition at 181 °C. Compound M2, interestingly, displayed no melting or crystallization transitions in the temperature regime used indicating that the incorporation of the phthalimide unit as a central acceptor in the Phth–Th–CORE–Th–Phth architecture has a large effect on the thermal properties of the resulting compound (M1vs.M2). Compound M3 showed multiple transitions in the DSC curve, two melting transitions at 144 °C and 254 °C, and two corresponding crystallization transitions at 126 °C and 209 °C. The lower temperature transitions are attributed to the inter chain stacking of the alkyl groups, while the higher temperature transitions are attributed to a lamellar melting of the aggregated π-conjugated molecules, this type of behaviour is often seen in large π-extended chromophores with appended alkyl chains.58,59 Compound M4 exhibited sharp melting and crystallization transitions at 259 °C and 209 °C, respectively. Small molecule M5 displays a melting transition in the DSC curve at 279 °C and does not display a crystallization transition. Small molecules M6 and M7 displayed no melting or crystallization peaks on the DSC. The lack of a melting point in the range of temperatures studied using the DSC for compounds M2, M6 and M7 was confirmed using a melting point apparatus, where compound M2 was found to melt at 334–335 °C, compound M6 began to melt at ∼250 °C and continued to become less viscous until approximately 270 °C. For compound M7 the melting transition was observed at 305–308 °C.Electronic characterization of small molecules M1–M7
With the UV-visible data in hand showing a dramatic difference in the optical band-gaps of M1–M7, caused by the substitution of the different core fragments to the phthalimide–thiophene architecture, cyclic voltammetry (CV) in CH2Cl2 solution was then used in order to gain rapid experimental insight on the ionization energy (IE) and electron affinity (EA) for small molecules M1–M7 (Fig. 3). Experimental details and tabulated oxidation and reduction potentials can be found in the ESI.† The use of cyclic voltammetry data for calculating frontier orbital energies may not provide exact results as discussed in a recent paper;60 however, the data can still provide useful insight into the relative energy levels of compounds that are studied using CV under the same conditions. As shown in Fig. 3, the EA can be systematically increased by increasing the electron affinity of the central core unit. M1 and M2, with no core and a phthalimide core possessed relatively low EAs of 3.03 and 2.98 eV, respectively. Introducing the more electron deficient DPP (M3) and IS (M4) cores increases the EA of the molecules to 3.44 and 3.64 eV, respectively. Clearly the IS unit is a stronger electron-accepting unit than DPP. Compounds M5 and M6 were found to have similar values for EA in the range from 3.8 to 3.9 eV. The very high EAs found for M5 and M6 are indicative of the strong electron accepting nature of the NDI and PDI building blocks. Small molecule M7 was found to have an EA of 3.29 eV which is to those values found for the IS and DPP containing molecules. An especially important feature of OSCs is the open circuit voltage of the cell (Voc), the maximum obtainable value of which is related to the energy level offset between the HOMO of the donor material and the LUMO of the acceptor material. This offset should be maximized; however, the LUMO level of the acceptor must still lie in an energetic position such that electron transfer from the excited donor material can take place.61 Small molecules M1–M7 all show promise in this regard, with LUMO levels that are low enough in energy to facilitate the transfer of electrons from donor materials material while still having potential for high Voc when paired with common donor materials such as P3HT.61,62 The CV plots (reduction only) are shown in Fig. 3B, full CV plots are given in the ESI† for this document (Fig. S22).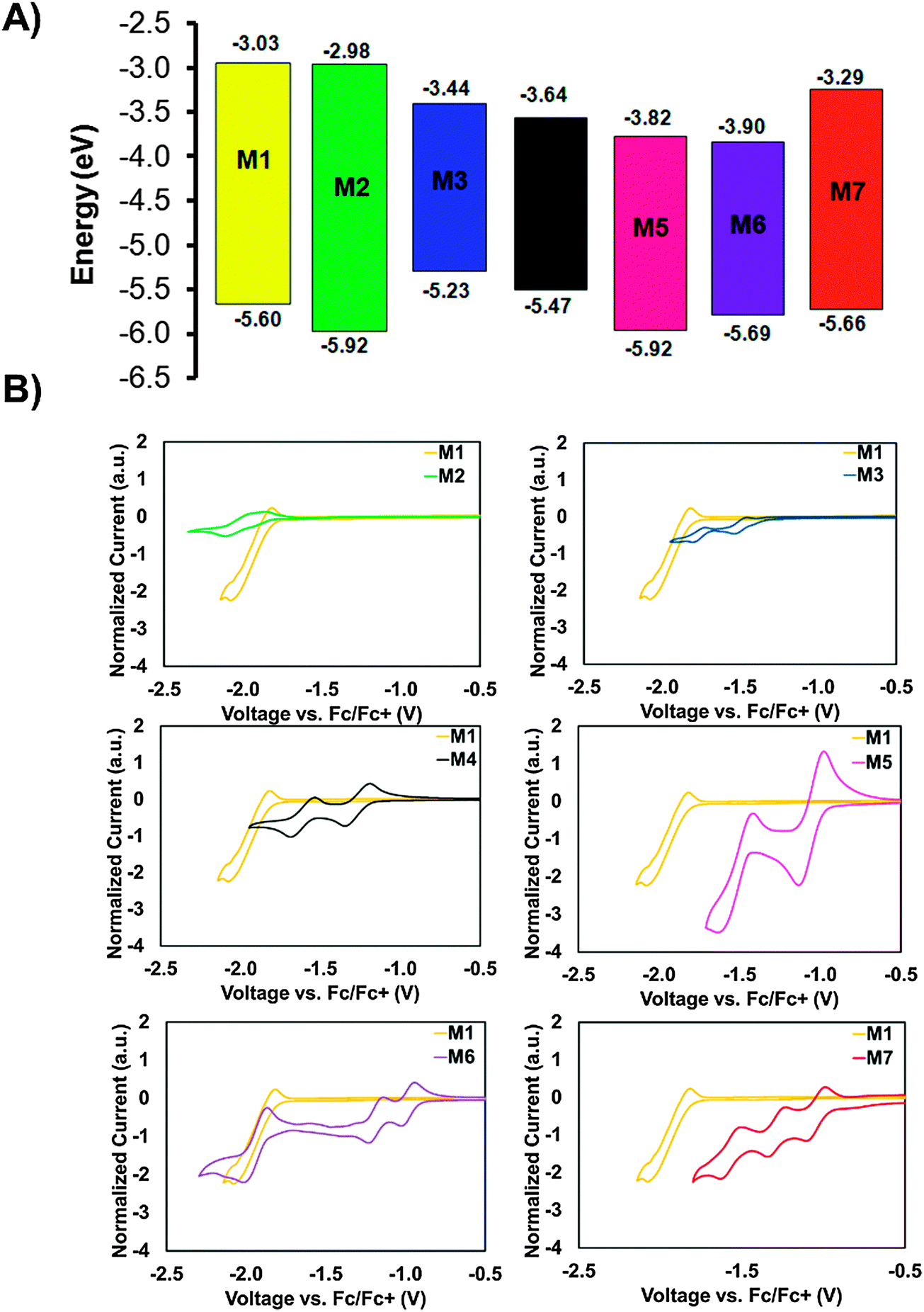 | ||
| Fig. 3 (A) Energy level diagram showing molecules M1–M7, data was collected using cyclic voltammetry in CH2Cl2 solution. For reference the energy levels of the donors P3HT and DTS(FBTTh2)2 are included in an expanded energy level diagram in the ESI† (Fig. S30). (B) Cyclic voltammetry plots for compounds M1–M7 in CH2Cl2 solution showing reduction only, full CV plots for M1–M7 can be found in the ESI,† (Fig. S22). | ||
Ultraviolet photoelectron spectroscopy (UPS) was used to measure the solid-state IEs of M1–M7. The results are summarized in Table 1, with spectra shown in the ESI† (Fig. S23). M1 exhibits an ionization energy (IE) of 6.0 eV, owing to the electron withdrawing nature of the terminal phthalimide groups. M2, with an addition central phthalimide has a slightly higher solid-state IE of 6.1 eV. Going from M2 to M5, the NDI core adds an electron withdrawing imide group in addition to an extra conjugated carbon ring, two opposing effects that balance out and result in the same IE of 6.1 eV. From M5 to M6, the addition of two carbon rings has the effect of lowering the IE to 6.0 eV. M3 has the lowest IE of the series. The two amides on the DPP core are less effective at stabilizing the π-electrons, resulting in an IE of 5.4 eV. M5, with an IS core contains the same stabilizing groups as M4, yet bears a higher IE of 5.7 eV. The two fluorine atoms on the F2BT core of M7 also act to withdraw electron density, resulting in an IE of 5.9 eV.
Theoretical evaluation of small molecules M1–M7
Molecules M1–M7 were investigated in the gas-phases using density functional theory (DFT)63–65 and time dependent DFT (TD-DFT)66 to further understand the impact of the core structure on molecular geometries, electronic structure and electronic transitions. Structures with truncated alkyl chains (i.e. methyl groups in place of the longer alkyl chains) were optimized to reduce computation time.67 All calculations were conducted using the Gaussian09 suit of programs.68,69 The structures for the rotational isomers of each compound were optimized to a local minimum. The lowest energy confirmation of each molecule is shown in Fig. 4. All subsequent calculations were performed on that isomer. The geometries were optimized with B3LYP/6-311+G(d)70,71 except M4 which would not converge with diffuse functionals. For M4 the geometry was optimized with B3LYP/6-31G(d) and a single point energy calculation was performed with B3LYP/6-311+G(d) to obtain energy and orbital diagrams. The frontier orbital energies are given in Fig. 5A. The calculated HOMO and LUMO energy values for the ground state molecules follow similar trends, to experimental values of IE and EA as determined from CV and UPS (see Fig. S25, ESI†).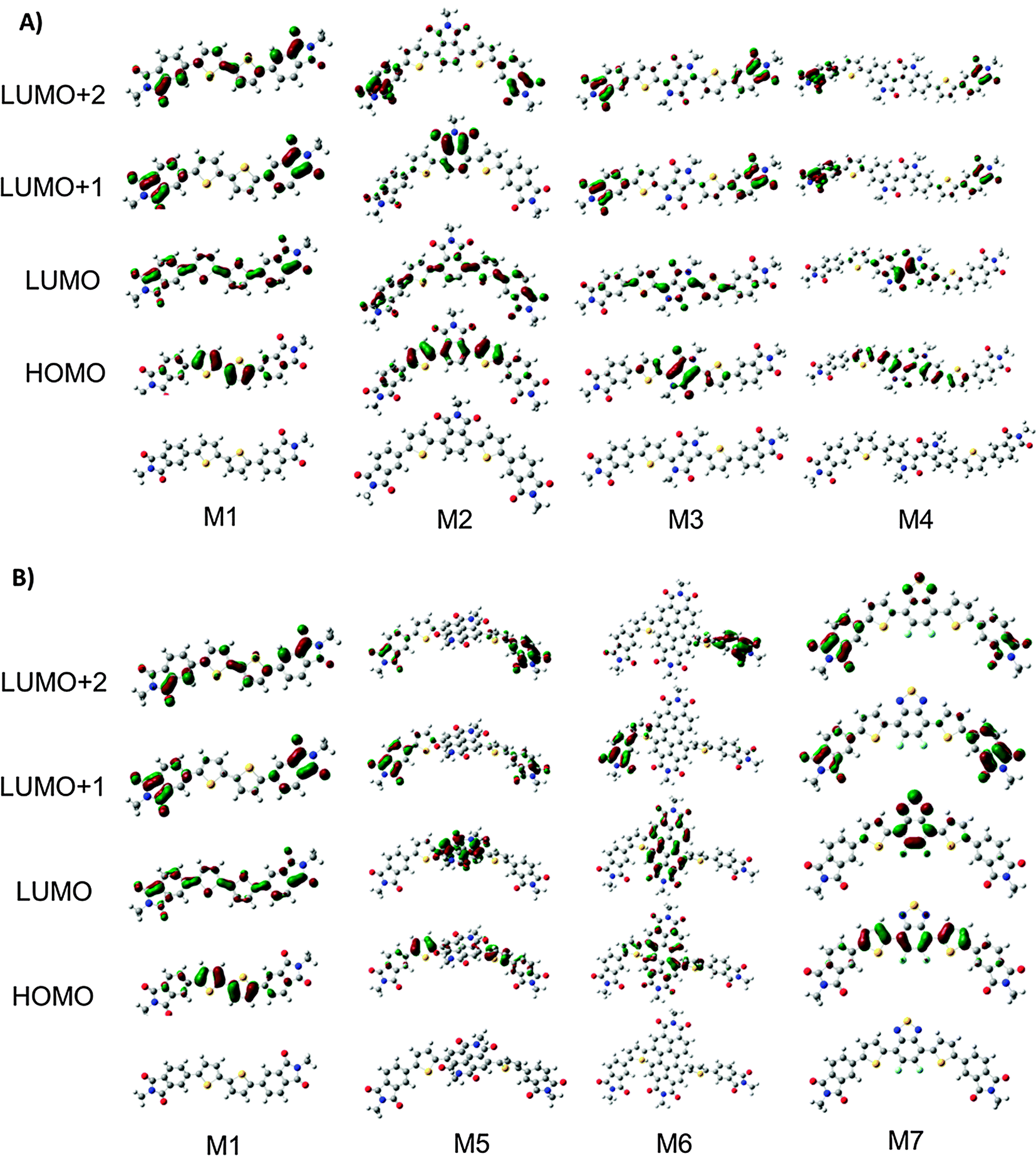 | ||
| Fig. 4 (A) Optimized structures and molecular orbital distributions for compounds M1–M4. (B) Optimized structures and molecular orbital distributions for compounds M1, M5–M7. | ||
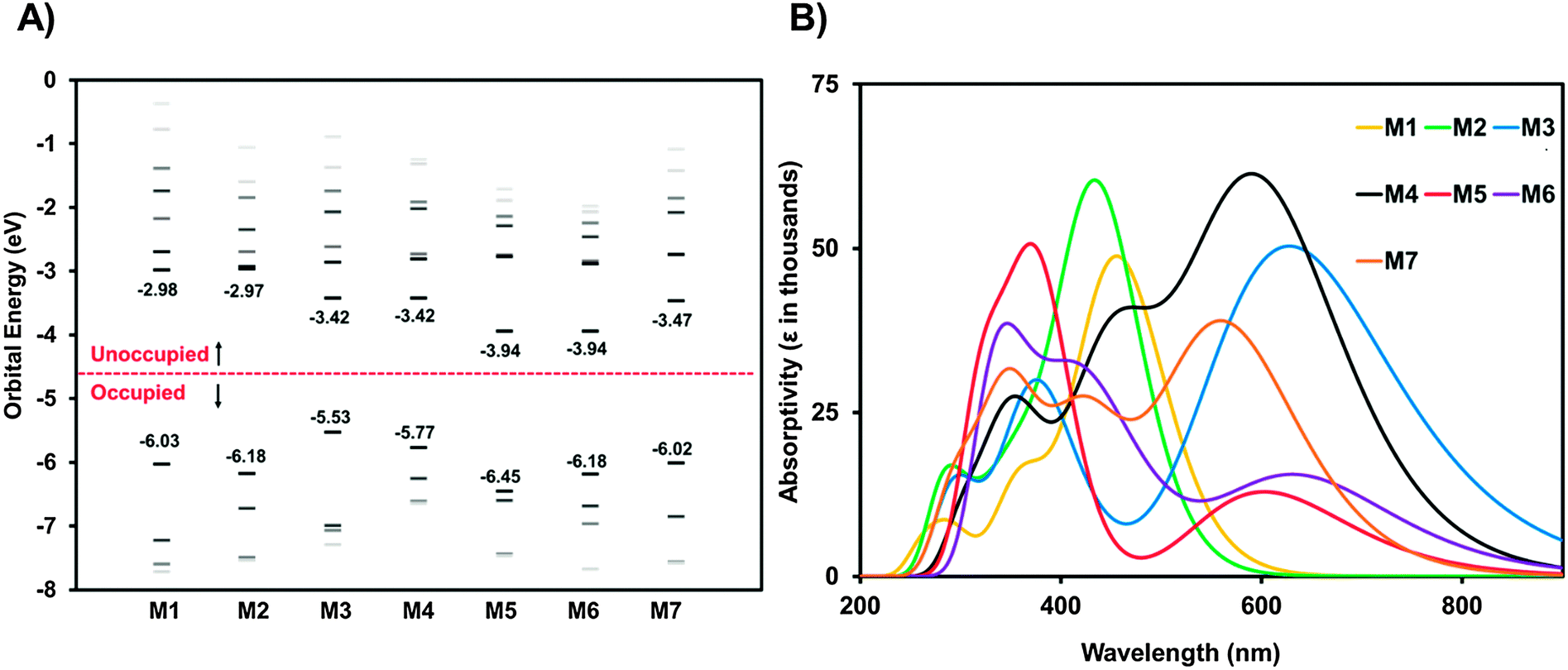 | ||
| Fig. 5 (A) Calculated energy level diagram for M1–M7. (B) Calculated absorption spectra for M1–M7 (gas phase). Absorption spectra are predicted from molecules in the gas phase modeled with DFT using B3LYP with the 6-31G(d) basis set. | ||
All compounds exhibit distortions from planarity along their respective π-conjugated backbones with compounds M1, M3, M4, and M7 having the most planar structures (Fig. 4, data tabulated in Table S3, ESI†). The imide based cores of M2, M5, and M6 cause a significant twist in the backbone due to ortho-hydrogen atom interactions. The HOMO and LUMO orbital distributions are shown in Fig. 5. Only for compounds M1 and M2 are the orbitals fully delocalized. Introduction of DPP (M3), II (M4), and F2BT (M7) results in both the HOMO and LUMO only being delocalized across the thiophene–core–thiophene portion of the molecules. For compounds M5 and M6 the high electron affinity of the NDI and PDI core along with the twisted backbone result in a complete localization of the LUMOs on the core and only partial delocalized HOMOs. These results can help explain the differences in optical absorption profiles (Fig. 2). M3 and M7 have near planar structures and favourable HOMO–LUMO overlap and both show λmax as the lowest energy transition. Compound M4 exhibits favourable HOMO–LUMO overlap but the distorted structure likely plays into the fact that both the low and high-energy bands in the optical absorption have similar intensity. For M5 and M6, the significant twisting of the π-conjugated backbone along with misalignment of electronic energy levels between the thiophene and core prevent strong HOMO–LUMO overlap resulting in weakly absorbing low energy bands. For M2, any increase in conjugation length afford by the phthalimide core is offset by disruption in conjugation, thus leading to the observed similar optical absorption spectra to M1. Indeed the predicted optical absorption profiles match with those determined experimentally. TD-DFT calculations were done on the optimized geometries with B3LYP/6-311+G(d). The calculated absorption spectra are shown in Fig. 5B with 0.3 eV FWHM for each transition. The TD-DFT calculations for M4 were done using the 6-31G(d) basis set. The electronic transitions for optimized structures of M1–M7 are tabulated in Table S5 (ESI†). The lowest energy transition for all is HOMO to LUMO with the exception of M2. For M2 the first excitation has a low oscillator strength (f = 0.099). The HOMO to LUMO for M2 is the second singlet state excitation, and has a higher oscillator strength (f = 1.2). The oscillator strength for the twisted M5 and M6 is much lower than the other compounds. Interestingly, the double peak absorption observed for M3 in solution is not reproduced theoretically, thus indicating a possible intermolecular effect.
It has recently been postulated that degeneracy of the LUMO energy levels and low reorganization energies upon one electron reduction are favourable properties for molecules intended to be used as electron acceptors.25 Thus, to further analyze these compounds and their potential to act as electron transporting materials in OPVs we investigated the energy of their frontier molecular orbitals (FMOs) in addition to the distortions caused to the aromatic backbone upon reduction. The frontier molecular orbital energies for all compounds from HOMO−3 to LUMO+6 are shown in Fig. 5A and tabulated in Table S4 (ESI†). For M1 the LUMOs are evenly spread out with none-being degenerate. Interestingly, M2 has nearly energetically degenerate LUMO and LUMO+1; however, the LUMO is delocalized across the molecule and the LUMO+1 is localized on the core. As the electron affinity of the core increases from M2–M7 the energy of the LUMO decreases and the gap between the LUMO and LUMO+1 increases. For M3–M7 the LUMO is localized on the core, indicating the energy of the LUMO is directly dictated by the electron affinity of the core. Conversely, the energy difference between LUMO+1 and LUMO+2 decreases from M1 to M7 with M6 showing the closest energy in these levels. Investigation of the MO distributions shows that in each case the core significantly contributes to the LUMO whereas the LUMO+1 and LUMO+2 are localized on the phthalimide end-capping units. In the extreme case of M6, the LUMO+1 and LUMO+2 are degenerate, and each resides on one phthalimide unit (Fig. 4). Thus in this series of molecules there are two competing effects determining the energy of the LUMOs. (1) Energy level alignment between the core and phthalimide end-capping unit. For M2, both the core and end-capping unit are phthalimide groups both having the same energy, thus resulting in similar energies for the LUMO and LUMO+1. As the electron affinity of the core increases from M2 to M7, the energy differences between the core and the end-capping phthalimide become greater, thus the LUMO becomes more localized on the core and the LUMO+1 on the phthalimide resulting in the greater energy differences between these molecular orbitals. (2) Twisting of the π-conjugated backbone. For all compounds the end-capping phthalimide groups contribute to the LUMO+1 and LUMO+2. From M3–M6, as the twisting of the π-conjugated backbone increases electronic communication between the two end-capping phthalimide groups decreases, thus localizing the LUMO+1 and LUMO+2 on one phthalimide group each, making them similar in energy. M7 is more planar, with good communication between phthalimide groups in the LUMO+1. The LUMO is localized on the core due to the high electron affinity from the fluorine groups. Based on these results, it is clear that none of these compounds poses a large density of states at the LUMO which may be detrimental to their ability to act as acceptors in BHJ type solar cells.
To examine the effect of reduction on M1–M7 the geometries of the reduced compounds were optimized (charge −1 doublet states). The change in bond length of a molecule upon addition of an electron in this manner corresponds to the delocalization of the injected electron (Fig. S24, ESI†).72 The smaller the magnitude of the change in bond length the higher the delocalization. Likewise, the location of the added electron can be determined by examining which bonds show the greatest change. Keeping this in mind and observing that for molecules M1 and M3 the bond length changes are roughly uniform across the entire conjugated backbone, we expect the injected electron to be delocalized across the entire backbone. For molecule M2 the bond length changes are greatest on the thiophene–phthalimide arms of the molecule indicating that the injected electron would be centred on the side arms of the molecule. For M4–M7 the greater bond length changes upon one electron injection occur on the conjugated core of the molecules, indicating that the electron would be localized on the core part of the molecules. Interestingly, the greatest changes in bond length for all molecules M1–M7 are approximately in line with the orbital distribution of the LUMO for each of M1–M7 (Fig. 4).
Single crystal X-ray diffraction study of M5
One of the well-recognized advantages of small molecule semiconductors compared to polymeric structures is that they are often more easily crystallized into large crystals suitable for single crystal X-ray diffraction. Single crystal diffraction experiments can lend insight towards the supramolecular organization of a molecular system and this in turn can lead to an understanding of the structure property relationships for this system. For both M5 and M6, the UV-visible profiles showed strong high-energy absorption bands and relatively weak low energy absorption bands. These molecules were further examined using DFT calculations where it was revealed that both M5 and M6 were expected to have large torsion angles between the core acceptor units and the thiophene–phthalimide side arms. Large torsion angles between the donor and acceptor parts of D–A type molecules can cause inefficient orbital mixing and therefore it might be expected that the low energy band that is attributed to intramolecular charge transfer would be relatively weak. It is also well known; however, that when π-conjugated molecules transition from the solution to the solid state they have a tendency to form more planar structures via π–π interactions between adjacent molecules.73–77 Therefore, we decided to investigate the single crystal structures of M5 and M6 to confirm their solid-state molecular geometry.Single crystals of M5 were grown by layering methanol carefully over a concentrated solution of M5 in CH2Cl2. Crystallization of M6 was unsuccessful. Small molecule M5 crystallized in the monoclinic space group C2/c as red needle-like crystals. Notably, the NDI core of each M5 unit is significantly twisted at ∼58° with respect to the thiophene–phthalimide arms (Fig. 6A). This was not unexpected, as the DFT calculations predicted a significant twisting of the flanking thiophene–phthalimide units and the UV-visible spectrum for this compound showed a distinctly weak absorption in the low energy region of the spectrum, indicating the possibility of inefficient orbital mixing which can occur due to loss of co-planarity between two adjacent conjugated fragments. The twisting of the side-arms with respect to the NDI core has been observed in other small molecules containing NDI and thiophene based building blocks, for example Patil et al. showed that NDI cores with benzofuran side arms display significant twisting of the core with respect to the side arms.78 For compound M5, there is a small torsion angle of 11.5° between the phthalimide and thiophene rings that flank the NDI core. Along the c axis direction of the unit cell (Fig. 6B) these thiophene phthalimide arms form π-stacked arrangements with a distance of 3.29 Å between stacked units. It can be seen as viewed down the c axis that the NDI cores do not lie atop of one another in a face-to-face fashion as the side arms do. Instead the NDI cores arranged in a ‘zig-zag’ fashion to one another, with an angle of ∼83° between the two different orientations for the NDI core (Fig. 6C and D). This packing arrangement is interesting because the NDI fragments are not interacting in a direct face-to-face fashion that is often seen in other single crystal studies of NDI based small molecules.56,79,80
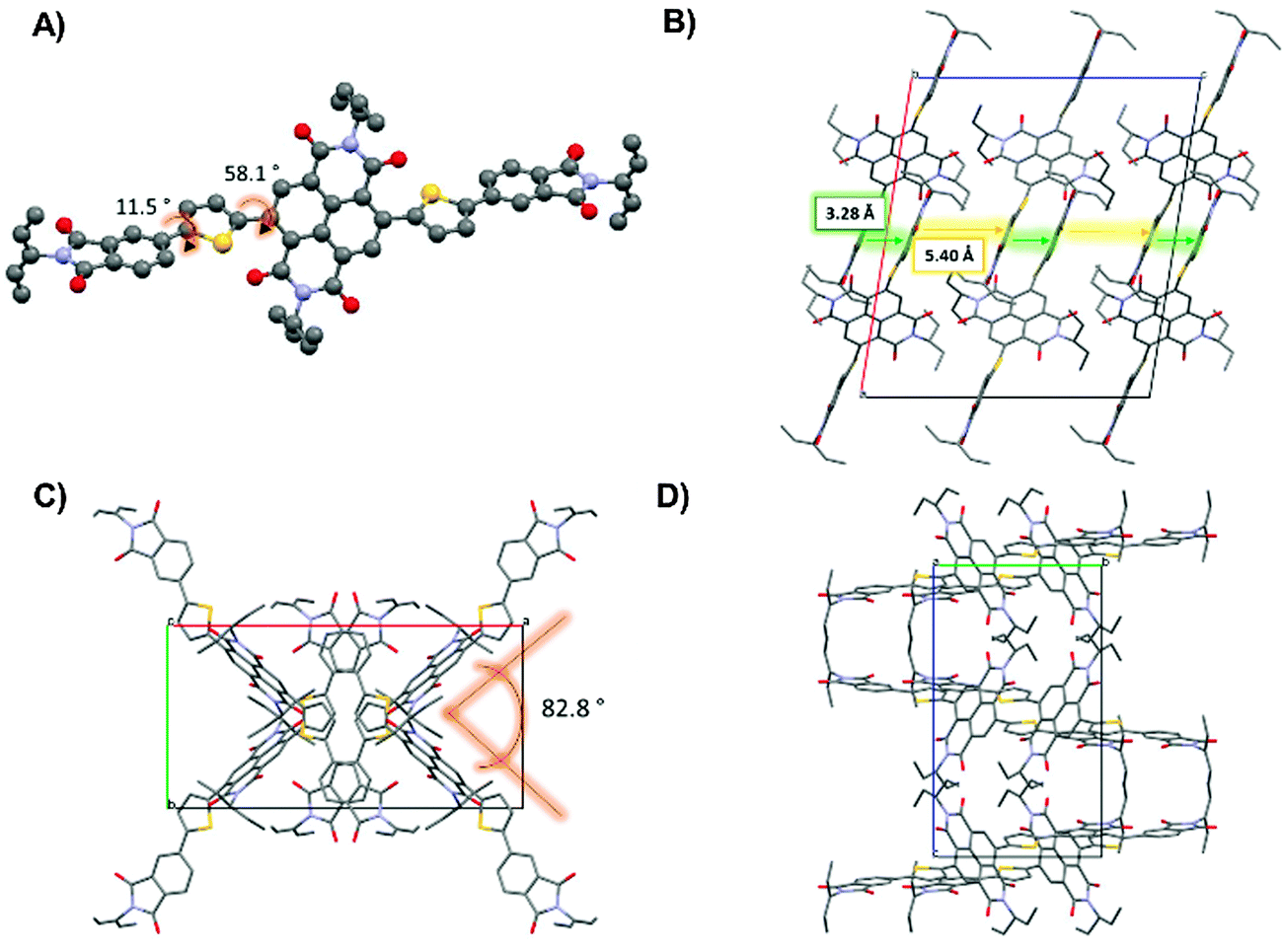 | ||
| Fig. 6 X-ray crystal structure of M5. (A) Single unit of M5 showing torsion angles. (B) Molecular packing as viewed along the ‘b’ axis of the unit cell showing π-stacking of the thiophene–phthalimide arms. (C) Viewed along the ‘c’ axis of the unit cell. (D) Viewed along the ‘a’ axis of the unit cell. | ||
Preliminary device characterization of small molecules M1–M7
Small molecules M1–M7 possess energy levels that make them appropriate candidates for electron transporting-light harvesting materials for use in small molecule BJH organic solar cells. In order to confirm their possible utility towards their application in BHJ–OPVs we conducted a preliminary screening of M1–M7 with two popular donor materials; DTS(FBTTh2)2 and P3HT.Previously, we have reported on DTS(FBTTh2)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M4 devices that showed the best performance when hot-cast at 90 °C from a 20 mg mL−1 solution (1:1 weight ratio) in chlorobenzene with 0.4% v/v DIO additive at 1000 rpm.39 To probe the photovoltaic performance of small molecules M1–M7, devices were made with the general architecture ITO/PEDOT:PSS/donor:acceptor/Ca/Al and using the above mentioned processing conditions for active layer formation. Both DTS(FBTTh2)2 and P3HT were investigated as the electron donating component; however, DIO was omitted for P3HT devices. Prior to cathode deposition films were annealed at 70 °C for 10 minutes to drive off residual solvents. Post-cathode-deposition annealing was also investigated for P3HT blend devices at 10 minute intervals. For P3HT blends with M3, M4, M5, and M6, post-cathode-deposition annealing resulted in reduced Voc and Jsc.
:M4 devices that showed the best performance when hot-cast at 90 °C from a 20 mg mL−1 solution (1:1 weight ratio) in chlorobenzene with 0.4% v/v DIO additive at 1000 rpm.39 To probe the photovoltaic performance of small molecules M1–M7, devices were made with the general architecture ITO/PEDOT:PSS/donor:acceptor/Ca/Al and using the above mentioned processing conditions for active layer formation. Both DTS(FBTTh2)2 and P3HT were investigated as the electron donating component; however, DIO was omitted for P3HT devices. Prior to cathode deposition films were annealed at 70 °C for 10 minutes to drive off residual solvents. Post-cathode-deposition annealing was also investigated for P3HT blend devices at 10 minute intervals. For P3HT blends with M3, M4, M5, and M6, post-cathode-deposition annealing resulted in reduced Voc and Jsc.
Fig. S29 (ESI†) shows the current density–voltage (J–V) plots for the as-fabricated DTS(FBTTh2)2 devices, and P3HT devices under the best annealing conditions. Device performance parameters are summarized in Table 2, with the best devices achieving power conversion efficiencies (PCEs) of ∼0.5%. The importance of ideal donor acceptor pairings is clearly illustrated in this work, a good example of this can be seen by comparing the devices made using M4 with each donor, for DTS(FBTTh2)2:M4 devices a modest PCE of 0.43% is achieved; however, for the P3HT:M4 devices a PCE that is essentially negligible was observed (Table 2). Overall the PCEs are significantly lower than related devices fabricated with fullerene acceptors37,39 but based on film morphology studies (see Fig. S31 and S32, ESI†) we believe the PCEs obtained for M1–M7 devices are currently limited by excessive phase separation. It is worthy to note; however, that molecule M1 with a relatively high-lying LUMO level of −3.03 eV achieved a Voc of 1.20 and 1.23 V in devices with DTS(FBTTh2)2 and P3HT, respectively. A plot of Vocvs. CV derived acceptor LUMO level is provided in Fig. 7. For both donors the Voc was not found to be strictly related to the energetic offset between donor HOMO and acceptor LUMO, although in general molecules with a larger offset displayed an expectedly larger Voc. This is important to note, because it clearly illustrates that other effects such as film morphology and donor–acceptor miscibility are more critical to the performance of the solar cell device. Fig. S26 (ESI†) shows the external quantum efficiency (EQE) plots of devices M1–M7 with both donors. The thin film absorbance of each donor is provided along with the EQE to demonstrate that channel II photocurrent is possible using several the new small molecules. For instance, in Fig. S26B (ESI†) it can clearly be seen that small molecule M3 contributes to the photocurrent generation of the device in the high (<450 nm) and low energy (>650 nm) regions of the visible spectrum where P3HT is not an effective light harvester. M1 also displays potential in this regard as it displays a contribution to the photocurrent generation in the high energy (<450 nm) region of the visible spectrum where P3HT has minimal absorbance. It is worth noting that EQE spectra are taken at very low illumination intensities (∼10−4 suns per monochromator spectral bandwidth), and tend to overstate the performance of devices where bimolecular recombination at 1 Sun is significant, as is likely the case of DTS(FBTTh2)2:M5 devices.
| Donor | Acceptor | Annealing temp. (°C) | V oc (V) | J sc (mA cm−2) | PCE (%) | FF | Active layer thickness (nm) |
|---|---|---|---|---|---|---|---|
| DTS(FBTTh2)2 | M1 | 70 | 1.20 | −0.0033 | 0.00051 | 0.13 | 120 |
| M2 | 70 | 0.86 | −0.028 | 0.0086 | 0.37 | 120 | |
| M3 | 70 | 0.92 | −0.64 | 0.27 | 0.46 | 110 | |
| M4 | 70 | 0.89 | −1.6 | 0.43 | 0.29 | 130 | |
| M5 | 70 | 0.72 | −1.3 | 0.15 | 0.17 | 120 | |
| M6 | 70 | 0.77 | −0.37 | 0.089 | 0.31 | 80 | |
| M7 | 70 | 0.99 | −0.58 | 0.17 | 0.30 | 110 | |
| P3HT | M1 | 130 | 1.23 | −1.2 | 0.46 | 0.32 | 150 |
| M2 | 130 | 0.92 | −1.7 | 0.50 | 0.31 | 140 | |
| M3 | 70 | 0.81 | −1.4 | 0.37 | 0.33 | 190 | |
| M4 | 70 | 0.63 | −0.15 | 0.027 | 0.28 | 120 | |
| M5 | 70 | 0.49 | −0.29 | 0.056 | 0.39 | 130 | |
| M6 | 70 | 0.78 | −0.23 | 0.054 | 0.30 | 150 | |
| M7 | 130 | 0.94 | −0.84 | 0.22 | 0.28 | 150 | |
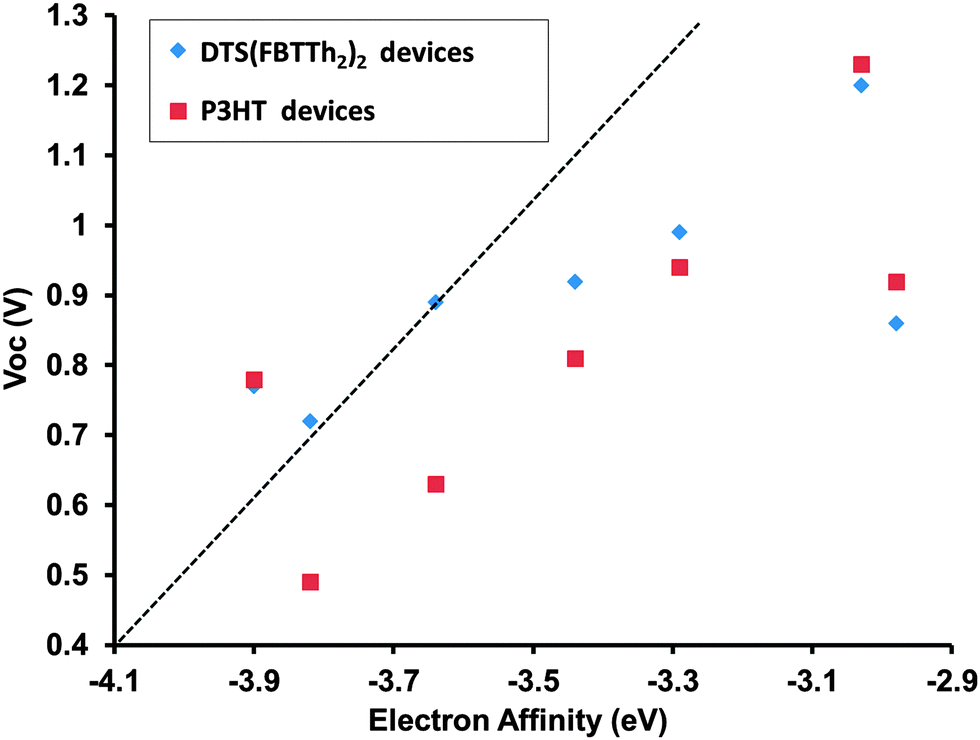 | ||
| Fig. 7 Acceptor LUMO vs. open circuit voltage of M1–M7 devices with DTS(FBTTh2)2 and P3HT as the donor. LUMO is estimated from CV EA. A line with slope = 1 V eV−1 is provided as a guide. | ||
Conclusions
In summary, we have presented a series of seven electron deficient small molecules with a Phth–Th–CORE–Th–Phth architecture. The resulting molecules had varying reduction potentials as calculated from the CV data, which could be tuned in the range of 2.98 to 3.90 eV, simply by exchanging the CORE component in the architecture. In addition, the different cores introduced had a strong impact on the optical, thermal and electronic properties of the series. The impacts of including each core were examined experimentally with theoretical results in good agreement. For instance, when DPP or isoindigo were included as cores a strong low energy absorption peak was observed, attributed to the strong donor–acceptor nature of the produced compounds. When phthalimide, NDI or PDI were included as cores; however, the effect of the donor–acceptor nature of the compounds was severely inhibited by steric interactions between the core units and side arms. This demonstrated that not only is the donor–acceptor nature of the compounds to be considered but also that the new geometries produced can have a strong impact on the properties of the new materials. Preliminary solar cell performance of M1–M7 was evaluated by screening each new acceptor with two different donor materials and it was found that the new materials were able to produce channel II photocurrent. It was also shown that for M1–M7 there was only a weak relation between the Voc obtained in solar cell devices and the energetic offset between donor HOMO and acceptor LUMO as determined using CV. The choice of donor acceptor pairings used in the bulk heterojunction had a strong effect on the performance of the devices, where some molecules showed similar performance with both donors, while others displayed functionality with only one donor or the other. Further investigation of this in relation to the concept of ideal donor acceptor pairings is underway in our research group.Acknowledgements
GCW acknowledges the NSERC Discovery Grants Program, Canada Research Chairs, and the Canadian Foundation for Innovation. IGH acknowledges support from the NSERC Discovery Grants program, the Canada Foundation for Innovation and the NSERC Photovoltaic Innovation Network. TM thanks Portland State University for funding. ADH is grateful for an NSERC scholarship. JPS acknowledges Killam Trusts, NSERC and NSERC CREATE DREAMS for financial support.References
- Z. Bao, Mater. Matters, 2007, 2, 4–6 CAS.
- C. J. Brabec and J. R. Durrant, MRS Bull., 2008, 33, 670–675 CrossRef CAS.
- H. E. Katz and J. Huang, Annu. Rev. Mater. Res., 2009, 39, 71–92 CrossRef CAS.
- J.-D. Chen, C. Cui, Y.-Q. Li, L. Zhou, Q.-D. Ou, C. Li, Y. Li and J.-X. Tang, Adv. Mater., 2015, 27, 1035–1041 CrossRef CAS PubMed.
- J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C.-C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446 CrossRef PubMed.
- C. Liu, C. Yi, K. Wang, Y. Yang, R. S. Bhatta, M. Tsige, S. Xiao and X. Gong, ACS Appl. Mater. Interfaces, 2015, 7, 4928–4935 CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293, DOI:10.1038/ncomms6293.
- Y. Fang, A. K. Pandey, D. M. Lyons, P. E. Shaw, S. E. Watkins, P. L. Burn, S.-C. Lo and P. Meredith, ChemPhysChem, 2014, 16, 1295–1304 CrossRef PubMed.
- K. Takimiya, I. Osaka and M. Nakano, Chem. Mater., 2014, 26, 587–593 CrossRef CAS.
- M. J. Robb, S.-Y. Ku, F. G. Brunetti and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1263–1271 CrossRef CAS PubMed.
- L. J. A. Koster, S. E. Shaheen and J. C. Hummelen, Adv. Energy Mater., 2012, 2, 1246–1253 CrossRef CAS PubMed.
- Y. Fang, A. K. Pandey, A. M. Nardes, N. Kopidakis, P. L. Burn and P. Meredith, Adv. Energy Mater., 2013, 3, 54–59 CrossRef CAS PubMed.
- Y. Zhang, A. K. Pandey, K. Tandy, G. K. Dutta, P. L. Burn, P. Meredith, E. B. Namdas and S. Patil, Appl. Phys. Lett., 2013, 102, 223302 CrossRef PubMed.
- Y. Lin, Z.-G. Zhang, H. Bai, J. Wang, Y. Yao, Y. Li, D. Zhu and X. Zhan, Energy Environ. Sci., 2015, 8, 610–616 CAS.
- Y. Liu, C. Mu, K. Jiang, J. Zhao, Y. Li, L. Zhang, Z. Li, J. Y. L. Lai, H. Hu, T. Ma, R. Hu, D. Yu, X. Huang, B. Z. Tang and H. Yan, Adv. Mater., 2015, 27, 1015–1020 CrossRef CAS PubMed.
- O. K. Kwon, J.-H. Park, D. W. Kim, S. K. Park and S. Y. Park, Adv. Mater., 2015, 27, 1951–1956 CrossRef CAS PubMed.
- J. T. Bloking, T. Giovenzana, A. T. Higgs, A. J. Ponec, E. T. Hoke, K. Vandewal, S. Ko, Z. Bao, A. Sellinger and M. D. McGehee, Adv. Energy Mater., 2014, 4, 1301426, DOI:10.1002/aenm.201301426.
- J. T. Bloking, X. Han, A. T. Higgs, J. P. Kastrop, L. Pandey, J. E. Norton, C. Risko, C. E. Chen, J.-L. Brédas, M. D. McGehee and A. Sellinger, Chem. Mater., 2011, 23, 5484–5490 CrossRef CAS.
- S. Holliday, R. S. Ashraf, C. B. Nielsen, M. Kirkus, J. A. Röhr, C.-H. Tan, E. Collado-Fregoso, A.-C. Knall, J. R. Durrant, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 898–904 CrossRef CAS PubMed.
- Y. Kim, C. E. Song, S.-J. Moon and E. Lim, Chem. Commun., 2014, 50, 8235–8238 RSC.
- K. N. Winzenberg, P. Kemppinen, F. H. Scholes, G. E. Collis, Y. Shu, T. Birendra Singh, A. Bilic, C. M. Forsyth and S. E. Watkins, Chem. Commun., 2013, 49, 6307–6309 RSC.
- Z. Zhu, Y. Bai, H. K. H. Lee, C. Mu, T. Zhang, L. Zhang, J. Wang, H. Yan, S. K. So and S. Yang, Adv. Funct. Mater., 2014, 24, 7357–7365 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- E. Ahmed, G. Ren, F. S. Kim, E. C. Hollenbeck and S. A. Jenekhe, Chem. Mater., 2011, 23, 4563–4577 CrossRef CAS.
- H. Li, T. Earmme, G. Ren, A. Saeki, S. Yoshikawa, N. M. Murari, S. Subramaniyan, M. J. Crane, S. Seki and S. A. Jenekhe, J. Am. Chem. Soc., 2014, 136, 14589–14597 CrossRef CAS PubMed.
- Y. Lin, Y. Wang, J. Wang, J. Hou, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2014, 26, 5137–5142 CrossRef CAS PubMed.
- W. Jiang, L. Ye, X. Li, C. Xiao, F. Tan, W. Zhao, J. Hou and Z. Wang, Chem. Commun., 2014, 50, 1024–1026 RSC.
- S. Rajaram, R. Shivanna, S. K. Kandappa and K. S. Narayan, J. Phys. Chem. Lett., 2012, 3, 2405–2408 CrossRef CAS.
- A. J. Kronemeijer, E. Gili, M. Shahid, J. Rivnay, A. Salleo, M. Heeney and H. Sirringhaus, Adv. Mater., 2012, 24, 1558–1565 CrossRef CAS PubMed.
- Y. Zang, C.-Z. Li, C.-C. Chueh, S. T. Williams, W. Jiang, Z.-H. Wang, J.-S. Yu and A. K.-Y. Jen, Adv. Mater., 2014, 26, 5708–5714 CrossRef CAS PubMed.
- P. Sonar, J. P. Fong Lim and K. L. Chan, Energy Environ. Sci., 2011, 4, 1558–1574 CAS.
- A. F. Eftaiha, J.-P. Sun, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2014, 2, 1201–1213 CAS.
- W. F. Fu Yu, Acta Chim. Sin., 2014, 72, 158–170 CrossRef.
- Y. Lin and X. Zhan, Mater. Horiz., 2014, 1, 470–488 RSC.
- S. M. McAfee, J. M. Topple, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2015 10.1039/C5TA04310G.
- J.-P. Sun, A. D. Hendsbee, A. F. Eftaiha, C. Macaulay, L. R. Rutledge, G. C. Welch and I. G. Hill, J. Mater. Chem. C, 2014, 2, 2612–2621 RSC.
- A. F. Eftaiha, J.-P. Sun, A. D. Hendsbee, C. Macaulay, I. G. Hill and G. C. Welch, Can. J. Chem., 2014, 92, 932–939 CrossRef CAS.
- A. D. Hendsbee, J.-P. Sun, L. R. Rutledge, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2014, 2, 4198–4207 CAS.
- S. M. McAfee, J. M. Topple, A.-J. Payne, J.-P. Sun, I. G. Hill and G. C. Welch, ChemPhysChem, 2015, 16, 1190–1202 CrossRef CAS PubMed.
- L. R. Rutledge, S. M. McAfee and G. C. Welch, J. Phys. Chem. A, 2014, 118, 7939–7951 CrossRef CAS PubMed.
- S. M. McAfee, J. S. J. McCahill, C. M. Macaulay, A. D. Hendsbee and G. C. Welch, RSC Adv., 2015, 5, 26097–26106 RSC.
- T. B. Singh, S. Erten, S. Günes, C. Zafer, G. Turkmen, B. Kuban, Y. Teoman, N. S. Sariciftci and S. Icli, Org. Electron., 2006, 7, 480–489 CrossRef CAS PubMed.
- A. Sharenko, C. M. Proctor, T. S. van der Poll, Z. B. Henson, T.-Q. Nguyen and G. C. Bazan, Adv. Mater., 2013, 25, 4403–4406 CrossRef CAS PubMed.
- R. Matsidik, J. Martin, S. Schmidt, J. Obermayer, F. Lombeck, F. Nübling, H. Komber, D. Fazzi and M. Sommer, J. Org. Chem., 2015, 80, 980–987 CrossRef CAS PubMed.
- J. Huang, C. Zhan, X. Zhang, Y. Zhao, Z. Lu, H. Jia, B. Jiang, J. Ye, S. Zhang, A. Tang, Y. Liu, Q. Pei and J. Yao, ACS Appl. Mater. Interfaces, 2013, 5, 2033–2039 CAS.
- P. Sonar, G.-M. Ng, T. T. Lin, A. Dodabalapur and Z.-K. Chen, J. Mater. Chem., 2010, 20, 3626–3636 RSC.
- K. H. Hendriks, G. H. L. Heintges, V. S. Gevaerts, M. M. Wienk and R. A. J. Janssen, Angew. Chem., Int. Ed., 2013, 52, 8341–8344 CrossRef CAS PubMed.
- Y. Ren, A. K. Hailey, A. M. Hiszpanski and Y.-L. Loo, Chem. Mater., 2014, 26, 6570–6577 CrossRef CAS PubMed.
- J. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Org. Lett., 2010, 12, 660–663 CrossRef CAS PubMed.
- J. Areephong, R. R. San Juan, A.-J. Payne and G. C. Welch, New J. Chem., 2015 10.1039/C5NJ01150G.
- E. Wang, W. Mammo and M. R. Andersson, Adv. Mater., 2014, 26, 1801–1826 CrossRef CAS PubMed.
- A. Luzio, D. Fazzi, F. Nübling, R. Matsidik, A. Straub, H. Komber, E. Giussani, S. E. Watkins, M. Barbatti, W. Thiel, E. Gann, L. Thomsen, C. R. McNeill, M. Caironi and M. Sommer, Chem. Mater., 2014, 26, 6233–6240 CrossRef CAS.
- T. Earmme, Y.-J. Hwang, N. M. Murari, S. Subramaniyan and S. A. Jenekhe, J. Am. Chem. Soc., 2013, 135, 14960–14963 CrossRef CAS PubMed.
- P. Piyakulawat, A. Keawprajak, A. Chindaduang, M. Hanusch and U. Asawapirom, Synth. Met., 2009, 159, 467–472 CrossRef CAS PubMed.
- J. Huang, Y. Wu, H. Fu, X. Zhan, J. Yao, S. Barlow and S. R. Marder, J. Phys. Chem. A, 2009, 113, 5039–5046 CrossRef CAS PubMed.
- L. M. Kozycz, D. Gao, A. J. Tilley and D. S. Seferos, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3337–3345 CrossRef CAS PubMed.
- B. Jiang, X. Zhang, C. Zhan, Z. Lu, J. Huang, X. Ding, S. He and J. Yao, Polym. Chem., 2013, 4, 4631–4638 RSC.
- W. S. Wong and A. Salleo, Flexible Electronics: Materials and Applications, Springer, 2009 Search PubMed.
- Y.-H. Lin, K. G. Yager, B. Stewart and R. Verduzco, Soft Matter, 2014, 10, 3817–3825 RSC.
- J.-L. Bredas, Mater. Horiz., 2014, 1, 17–19 RSC.
- C. L. Chochos, N. Tagmatarchis and V. G. Gregoriou, RSC Adv., 2013, 3, 7160–7181 RSC.
- J. Rawson, A. C. Stuart, W. You and M. J. Therien, J. Am. Chem. Soc., 2014, 136, 17561–17569 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- T. M. McCormick, C. R. Bridges, E. I. Carrera, P. M. DiCarmine, G. L. Gibson, J. Hollinger, L. M. Kozycz and D. S. Seferos, Macromolecules, 2013, 46, 3879–3886 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision D.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 5, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS PubMed.
- P. M. DiCarmine, T. B. Schon, T. M. McCormick, P. P. Klein and D. S. Seferos, J. Phys. Chem. C, 2014, 118, 8295–8307 CAS.
- T. Ghosh, A. Gopal, A. Saeki, S. Seki and V. C. Nair, Phys. Chem. Chem. Phys., 2015, 17, 10630–10639 RSC.
- M. Chandrasekharam, M. A. Reddy, K. Ganesh, G. D. Sharma, S. P. Singh and J. L. Rao, Org. Electron., 2014, 15, 2116–2125 CrossRef CAS PubMed.
- M. Scarongella, A. A. Paraecattil, E. Buchaca-Domingo, J. D. Douglas, S. Beaupre, T. McCarthy-Ward, M. Heeney, J.-E. Moser, M. Leclerc, J. M. J. Frechet, N. Stingelin and N. Banerji, J. Mater. Chem. A, 2014, 2, 6218–6230 CAS.
- R. Fitzner, C. Elschner, M. Weil, C. Uhrich, C. Körner, M. Riede, K. Leo, M. Pfeiffer, E. Reinold, E. Mena-Osteritz and P. Bäuerle, Adv. Mater., 2012, 24, 675–680 CrossRef CAS PubMed.
- M. J. Hollamby, M. Karny, P. H. H. Bomans, N. A. J. M. Sommerdjik, A. Saeki, S. Seki, H. Minamikawa, I. Grillo, B. R. Pauw, P. Brown, J. Eastoe, H. Möhwald and T. Nakanishi, Nat. Chem., 2014, 6, 690–696 CrossRef CAS PubMed.
- H. Patil, A. Gupta, A. Bilic, S. Jackson, K. Latham and S. Bhosale, J. Electron. Mater., 2014, 43, 3243–3254 CrossRef CAS.
- E. Ahmed, G. Ren, F. S. Kim, E. C. Hollenbeck and S. A. Jenekhe, Chem. Mater., 2011, 23, 4563–4577 CrossRef CAS.
- T. He, M. Stolte, C. Burschka, N. H. Hansen, T. Musiol, D. Kälblein, J. Pflaum, X. Tao, J. Brill and F. Würthner, Nat. Commun., 2015, 6, 5954, DOI:10.1038/ncomms6954.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, mass spectra, absorbance spectra, UPS spectra, EQE spectra, AFM images and current density vs. voltage curves. CCDC 1408228 (M5). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5tc01877c |
| This journal is © The Royal Society of Chemistry 2015 |