
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOpto-electronics of PbS quantum dot and narrow bandgap polymer blends†
Simon
Kahmann
ab,
Andrea
Mura
ac,
Loredana
Protesescu
de,
Maksym V.
Kovalenko
de,
Christoph J.
Brabec
bf and
Maria A.
Loi
*a
aZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, Groningen, NL-9747 AG, The Netherlands. E-mail: s.kahmann@rug.nl; m.a.loi@rug.nl
bInstitute of Materials for Electronics and Energy Technology (i-MEET), Friedrich-Alexander University Erlangen-Nürnberg, Martensstraße 7, D-91058 Erlangen, Germany. E-mail: christoph.brabec@fau.de
cDipartimento di Fisica, Università di Cagliari, I-09042 Monserrato (CA), Italy. E-mail: andrea.mura@dsf.unica.it
dDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir Prelog Weg 1, Zürich, CH-8093, Switzerland. E-mail: mvkovalenko@ethz.ch
eEMPA-Swiss Federal Laboratories for Materials Science and Technology, überlandstraße 129, Dübendorf, CH-8600, Switzerland
fBavarian Center for Applied Energy Research (ZAE Bayern), Haberstraße 2a, D-91058, Erlangen, Germany
First published on 28th April 2015
Abstract
Here we report on the interaction between the narrow bandgap polymer [2,6-(4,4-bis(2-ethylhexyl)-4H-cyclopenta-[2,1-b;3,4-b]dithiophene)-alt-4,7-(2,1,3-benzothiadiazole)] (PCPDTBT) and lead sulphide (PbS) colloidal quantum dots (CQDs) upon photoexcitation. We show that the presence of both materials in a blend leads to a significant reduction of photoluminescence (PL) lifetime of the polymer. This observation is attributed, supported by transient absorption (TA) data, to an efficient electron transfer towards the QDs for excitons generated on the polymer. Furthermore, the ligand capping the QD surface exhibits a great impact on the dynamics of the PL, with the long-chain oleic acid (OA) largely suppressing any kind of interaction. By means of external quantum efficiency (EQE) measurements we find evidence that both components give rise to a contribution to the photocurrent, making this an interesting blend for future applications in hybrid organic–inorganic solar cells.
1 Introduction
The so-called third generation of photovoltaic technologies aims at a reduction of manufacturing costs and material consumption, while maintaining high efficiencies. A plethora of new materials and concepts is currently investigated in order to make future products more efficient and less expensive. Amongst them organic photovoltaics (OPV) especially have taken a leap from the proof of concept of a heterojunction by Tang in 19861 up to 10.6% power conversion efficiency (PCE) in 2013.2 The relatively low permittivities of materials involved in organic solar cells demand an electron donor (D) and acceptor (A) couple to be applied in order to achieve an efficient photocurrent generation through separation of electron and holes. The most widely spread D–A combination consists of a fullerene derivative as acceptor and a polymer as donor, forming a type-II heterojunction. Since most acceptors tend to exhibit low extinction coefficients, it is common that just the donor component gives rise to a significant solar light harvesting in these blends.Colloidal quantum dot solar cells (QDSC) are a new class of devices that has been attracting interest for several years now. Here the active layer consists of a film of quantum dots – most commonly cadmium- or lead chalcogenides.3–8 The absorption profile of these QDs can be tuned due to the quantum size effect.9 Lead sulphide is of particular interest in QDSCs since its rather narrow bulk bandgap (0.41 eV) allows for harvesting photons in the near infrared region (IR) of the solar spectrum – a spectral region that is beyond reach for many other materials.
In order to fabricate working QDSCs the long insulating ligands (commonly oleic acid (OA)) that stabilise the QDs in solution, need to be exchanged for shorter entities, which establish a more efficient interconnection between the particles (without losing their confined nature). The exchange is often carried out as a solid state treatment involving thiol-containing short molecules such as 1,4-benzenedithiol (BDT), 1,2-ethanedithiol (EDT), or 3-mercaptopropionic acid (MPA) acting as bidentate linkers.
By intermixing both a narrow bandgap polymer with good performance in OPVs and QDs, one might overcome drawbacks of the two respective concepts.10–15 Namely, the rather poor absorption of the acceptor species in OPV may be improved by using lead chalcogenides. From the QDSC point of view, the addition of the polymer into the inorganic matrix might be able to suppress the formation of cracks upon exchanging the ligand, hence offering the possibility for large-scale compatible manufacturing techniques. Furthermore, the confinement of the QDs leads to the formation of a “valley” in the absorption profile which might be closed efficiently by adding a second absorbing component to the active layer.16
Here we report on the well-known narrow bandgap polymer PCPDTBT17 blended with PbS QDs and investigate their interaction upon photoexcitation using steady state and transient spectroscopy. To our knowledge this is the first investigation using this polymer with PbS QDs. We also include a variation of the ligand capping the QDs in order to elucidate the impact of these molecules on the system.
Our findings reveal that an efficient electron transfer from the polymer occurs upon exciton generation on the PCPDTBT, but only if the short ligand BDT is applied. In the same way evidence for a hole transfer can be found albeit less pronounced. No favourable interaction between the materials seems to occur when employing the longer oleic acid ligand.
2 Experimental
PbS QDs were synthesised analogously to previous reports.18 In short, a lead precursor PBAc2·H2O is dissolved in octadecene (ODE) and OA and heated to 145 °C. The sulphur precursor is also dissolved in ODE and rapidly injected into the lead-solution to evoke a burst nucleation event. After cooling the mixture down 4 washings are carried out and the QDs are eventually dissolved in chloroform. All employed QDs stem from the same synthesis batch. PCPDTBT was purchased from Konarka Technologies GmbH and used as received. All samples were cast under nitrogen atmosphere from solutions of 10 mg mL−1 concentration. Films comprising OA capped QDs were drop cast on a hot plate (approx. 60 °C) and both pristine PbS_BDT and polymer comprising films were fabricated via spin-coating in a layer-by-layer fashion. For the ligand exchange the cast layer was immersed in a solution of BDT in acetonitrile (2.85 mg mL−1) for 30 s. The film was spun dry at 4000 rpm for 60 s and the next layer deposited. A complete removal of OA ligands for this method has previously been confirmed.19,20 QD comprising films were annealed at 140 °C analogously to QDSCs.All samples for spectroscopy were cast on quartz substrates and sealed using a cover glass and epoxy glue in order to avoid oxygen exposure. Absorption spectra were recorded with a Shimadzu 3600 UV-vis-NIR spectrometer. For PL investigations the samples were excited at 400 nm by the second harmonic of a mode-locked Ti:sapphire (Mira 900) laser delivering pulses of 150 fs width, with a repetition rate of approximately 76 MHz. Steady-state spectra were recorded with an InGaAs detector from Andor. Time-resolved traces were taken with a Hamamatsu streak camera working in synchroscan mode for fast decaying samples. An optical pulse selector was used to vary the repetition rate of the exciting pulses for OA capped QDs and the streak camera was driven in the single sweep mode.
Transient absorption spectra were recorded using a super continuum laser source (SuperK Extreme, NKT Photonics) that provided both the pump and probe at a MHz repetition rate. The pulse duration was approximately 1 ps and the pump power was always kept below 4 μJ cm−2 in order to avoid, thermal effects, sample degradation and non-linear higher order processes. The signals were measured with an auto-balanced photoreceiver (Nirvana 2017, New Focus) and amplified with a Lock-In amplifier (SR 830 DSP, Standford Research Systems).21 All measurements were carried out at room temperature.
Solar cell active layers were deposited as explained above, with a thickness of approximately 150 nm on pre-patterned indium tin oxide (ITO) substrates. The devices were annealed at 140 °C for 10 min before thermal evaporation of 1 nm LiF and 100 nm Al as top contact. Device areas were defined by the overlap of the Al and ITO electrodes to be 16 mm2. External quantum efficiencies were measured near short circuit conditions using a 250 W quartz tungsten halogen lamp (6334NS, Newport) with lamp housing (67009, Newport). Monochromatic light was generated using narrow band pass filter (Thorlabs) with a full width at half maximum (FWHM) of 10 ± 2 nm from 400 to 1300 nm and a FWHM of 12 ± 2.4 nm from 1300 to 1400 nm. The light intensity was determined by calibrated photodiodes (PD300 and PD300IR, Ophir Optics).
3 Results
The molecular structure of PCPDTBT and the energy levels of the materials are depicted in Fig. 1.22,23 It is crucial to note that these levels are determined collectively, i.e. for the bulk polymer and an ensemble of dissolved OA capped QDs. The values may differ on a local level and the determination for the QDs involves a particular amount of uncertainty. Furthermore, whereas the bandgap is generally not affected by the ligands, it has been shown that they may induce a significant shift of the HOMO and LUMO energies.24 | ||
| Fig. 1 Energy levels and schemes of the employed materials. The interactions discussed here, i.e. electron, hole and energy transfer are depicted in blue. The two different ligands on the PbS surface, OA and BDT are shown on the right hand side. | ||
Nevertheless, when assuming the here depicted levels, one would expect a type-II heterojunction to form, albeit with a relatively small energy offset between the two HOMO levels. As depicted, there are three processes of interest which might give rise to efficient photocurrent generation. In the following it will be convenient to distinguish between photons absorbed by the polymer and those that are absorbed by the QDs. Both events offer the opportunity for a charge carrier transfer towards the other material, i.e. an electron transfer from the polymer towards the QDs and a hole transfer vice versa. Moreover, photons absorbed by the polymer may lead to a Förster-like energy transfer since the polymer PL and QD absorption overlap (vide infra).
Absorption spectroscopy
As a first step we investigated the absorption spectra of the pristine materials and blends. Their spectra are depicted in Fig. S1.† The QDs dissolved in chloroform exhibit a first transition located at 1022 nm and an increasing absorption at shorter wavelengths, as it is common for these quantum-confined particles.9 Using sizing curves previously determined, the QD diameter can be estimated to roughly 3.5 nm.23 For a film of QDs on glass (still capped with oleic acid, PbS_OA) the excitonic peak shifts to 1076 nm which is generally attributed to a change in the permittivity of the effective medium surrounding the QDs (our QD film exhibit a εr between 21–24,25 polymers typically lie between two and three). Upon exchanging the ligands from OA to the shorter bidentate linker BDT, the excitonic peak further shifts to the red. The decrease in energy is a consequence of both an improved coupling of the QDs, i.e. a delocalisation of the carriers' wavefunctions and a further increase in the permittivity of the effective medium surrounding the QDs. Furthermore, the width of the excitonic peak significantly increases when comparing the BDT capped film with the OA-capped QDs in solution, which we attribute to the increased degree of disorder in the former.The pristine polymer displays two absorption bands, a weaker one at short wavelengths (419 nm) and a more pronounced band ranging from roughly 600 to 840 nm. The latter coincides with the absorption valley of BDT capped QDs. Blending the two components (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 wt%; polymer:QDs) thus offers an increased absorbance in this area.
:9 wt%; polymer:QDs) thus offers an increased absorbance in this area.
Photoluminescence
Steady state PL spectra of both pristine and blended films are depicted in Fig. 2. The polymer (a) exhibits a rather broad emission with two distinct peaks (at 886 and 974 nm), analogously to the absorption in Fig. S1.† For the pristine PbS_BDT film (d) there is a rather symmetric emission with a peak at 1205 nm, i.e. on the low energy side of the polymer maximum emission, but coinciding with its low energy tail. The blend of these two components (b) exhibits a superposition of the emission of the pristine materials; a pronounced contribution of PbS on the low energy side and a smaller, but still visible, emission of PCPDTBT around 885 nm. Notably, the QD emission is blue-shifted by approximately 40 nm with the peak located at 1163 nm. This might be ascribed to the reduced permittivity of the effective medium surrounding the QDs. | ||
| Fig. 2 Steady state PL spectra of pristine PCPDTBT (a), PCPDTBT:PbS_BDT (b), PCPDTBT:PbS_OA (c), PbS_BDT (d), PbS_OA (e) normalised to the respective maxima. There are two distinct spectral regions of which one is characterised by the polymer emission (I) and the other mainly by the QD PL (II). | ||
A similar behaviour is observed for the spectra of the sample involving OA capped PbS. The blue-shift for the QD emission takes place from 1237 to 1179 nm upon blending with the polymer (c). Strikingly, the pristine PbS_OA film (e) offers a significantly narrower emission than the other samples – an entity generally associated with the disorder in a QD film (vide supra). We thus conclude that the degree of disorder induced by blending with the polymer and/or by cross-linking with BDT becomes larger than for an as-cast film of pristine PbS_OA.
In order to further elucidate the interaction between the two components, we measured the time resolved photoluminescence (TRPL) of the films above. In order to quantitatively determine the lifetimes and examine possible differences, we investigated the respective traces in the windows (I) and (II) indicated in Fig. 2. These data are depicted in Fig. 3 and 4. Fitting the decay of the pristine polymer's emission (Fig. 3, red) with a monoexponential function y = A1·eτ1/t reveals a lifetime of approximately τ1 = 118 ps, which is in good accordance with previous investigations involving this material.26 The blended sample with OA capped PbS (dark blue) offers a similar lifetime for the polymer (128 ps; a summary is given in Table 1). However, upon blending the polymer with QDs that are capped with the short BDT ligand (light blue), the lifetime is significantly reduced and must be fitted with a biexponential function y = A1·eτ1/t + A2·eτ2/t. By doing so, we obtain two fast components of approximately τ1 = 5 and τ2 = 75 ps of which the former approaches the resolution of our set-up in this configuration (about 4 ps). Upon exposing pristine PCPDTBT to BDT we do not observe any changes in lifetime (Fig. S2†) and thus conclude that the quenching arises from the presence of the QDs.
 | ||
| Fig. 3 TRPL in the polymer region (I) of a pristine PCPDTBT film and blends with PbS QDs. The pristine PbS_OA containg films can be fitted with a monoexponential decay whereas a biexponential fit is necessary for the BDT comprising blend. | ||
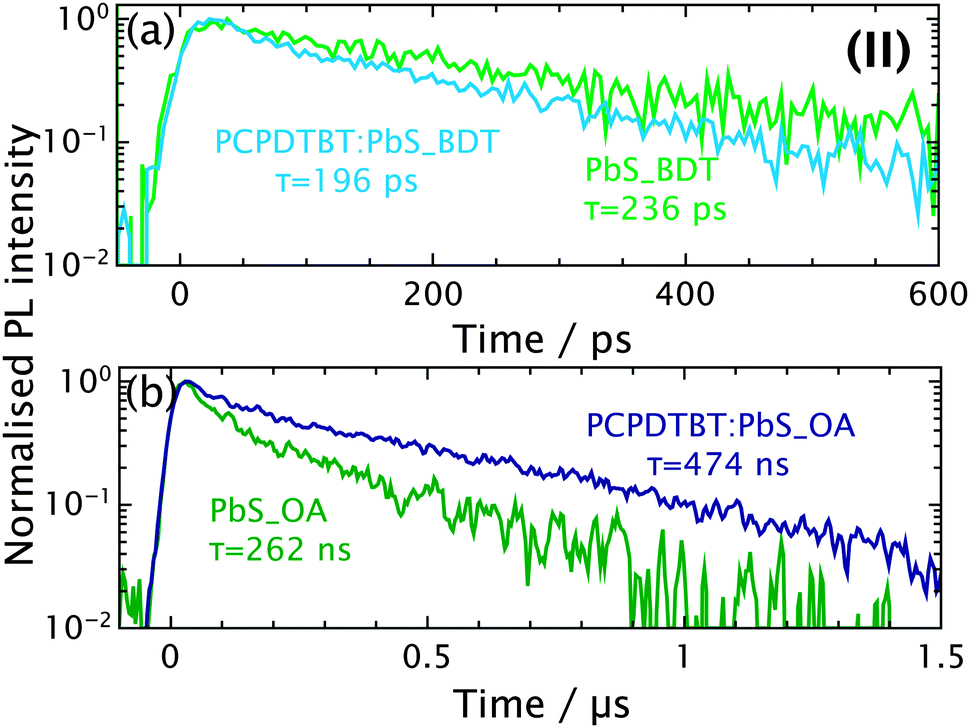 | ||
| Fig. 4 TRPL (II) of films comprising BDT (a) and OA (b) capped PbS. In order to suppress possible contributions from the polymer only monoexponential fits in the QD regime are considered. | ||
| Emission | Sample | λ max/nm | τ 1 | τ 2 |
|---|---|---|---|---|
| Polymer | Pristine | 886 | 118 ps | |
| Blend_OA | 891 | 128 ps | ||
| Blend_BDT | 885 | 5.0 ps | 75.3 ps | |
| QDs | Pristine_OA | 1237 | 45.7 ns | 275 ns |
| Blend_OA | 1179 | — | 474 ns | |
| Pristine_BDT | 1205 | — | 236 ps | |
| Blend_BDT | 1163 | — | 196 ps | |
The QD emission is depicted in Fig. 4. The lifetimes are longer than for the polymer. Here the difference between the pristine and the blended film is less pronounced.27 Fitting the decays of pristine PbS_BDT and of the PCPDTBT containing blend reveals lifetimes of 236 and 196 ps, respectively. Note that the initial decay of the QDs coincides with the polymer tail (vide supra), we therefore do not consider the early decay in order to suppress the polymer contribution (hence only one lifetime is given). The trend suggests a slight lifetime quenching for the blend. Conclusively, an interaction between the components appear present, albeit rather weak.
The OA capped QDs display significantly longer lifetimes than their BDT capped counterparts. This is a well-known phenomenon6 mostly attributed to either a reduced surface passivation or the improved carrier delocalisation when cross-linked; both phenomena reduce the probability of radiative decay. For a better comparison of the pristine and blended film, both spectra are plotted on a μs-range in Fig. 4(b). Curiously, the lifetime of the blended film exceeds the one of the neat QDs. This is in stark contrast to the observations for the short ligands above. Again, this might be evoked by both change in morphology and permittivity.
Transient absorption
Transient absorption (TA) can offer valuable insight into carrier dynamics. It may especially allow to determine which species of carrier is present in a system.From literature we know that hole polarons, singlet- and triplet-excitons can be tracked on PCPDTBT – all give rise to signals in a wavelength region around 1200 nm.28–31 We thus tracked the dynamics of pristine PCPDTBT, a blend with PbS_OA and one with PbS_BDT when pumping at 700 nm, i.e. in the maximum of the polymer absorption, and probing at 1200 nm. For samples of pristine PbS – both with OA and BDT – we could not observe any signal which is possibly due to the low excitation power we are limited to. We can thus assume that all signals discussed below derive from the polymer.
The decay for pristine PCPDTBT depicted in Fig. 5 (red) can be fitted with a monoexponential function and a lifetime of roughly 90 ps. This value is of the magnitude of the lifetime observed for the PL and suggests the measured signal to be the photo-induced absorption (PIA) originating from the polymer singlet exciton (S1 → Sn). Results from literature support this assumption.28,29,32 The same behaviour is obtained for the blend comprising OA capped QDs. Given the striking resemblance between these two samples (and their PL; Table 1) we conclude that the probed signal here is also the polymer singlet exciton.
 | ||
| Fig. 5 Transient absorption of respective films pumped at 700 nm and probed at 1200 nm. | ||
For the BDT capped PbS blended with PCPDTBT there is a strikingly different behaviour observable. The signal stays virtually constant within the measurement window of approximately 800 ps. Such a long lifetime signal could either be evoked by a triplet exciton or by a hole polaron on PCPDTBT.28–30,33 Though we cannot fully exclude the former, we have indirect evidence for this to be a polaron – namely the fact that we measured at room temperature and that under such conditions Etzold et al.29 needed a sensitiser to generate sufficient triplet population on PCPDTBT in a blend with PCBM. Finally, the pronounced contribution of the polymer in the EQE (Fig. 6) suggests an efficient charge carrier generation.
 | ||
| Fig. 6 External quantum efficiency of a pristine QD cell and a blend with PCPDTBT (1:9) (lines with symbols) and the respective absorption spectra (solid lines). The inset depicts the employed device architecture with the active layer (AL) sandwiched between the ITO and LiF/Al electrode. | ||
Solar cells
Based on the observations above, it is promising to use a blend of PCPDTBT with BDT capped PbS as active layer (AL) of a solar cell. We fabricated cells in the simple Schottky-like architecture (vide inset of Fig. 6) which has previously lead our group to achieve power conversion efficiencies of up to 5.2% employing pristine PbS.19 The measured external quantum efficiencies of both types of cells are depicted in Fig. 6 (for the J/V curves and electrical parameters vide Fig. S3 and Table S1†) where the black curve, referring to the QD-only device, exhibits a pronounced contribution of the excitonic peak centred around 1050 nm and a steep increase at shorter wavelengths ultimately exceeding 60% at 420 nm. Furthermore, a dip in the intermediate spectral range is observable, roughly between 700 and 950 nm. The blended device exhibits a pronounced increase of EQE in this spectral range analogously to the absorption spectrum of the pristine polymer (Fig. S1†). Consequently, both materials take part in the photocurrent generation.A significant drawback, however, arises when considering the spectral regions of pronounced QD contribution. Adding the polymer leads to a reduction of EQE for the QDs' first excitonic peak and at shorter wavelengths. The extent cannot be justified by the difference in QD content (the QD content in the blend should amount to roughly 90 wt%). A possible explanation for this might be the increased disorder in the QD phase (vide supra) that may hamper the carrier transport. Additionally, we find evidence that upon annealing a polymer rich layer forms near the LiF/Al backcontact (vide Fig. S4†). Such a layer could reduce the efficiency of electron extraction. The improvement in the intermediate spectral region is hence accompanied and compensated by a reduction of the QD contribution. Calculating the theoretically achievable JSC leads to 11.06 and 11.15 mA cm−2 for the pristine and the blended device, respectively.
4 Discussion
We sought to elucidate the behaviour of PCPDTBT and PbS QDs blended in a film upon photoexcitation for the eventual use in hybrid organic–inorganic solar cells. In the following, all possible steps from the absorption of an incident photon up to the successful extraction of a free carrier at an electrode shall be discussed.Starting from photons absorbed by the polymer, i.e. predominantly from approximately 650 to 840 nm, there are several processes possible. For a pristine film of PCPDTBT it is known that photons lead to the formation of strongly bound Frenkel excitons with a typical diffusion length of about 10 nm.34 These excitons percolate within the material and eventually recombine either radiatively or non-radiatively. Bringing the polymer chains into a blend with the QDs could lead to a charge transfer, i.e. an electron may be transferred to the lower lying LUMO level of the QDs – thereby leaving a free hole in the organic phase. This interaction is the prototypical donor–acceptor interaction which should provide the best performance in solar cells. Furthermore, given the overlap of polymer fluorescence and QD absorption, an energy transfer between the components might also be possible. Finally, blending the materials may lead to an increased (non-favourable) recombination of excitons, e.g. via surface traps of the QDs.
Though the latter cannot fully be excluded, the magnitude of the polymer contribution to the photocurrent depicted in the EQE (Fig. 6) suggests that this is by far not a dominant process for BDT capped QDs.
Both the energy (E) and electron (e−) transfer should lead to a reduction of the PL lifetime of the polymer. As can be seen from the respective TRPL depicted in Fig. 3, there is a significant quenching of the PL lifetime for PCPDTBT when BDT is applied. In presence of OA capped PbS the lifetime is essentially identical and the TA absorption in Fig. 5 also reveals the same spectra for pristine PCPDBT and the blend comprising PbS_OA. We thus exclude transfers between these two phases.
When considering the BDT capped QDs, the behaviour is strikingly different. The reduction in PL lifetime of the polymer and the presence of its EQE signal adduces evidence for the successful extraction of charge carriers that arose from polymer excitations. In order to distinguish between E- or e−-transfer we find evidence via TA as depicted in Fig. 5. There is a clear difference for the behaviour of pristine PCPDTBT and when blended with PbS_BDT. The blend exhibits a signal that virtually stays constant over the observed time range. This can be attributed to the formation of long-lived hole polarons on the polymer28,30 and thereby proves the presence of a charge transfer in this system.
For the case of a photon absorbed by PbS, which is predominantly the case for wavelengths shorter than approximately 650 or longer than 800 nm (vide Fig. S1†), several further steps are possible: initially, the photon will lead to the formation of an exciton on the same QD it was absorbed by. Depending mostly on the nano-morphology this might either lead to a dissociation of the exciton into free charge carriers promoted by thermal energy or the exciton can percolate within the material until it either recombines or reaches an interface. For long chain ligands such as OA it is generally assumed that bound excitons form.35 In this case the exciton would have to diffuse towards an interface in order to dissociate.
The energy levels depicted in Fig. 1 allow for a hole transfer from the QDs. This, however, should lead to a reduction of the lifetime of the QDs PL (and the formation of a signal in the TA spectra) which we do not observe. On the contrary, the PL lifetime of the PbS_OA QDs is even prolonged in presence of the polymer (Fig. 4 bottom). We therefore conclude that for photons absorbed by PbS_OA there is no favourable interaction between the two materials.
A possible explanation for the prolonged lifetime in the blend might be due to the reduced permittivity. Following the explanation of Wehrenberg et al.36 the lifetime of a spherical particle is proportional to  with the (optical) permittivity ε of the particle p and the host h. Reducing the matrix' permittivity by introducing the polymer thus leads to an increase in the radiative lifetime. It should be considered however, that this theory is strictly only valid for isolated particles in a homogeneous matrix.
with the (optical) permittivity ε of the particle p and the host h. Reducing the matrix' permittivity by introducing the polymer thus leads to an increase in the radiative lifetime. It should be considered however, that this theory is strictly only valid for isolated particles in a homogeneous matrix.
The same pathways are possible for BDT capped QDs. In this case we find a slightly reduced PL lifetime of the blend in comparison to the pristine film (Fig. 4 top) that suggests a charge transfer. It is worth mentioning here that the QD content of the film strongly exceed the amount of polymer which leads to a plethora of quenching sites for polymer excitons, whereas the smaller impact on the QD PL might be due to a lack of sufficient quenching sites. Therefore a bulk of QD excitations might lead to the same processes occurring in a pristine QD film. From above considerations for photons absorbed in pristine PbS_BDT, we know that the QDs there act as both the hole and electron conducting phase. Obviously, the QDs also act as the electron transporting phase in the case of absorption by the polymer in a blend, but the question remains how the hole is driven towards the electrode. The TA measurement offers important information on this question, namely the long lifetime of the hole polaron on the polymer. If there were any second transfer, i.e. a successive hole transfer towards the QDs, the polymer polaron signal should decay on a relatively short time range. Since this is not the case we conclude that the lifetime is sufficient to allow the holes to percolate towards the electrodes via the polymer phase.
For absorption by the QDs the electron again is transported via the particles. Whether the hole also remains on the QDs cannot fully be answered – we cannot exclude it.
5 Conclusion
We investigated the optical properties and charge carrier dynamics for a hybrid organic–inorganic system comprising a narrow bandgap polymer and lead sulphide quantum dots. We observed that their interaction upon photoexcitation strongly depends on both the initially absorbing component and the ligand capping the QDs.For light absorbed by OA capped QDs we could not track any interaction with the polymer phase which we attribute to the ligand length that successfully suppresses any transfer. When applying the shorter BDT, we observe a reduced lifetime which we attribute to a hole transfer. Nevertheless, given the unequal amounts of QDs and polymer in the blend we note that there are probably many holes that are not transferred to the polymer. Carrier transport following photon absorption by QDs thus predominantly follows the behaviour of a pristine QD film. Since, however, the degree of disorder is increased, the EQE in the spectral regions of pronounced QD absorption is reduced compared to a pristine film.
For photons absorbed by the polymer we found essentially the same behaviour, but more pronounced. For BDT we observed a significant quenching of the PL lifetime and a TA signal of a long-lived species, which we ascribed to an efficient exciton dissociation at the heterointerface leading to the formation of a hole polaron on the polymer and an electron transferred to the QD phase. Accordingly, we found both components to offer a contribution to the EQE.
Given that fabricated solar cells did not outperform their pristine inorganic counterparts, we conclude that although there is a promising interaction between the two materials, further effort is required to optimise the film deposition for efficient devices.
Acknowledgements
Widianta Gomulya and Tayebeh Ameri are kindly thanked for helpful discussions. Arjen Kamp is greatly acknowledged for technical support.References
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183 CrossRef CAS PubMed.
- J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C.-C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446 CrossRef PubMed.
- A. H. Ip, S. M. Thon, S. Hoogland, O. Voznyy, D. Zhitomirsky, R. Debnath, L. Levina, L. R. Rollny, G. H. Carey, A. Fischer, K. W. Kemp, I. J. Kramer, Z. Ning, A. J. Labelle, K. W. Chou, A. Amassian and E. H. Sargent, Nat. Nanotechnol., 2012, 7, 577–582 CrossRef CAS PubMed.
- J. M. Luther, M. Law, M. C. Beard, Q. Song, M. O. Reese, R. J. Ellingson and A. J. Nozik, Nano Lett., 2008, 8, 3488–3492 CrossRef CAS PubMed.
- B. Ehrler, K. P. Musselman, M. L. Böhm, F. S. F. Morgenstern, Y. Vaynzof, B. J. Walker, J. L. Macmanus-Driscoll and N. C. Greenham, ACS Nano, 2013, 7, 4210–4220 CrossRef CAS PubMed.
- K. Szendrei, M. Speirs, W. Gomulya, D. Jarzab, M. Manca, O. V. Mikhnenko, M. Yarema, B. J. Kooi, W. Heiss and M. A. Loi, Adv. Funct. Mater., 2012, 22, 1598–1605 CrossRef CAS PubMed.
- D. C. J. Neo, C. Cheng, S. D. Stranks, S. M. Fairclough, J. S. Kim, A. I. Kirkland, J. M. Smith, H. J. Snaith, H. E. Assender and A. A. R. Watt, Chem. Mater., 2014, 26, 4004–4013 CrossRef CAS.
- C.-H. M. Chuang, P. R. Brown, V. Bulović and M. G. Bawendi, Nat. Mater., 2014, 13, 796–801 CrossRef CAS PubMed.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CAS.
- C. Piliego, M. Manca, R. Kroon, M. Yarema, K. Szendrei, M. R. Andersson, W. Heiss and M. A. Loi, J. Mater. Chem., 2012, 22, 24411 RSC.
- H.-Y. Chen, J. Hou, S. Dayal, L. Huo, N. Kopidakis, M. C. Beard and J. M. Luther, Adv. Energy Mater., 2011, 1, 528–533 CrossRef CAS PubMed.
- Z. Liu, Y. Sun, J. Yuan, H. Wei, X. Huang, L. Han, W. Wang, H. Wang and W. Ma, Adv. Mater., 2013, 25, 5772–5778 CrossRef CAS PubMed.
- J. Yuan, A. Gallagher, Z. Liu, Y. Sun and W. Ma, J. Mater. Chem. A, 2015, 3, 2572–2579 CAS.
- C. Giansante, R. Mastria, G. Lerario, L. Moretti, I. Kriegel, F. Scotognella, G. Lanzani, S. Carallo, M. Esposito, M. Biasiucci, A. Rizzo and G. Gigli, Adv. Funct. Mater., 2014, 25, 111–119 CrossRef PubMed.
- L. Yang, M. Tabachnyk, S. L. Bayliss, M. L. Bo, K. Broch, N. C. Greenham, R. H. Friend and B. Ehrler, Nano Lett., 2014, 15, 354–358 CrossRef PubMed.
- D. T. Moore, B. Gaskey, A. Robbins and T. Hanrath, J. Appl. Phys., 2014, 115, 054313 CrossRef PubMed.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed.
- L.-H. Lai, L. Protesescu, M. V. Kovalenko and M. A. Loi, Phys. Chem. Chem. Phys., 2014, 16, 736–742 RSC.
- C. Piliego, L. Protesescu, S. Z. Bisri, M. V. Kovalenko and M. A. Loi, Energy Environ. Sci., 2013, 6, 3054 CAS.
- K. Szendrei, W. Gomulya, M. Yarema, W. Heiss and M. A. Loi, Appl. Phys. Lett., 2010, 97, 203501 CrossRef PubMed.
- Additional information to the TA set-up will be published elsewhere.
- T. Ameri, P. Khoram, J. Min and C. J. Brabec, Adv. Mater., 2013, 25, 4245–4266 CrossRef CAS PubMed.
- G. Itskos, P. Papagiorgis, D. Tsokkou, A. Othonos, F. Hermerschmidt, S. P. Economopoulos, M. Yarema, W. Heiss and S. Choulis, Adv. Energy Mater., 2013, 3, 1490–1499 CrossRef CAS PubMed.
- P. R. Brown, D. Kim, R. R. Lunt, N. Zhao, M. G. Bawendi, J. C. Grossman and V. Bulovic, ACS Nano, 2014, 8, 5863–5872 CrossRef CAS PubMed.
- M. J. Speirs, D. M. Balazs, H.-H. Fang, L.-H. Lai, L. Protesescu, M. Kovalenko and M. A. Loi, J. Mater. Chem. A, 2014, 3, 1–8 Search PubMed.
- D. Jarzab, F. Cordella, J. Gao, M. Scharber, H.-J. Egelhaaf and M. A. Loi, Adv. Energy Mater., 2011, 1, 604–609 CrossRef CAS PubMed.
- D. Jarzab, K. Szendrei, M. Yarema, S. Pichler, W. Heiss and M. A. Loi, Adv. Funct. Mater., 2011, 21, 1988–1992 CrossRef CAS PubMed.
- I.-W. Hwang, C. Soci, D. Moses, Z. Zhu, D. Waller, R. Gaudiana, C. J. Brabec and A. J. Heeger, Adv. Mater., 2007, 19, 2307–2312 CrossRef CAS PubMed.
- F. Etzold, I. A. Howard, N. Forler, D. M. Cho, M. Meister, H. Mangold, J. Shu, M. R. Hansen, K. Müllen and F. Laquai, J. Am. Chem. Soc., 2012, 134, 10569–10583 CrossRef CAS PubMed.
- E. Couderc, M. J. Greaney, R. L. Brutchey and S. E. Bradforth, J. Am. Chem. Soc., 2013, 135, 18418–18426 CrossRef CAS PubMed.
- G. Grancini, M. Maiuri, D. Fazzi, A. Petrozza, H.-J. Egelhaaf, D. Brida, G. Cerullo and G. Lanzani, Nat. Mater., 2013, 12, 29–33 CrossRef CAS PubMed.
- G. Grancini, M. De Bastiani, N. Martino, D. Fazzi, H.-J. Egelhaaf, T. Sauermann, M. R. Antognazza, G. Lanzani, M. Caironi, L. Franco and A. Petrozza, Phys. Chem. Chem. Phys., 2014, 16, 8294–8300 RSC.
- M. Koppe, H.-J. Egelhaaf, G. Dennler, M. C. Scharber, C. J. Brabec, P. Schilinsky and C. N. Hoth, Adv. Funct. Mater., 2010, 20, 338–346 CrossRef CAS PubMed.
- O. V. Mikhnenko, H. Azimi, M. Scharber, M. Morana, P. W. M. Blom and M. A. Loi, Energy Environ. Sci., 2012, 5, 6960 CAS.
- S. Crooker, J. Hollingsworth, S. Tretiak and V. Klimov, Phys. Rev. Lett., 2002, 89, 186802 CrossRef CAS.
- B. L. Wehrenberg, C. Wang and P. Guyot-Sionnest, J. Phys. Chem. B, 2002, 106, 10634–10640 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S4. See DOI: 10.1039/c5tc00754b |
| This journal is © The Royal Society of Chemistry 2015 |