
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of the structure–property relationship of thiadiazoloquinoxaline-based copolymer semiconductors via molecular engineering†
Cunbin
An
,
Mengmeng
Li
,
Tomasz
Marszalek
,
Xin
Guo
 ,
Wojciech
Pisula
and
Martin
Baumgarten
*
,
Wojciech
Pisula
and
Martin
Baumgarten
*
Max Planck Institute for Polymer Research, Ackermannweg 10, Mainz, D-55128, Germany. E-mail: martin.baumgarten@mpip-mainz.mpg.de
First published on 6th March 2015
Abstract
Six thiadiazoloquinoxaline (TQ) based copolymers (P1–P6) have been synthesized using Stille coupling reaction upon varying donor moieties, substitution positions and architectures of polymer side chains. UV-vis-NIR absorption spectra indicated that all of these polymers exhibited low optical bandgaps from 0.96 to 0.75 eV. The electron affinities of these six polymers with values from −3.92 to −3.99 eV slightly changed due to the contribution of the same TQ core. However, the ionization potentials were tuned from −4.95 eV in P1 to −5.28 eV in P3 by introducing different donors. Comparing polymers P1–P4 with different donor parts, two dimensional wide-angle X-ray scattering measurements implied that P4 had a higher crystallinity because its coherence length (5.8 nm) was 2–3 times higher than those of P1–P3 (1.7–2.9 nm). This led to a best field-effect hole mobility of 0.1 cm2 V−1 s−1 for P4 among the four polymers. The polymers P4–P6 had identical molecular formulae of side chains but significant differences in device performance. In comparison with P4, the two linear alkyl chains were moved onto head to head positions of bithiophene in P5, resulting in a hole mobility of only 0.007 cm2 V−1 s−1. However, a pair of 2-decyl-tetradecyl alkyl chains was grafted onto thiophene units adjacent to the TQ core in P6, leading to the highest hole mobility up to 0.24 cm2 V−1 s−1 in this series of polymers.
Introduction
Donor–acceptor (D–A) alternating π-conjugated copolymers have attracted attention in organic light-emitting diodes (OLEDs), polymer solar cells (PSCs) and organic field-effect transistors (OFETs).1–4 Combination of different D and A units can easily tune optoelectronic properties, thereby also affecting the device performances of the D–A copolymers. Investigation of the structure–property relationship of such polymers is an important method to understand and design high-performance optoelectronic materials.5,6 Some strong acceptors have been reported, like diketopyrrolopyrrole (DPP),7–9 naphthalene diimide (NDI),10–12 benzobisthiadiazole (BBT)13 and isoindigo (IID).14,15 Thiadiazoloquinoxalines (TQs) possess outstanding electron affinity and variable chemical structures,16–20 thereby becoming a class of important building blocks for polymer semiconductors. However, TQ polymers usually exhibit relatively low FET performances due to their disorder in thin films.21–25 It is therefore necessary to systematically study the structure and charge transport correlation of TQ polymers in order to identify the essential parameters for highly ordered microstructures which could further improve their charge carrier mobility.A TQ derivative, 6,7-diphenyl-[1,2,5]thiadiazolo[3,4-g]quinoxaline (PTQ, Fig. 1), has been widely investigated as an acceptor for creating polymer semiconductors, because of their ease of synthesis and good charge carrier mobilities compared to other TQ derivatives.26–28 A D–A–D model of PTQ, 6,7-diphenyl-4,9-di(thiophen-2-yl)-[1,2,5]thiadiazolo[3,4-g]quinoxaline (TQ-2T, Fig. 1), was also reported by modifying the PTQ core with two flanking thiophenes, which can offer more alkylation positions to improve their solubility.24,29–32 In view of the lack of study on the structure–property relationship of TQ polymers, we herein investigate this topic based on TQ-2T polymers from two crucial aspects. Firstly, introduction of unsubstituted donor units with different electron-donating abilities, which enhances the backbone coplanarity of the TQ-2T polymers and potentially leads to ordered arrangement in the polymer films. Secondly, tuning substitution positions and architectures of the alkyl chains in the polymer repeating units improves the polymer solubility, the molecular weights as well as the device performance.
 | ||
| Fig. 1 Structures of PTQ and TQ-2T reported in literature. | ||
In this work, we report six new TQ-2T based copolymers P1–P6 (Fig. 2). Different unsubstituted donors, namely, thiophene (T), thieno[3,2-b]thiophene (TT), benzo[1,2-b:4,5-b′]dithiophene (BDT) and 2,2′-bithiophene (BT) were introduced alternatingly with TQ-2T providing the corresponding polymers P1–P4. These donor units are expected to decrease the torsion angles along the polymer backbone. P5 was designed and synthesized for comparison with P4 by varying the substitution positions of the alkyl chains. This design aims to improve the solubility and molecular weight but at the cost of sacrificing coplanarity of the polymer main chain. In order to possess better solubility, higher molecular weight, and similar backbone coplanarity in comparison with P4, the architecture and the bulkiness of the alkyl chains were modified by replacing four dodecyl linear chains in P4 with a pair of 2-decyl-tetradecyl alkyl chains to obtain P6. The optical, electrochemical, charge transport and self-organization properties of these copolymers were comparatively characterized and studied in detail.
 | ||
| Fig. 2 Structures of P1–P6 synthesized and studied in this work. | ||
Results and discussion
Synthesis and characterization
The synthesis of the six polymers is outlined in Scheme 1. Monomer 3 was obtained by condensation between 4,7-bis(4-dodecylthiophen-2-yl)benzo[c][1,2,5]thiadiazole-5,6-diamine (1) and 1,2-bis(4-dodecylphenyl)ethane-1,2-dione (2), and then it was dibrominated to produce compound 4 for polymerization. Compounds 7 and 12 were obtained using a procedure similar to that for 4. The Stille coupling reaction was performed to synthesize P1–P6. The number-average molecular weight (Mn) and the polydispersity index (PDI) of these polymers were determined by GPC using polystyrene as standard and 1,2,4-trichlorobenzene as eluent at 135 °C. The data are listed in Table 1. The low Mn of P1–P4 could be attributed to their poor solubility. Similar results were also observed in other TQ polymers.33,34 After tuning the substitution positions and architectures of the side chains, P5 and P6 yielded much better degrees of polymerization and both exhibit excellent solubility in common solvents such as chloroform and tetrahydrofuran at room temperature. Additionally, all copolymers exhibited excellent thermal stability, with 5% weight loss up to 415 °C, measured by thermogravimetric analysis (TGA) (Fig. S1, ESI†). | ||
| Scheme 1 Synthetic routes for TQ-2T-based polymers. | ||
| Polymers | M n (kg mol−1) | PDIa | λ max (nm) soln | λ max (nm) film |
E
optg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (eV) c (eV) |
IPd (eV) | EAd (eV) | μ h, max, ave (cm2 V−1 s−1) | I on/Ioff | V T (V) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by GPC in 1,2,4-trichlorobenzene using polystyrene standards at 135 °C. b Dissolved in toluene. c Drop-cast from toluene solution (2 mg mL−1), and Eopt = 1240 nm/λonset. d IP and EA values were estimated from the onsets of the first oxidation and reduction peaks, while the potentials were determined using ferrocene (Fc) as standard using empirical formulae IP/EA = −(EOx/Red onset − EFc/Fc+(1/2) + 4.8) eV, wherein EFc/Fc+1/2 = 0.40 eV. | ||||||||||
| P1 | 7.0 | 2.68 | 1140, 1330 | 1165 | 0.75 | −4.95 | −3.92 | 0.063, 0.046 | ∼10 | 0 |
| P2 | 5.4 | 2.15 | 1130, 1305 | 1135, 1313 | 0.81 | −5.04 | −3.99 | 0.081, 0.066 | 50–100 | −2 |
| P3 | 8.0 | 2.34 | 910–1090 | 916–1100 | 0.96 | −5.28 | −3.93 | 0.042, 0.033 | 100–150 | −6 |
| P4 | 9.1 | 2.33 | 975, 1160 | 962, 1169 | 0.94 | −5.19 | −3.94 | 0.10, 0.097 | 100–150 | −1 |
| P5 | 12.7 | 4.05 | 890, 1150 | 930, 1184 | 0.91 | −5.21 | −3.98 | 0.012, 0.007 | 100–150 | −20 |
| P6 | 18.8 | 3.48 | 900–1060 | 926, 1095 | 0.95 | −5.23 | −3.99 | 0.24, 0.19 | ∼104 | −15 |
Optical and electrochemical properties
UV-vis-NIR absorption spectra of the polymers were recorded in toluene solution as well as in thin films (Fig. 3). The relevant data are summarized in Table 1. In dilute toluene solution, all polymers exhibit typical dual-band absorptions as other TQ polymers. The first one ranges from 300 to 600 nm and the second one covers from 650 to 1650 nm. The former band corresponds to the π–π* transition of the conjugated backbone, whereas the latter could be attributed to intramolecular charge transfer (ICT) between the donor and the acceptor within the polymer backbone.25 It is worthy to mention that the intensity of the second absorption band of these polymers is close to the first one, implying that these polymers possess strong ICT. Due to variable donor moieties of P1–P4, their absorption band edges exhibit significantly different red-shifts between 1200–1600 nm. The electron-donating ability of the donor moieties was estimated from their HOMO levels (Table S1, ESI†), which were calculated using density functional theory (DFT) with the B3LYP functional and the 6-31 G* basis set.35 The HOMO levels follow the order: T (−6.34 eV) < TT (−6.03 eV) < BT (−5.54 eV) < BDT (−5.47 eV). The HOMO energy of the TQ-2T moiety itself, however, is already higher lying within −5.02 eV since the two thiophenes already act as donors strongly increasing the HOMO of unsubstituted PTQ (HOMO = −6.08 eV) by more than 1 eV. Therefore the additional thiophene bridges and their different donor strengths not influence the absorption band edges as often assumed.36 Much more important, therefore is the extent of conjugation in the combined molecules and that can already nicely be predicted by considering the HOMO and LUMO values of the monomeric subunits M1–M6 (Table S1, ESI†). This can then explain why P1 and P2 show the longest wavelength absorption followed by P4, P6 = P3, and P5.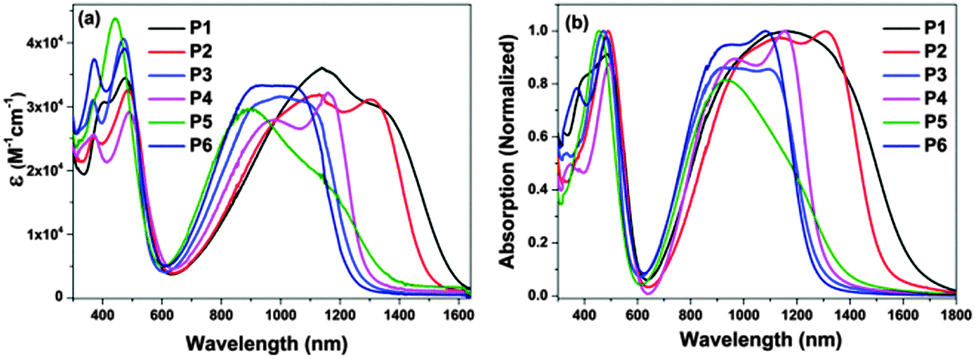 | ||
| Fig. 3 UV-visible-NIR absorption spectra of P1–P6 investigated in toluene solution (a) and in thin film (b). | ||
Compared to P4, P5 shows a blue-shift around 10 nm of the maximum wavelength (λmax) and significantly low molar extinction coefficient. The λmax value of P6 reveals a blue shift of 100 nm in comparison with that of P4. These were related to different alkylation positions and architectures, which affected intramolecular interaction in the polymer backbone, thereby changing the optical behavior of the TQ-2T polymers. On the other hand, P5 exhibits a long tail extending to 1500 nm, suggesting a high tendency to aggregate even in diluted solution.37 This is in agreement with the high PDI of P5.
The films were prepared by drop-casting toluene solutions of these polymers onto glass slides. P1–P6 displayed slightly broadened spectra but with only small red-shifts from 8 to 35 nm at λmax compared with those in solution, indicating some aggregation in thin film. The optical band gaps calculated according to the absorption onset of the solid films are listed in Table 1. These results demonstrated that changing the donors and side chains can be effective for tuning the optical behavior of the polymers.
The cyclic voltammetry was carried out to investigate the ionization potentials (IPs) and electron affinities (EAs) of the polymers.38 The EA values of three monomers 3, 6 and 11 were also studied to clarify the influence of the side chains on the electron-withdrawing ability of the acceptors. The reduction curves of monomers are shown in Fig. S2 (ESI†). The corresponding values of EA were calculated to be −3.77, −3.78 and −3.77 eV, according to the onset of the first reduction peak. These results implied that the number and architecture of side chains grafted onto the TQ-2T core induce a very weak change in their electron-withdrawing ability. Fig. 4 presents the reduction and oxidation curves of all TQ-2T copolymers. The corresponding data are calculated based on the onset potentials and listed in Table 1. For P1–P4, we found that the IP values decreased gradually with the increased donor strength and this also has opposite tendency in contrast to other acceptors.36,39,40 This result implied that the significant difference in IP of P1–P4 originated not only from different donors, but also from the conformation of the polymer backbone. Compared to IP, the slightly different EA of P1–P4 was attributed to the dominant contribution of the same TQ core. The variation of the IP and EA values for P4–P6 was very small. These results suggested that the adjustment of the substitution positions and architectures has a weak influence on the IP and EA values of the polymers, while variation of the donors in P1–P4 bear a stronger effect on the IP of the polymers.
 | ||
| Fig. 4 The reduction and oxidation curves of P1–P6. The films were deposited from chlorobenzene solutions. | ||
OFET properties
In order to evaluate the charge carrier transport of the polymers, OFET devices with bottom-gate bottom-contact architecture were fabricated onto the heavily doped silicon wafers with 300 nm thick thermally grown SiO2 as the dielectric. The dielectric was functionalized by hexamethyldisilazane (HMDS) to minimize interfacial trapping sites. Au electrodes with 60 nm in thickness were evaporated, which act as the source and the drain. All polymers were drop-cast from 2 mg mL−1 of chlorobenzene solutions and subsequently annealed at 180 °C for 30 min to remove the residual solvent. The field-effect measurements were carried out under nitrogen atmosphere, and the corresponding data including charge carrier mobilities (μ), current on/off ratios (Ion/Ioff) and threshold voltages (VT) are summarized in Table 1. The transistors of all polymers, P1–P6, only exhibit p-type field-effect behavior due to many thiophenes in the conjugated backbones. Indeed, weak donors like alkylated 2,5-diethynylthiophene26 and unsubstituted thiophene27 combined with PTQ can be applied for ambipolar polymer semiconductors. The transfer and output characteristics of P4 and P6 are presented in Fig. 5, while the plots of the other four polymers, P1–P3 and P5, are provided in ESI,† Fig. S3. Among P1–P4, P4 showed the highest mobility of 0.1 cm2 V−1 s−1 with an extremely low threshold voltage of −1 V. Due to the incorporation of the BDT unit, the transistor performance of P3 is significantly reduced with the mobility of 0.033 cm2 V−1 s−1 and the threshold voltage of −6 V. In comparison to P3, P1 and P2 exhibited a minor improvement in hole transport with mobilities of 0.046 and 0.066 cm2 V−1 s−1. Meanwhile their on/off ratio is lowered to <102. This behavior suggested that the donor segments play a crucial role in the charge carrier transport of TQ polymers. P5 and P6 were designed and synthesized with higher molecular weight and increased solubility. Unfortunately, P5 showed a hole mobility of approximately one order of magnitude lower than P4, most probably because of the increased torsion between the two neighboring thiophene units. Replacing four linear side chains on the TQ-2T core (P4) by a pair of branched ones as in P6 led to a dramatic improvement of the transistor performance with a maximum hole mobility of 0.24 cm2 V−1 s−1 and an on/off ratio of 104, although the threshold voltage is slightly increased to −15 V. This result proved that the substitution positions and architectures of the side chains critically affect the charge carrier transport in TQ-2T-based polymers. The nonlinear behavior of the drain current (ID) at low source-drain voltage (VD) in the output characteristics of P4 and P6 was attributed to the contact resistance and charge injection limitation.41 Additionally, the relatively low on/off ratio of P1–P6 might be related to adventitious doping, due to the high-lying IP of these polymers.42,43 In spite of low on/off ratios for these polymers, it has to be emphasized that the reproducibility of OFET performances is relatively good. As shown in Table 1, the difference between maximum mobility and average values is only ranging from 3% to 27% for P1–P4 and P6. For P5, this difference is slightly increased to 41%.| Polymers | FWHM (°) | Coherence length (nm) | π–π spacing (nm) | Interlayer distance (nm) |
|---|---|---|---|---|
| P1 | 4.7 | 1.7 | 0.38 | 3.06 |
| P2 | 3.1 | 2.6 | 0.36 | 3.02 |
| P3 | 2.8 | 2.9 | 0.36 | 2.80 |
| P4 | 1.4 | 5.8 | 0.37 | 3.03 |
| P5 | 4.2 | 1.9 | 0.37 | 2.87 |
| P6 | 1.2 | 6.6 | 0.38 | 2.94 |
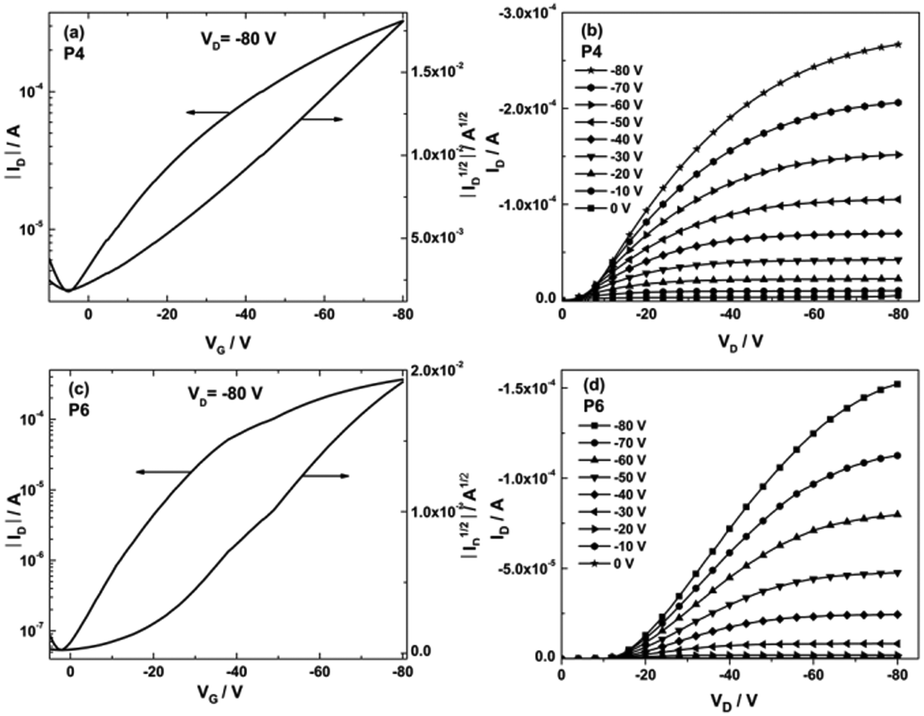 | ||
| Fig. 5 Transfer (a) and (c) and output (b) and (d) curves of P4 and P6. | ||
Self-organization in bulk
To understand the variation in device performance, two dimensional wide-angle X-ray scattering (2DWAXS) was used to investigate the molecular organization in bulk. The 2DWAXS measurements were performed on mechanical extruded fibers, which were subsequently thermally annealed at 180 °C for 30 min. For the measurements, the fibers were mounted vertically towards the 2D detector and the scattering was recorded at 30 °C. The 2D patterns confirm slight variations between P1, P2, P5 and P3. Significant differences are observed for P4 and P6.In the equatorial plane of the patterns reflections at small-angles are ascribed to the layer structure of conjugated polymer backbones oriented along the alignment direction of the fiber (Fig. 6). The interlayer distance between lamellar structures of 3.06 nm for P1, 3.02 nm for P2 and 2.87 nm for P5 were determined from the main peak position. The scattering intensities on the meridional plane with d-spacings of 1.45 nm for P1, 1.65 nm for P2 and 1.85 nm for P5 are related to the length of a single repeating unit. These values are in agreement with the theoretical lengths of 1.46 nm for P1, 1.62 nm for P2 and 1.80 nm for P5 as calculated by Cerius2 simulations.44 More crucially, wide-angle equatorial scattering intensities are assigned to π-stacking distances of 0.38 nm for P1, 0.36 nm for P2 and 0.37 nm for P5. Additionally, the π-stacking reflection for P2 showed a slightly smaller full width at half maximum (FWHM)45 than those for P1 and P5, indicating a larger coherence length along the stacking direction for P2. Table 2 summarizes the FWHM value and the coherent length for the π-stacking direction for all investigated polymers. Interestingly, due to an improved overall crystallinity, the alkyl side chains of P2 exhibit a preferential ordered direction as evident from off-meridional reflections in the middle-angle region (dashed circles in Fig. 6b). In contrast, the patterns for P1 and P5 display only a broad amorphous halo characteristic for disordered alkyl chains.
 | ||
| Fig. 6 2DWAXS patterns of (a) P1, (b) P2, (c) P3, (d) P4, (e) P5 and (f) P6 (insets show the small-angle scattering range). Off-meridional reflections related to a preferential organization of the alkyl chains are indicated in (b) and (d) by dashed circles. | ||
The interlayer distance of P3 compared to P1 and P2, however decreased to 2.80 nm, due to a slightly smaller backbone curvature, while a π-stacking distance of 0.36 nm is found. The amorphous halo related to a d-spacing of 0.45 nm is correlated also to the isotropic arrangement of alkyl chains. The interlayer distance observed for P4 and P6 is 3.03 nm and 2.94 nm, respectively. In comparison to the other polymers within this series, both compounds show more distinct π-stacking reflections corresponding to distances of 0.37 nm for P4 and 0.38 nm for P6. The variations in the interlayer and the π-stacking distance within this polymer series are due to the modifications in the backbone composition and substitution positions. A less planar donor unit in P4 and P6 in comparison to P2 and P3 increases the π-stacking distance. On the other hand, avoiding alkyl chains at the TQ acceptor and replacing the linear to branched alkyl chains for P6 slightly reduced the interlayer distance and increased the π-stacking distance. Surprisingly, at the same time the degree of order in P4 and P6 increased as evident from a higher coherence length of π-stacking for both polymers in comparison to the other polymers (Table 2). Reflections in the off-meridional middle-angle region corresponding to the d-spacings of 1.23 nm for P4 and 2.00 nm for P6 suggest that the repeating units of the polymer chains in neighbouring layers are shifted in the extrusion direction with respect to each other. Cerius2 simulations confirmed this effect on the scattering pattern (Fig. S4, ESI†). In such organization P6 with branched alkyl chains is arranged in a more energetically favourable fashion. The shift is induced by the steric demand for the branched substituents allowing a more efficient space filling in the periphery layer. In this configuration, the donor units occupy positions adjacent to the acceptor units in the neighboring layers. This packing of P6 induces higher crystallinity than for P1, P2, P3 and P5. Additional anisotropic off-meridional reflections for P4 corresponding to a d-spacing of 0.45 nm are assigned to alkyl chain ordering and confirm higher overall order in comparison to P1 and P3.
For this polymer series, the charge carrier transport in field-effect transistors is insensitive in the interlayer and π-stacking distances. It has been found that the coherence length in the π-stacking direction is more crucial for the electrical properties. In other words, a more ordered packing of the polymers chains in the layer structures ensures a more unhindered migration of charges as observed in the cases of P4 and P6.
Conclusions
Six new TQ-2T-based polymers have been synthesized and characterized. Varying donor moieties, substitution positions and architectures of the alkyl chains in TQ-2T polymers can facilitate the tuning of the optoelectronic behavior, self-organization in bulk and charge carrier transport. Compared to P1–P3, P4 bears higher crystallinity leading to a hole mobility as high as 0.1 cm2 V−1 s−1. Although P5 shows an enhanced solubility and molecular weight than P4, it produces a low crystallinity due to the increased torsion in the conjugated backbone and leads to therefore a low mobility of 0.007 cm2 V−1 s−1. Interestingly, a pair of 2-decyltetradecyl chains used to replace the linear side chains does not significantly change the planarity of the polymers, but improves its molecular weight and solubility. Due to a higher coherence length in the π-stacking direction, the charge carrier mobility of P6 is increased up to 0.24 cm2 V−1 s−1. The structure–property relationship of TQ-2T-polymers is beneficial to broaden the understanding of TQ-containing polymers and further design new TQ-polymers towards high performance.Acknowledgements
This work is supported by SFB-TR49. C. A. gratefully acknowledges the China Scholarship Council (CSC) for offering a 3 years Scholarship. M. Li and T. M. acknowledge the ERC Advanced Grant NANOGRAPH (AdG-2010-267160).Notes and references
- A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed.
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS PubMed.
- T. Lei, J.-Y. Wang and J. Pei, Chem. Mater., 2013, 26, 594–603 CrossRef.
- J. Mei and Z. Bao, Chem. Mater., 2013, 26, 604–615 CrossRef.
- A. T. Yiu, P. M. Beaujuge, O. P. Lee, C. H. Woo, M. F. Toney and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 134, 2180–2185 CrossRef PubMed.
- I. Meager, R. S. Ashraf, S. Mollinger, B. C. Schroeder, H. Bronstein, D. Beatrup, M. S. Vezie, T. Kirchartz, A. Salleo, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2013, 135, 11537–11540 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, A. Furlan, W. S. C. Roelofs, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2013, 135, 18942–18948 CrossRef CAS PubMed.
- X. Guo, F. S. Kim, M. J. Seger, S. A. Jenekhe and M. D. Watson, Chem. Mater., 2012, 24, 1434–1442 CrossRef CAS.
- Y. Kim, J. Hong, J. H. Oh and C. Yang, Chem. Mater., 2013, 25, 3251–3259 CrossRef CAS.
- T. Earmme, Y.-J. Hwang, N. M. Murari, S. Subramaniyan and S. A. Jenekhe, J. Am. Chem. Soc., 2013, 135, 14960–14963 CrossRef CAS PubMed.
- J. D. Yuen, R. Kumar, D. Zakhidov, J. Seifter, B. Lim, A. J. Heeger and F. Wudl, Adv. Mater., 2011, 23, 3780–3785 CAS.
- T. Lei, Y. Cao, X. Zhou, Y. Peng, J. Bian and J. Pei, Chem. Mater., 2012, 24, 1762–1770 CrossRef CAS.
- W. Zhuang, M. Bolognesi, M. Seri, P. Henriksson, D. Gedefaw, R. Kroon, M. Jarvid, A. Lundin, E. Wang, M. Muccini and M. R. Andersson, Macromolecules, 2013, 46, 8488–8499 CrossRef CAS.
- H. Li, T. L. Tam, Y. M. Lam, S. G. Mhaisalkar and A. C. Grimsdale, Org. Lett., 2011, 13, 46–49 CrossRef CAS PubMed.
- T. Dallos, M. Hamburger and M. Baumgarten, Org. Lett., 2011, 13, 1936–1939 CrossRef CAS PubMed.
- S. Ellinger, K. R. Graham, P. Shi, R. T. Farley, T. T. Steckler, R. N. Brookins, P. Taranekar, J. Mei, L. A. Padilha, T. R. Ensley, H. Hu, S. Webster, D. J. Hagan, E. W. Van Stryland, K. S. Schanze and J. R. Reynolds, Chem. Mater., 2011, 23, 3805–3817 CrossRef CAS.
- H. Li, F. Zhou, T. L. D. Tam, Y. M. Lam, S. G. Mhaisalkar, H. Su and A. C. Grimsdale, J. Mater. Chem. C, 2013, 1, 1745–1752 RSC.
- T. L. Tam, H. Li, Y. M. Lam, S. G. Mhaisalkar and A. C. Grimsdale, Org. Lett., 2011, 13, 4612–4615 CrossRef CAS PubMed.
- C. An, M. Li, T. Marszalek, D. Li, R. Berger, W. Pisula and M. Baumgarten, Chem. Mater., 2014, 26, 5923–5929 CrossRef CAS.
- M. X. Chen, E. Perzon, N. Robisson, S. K. M. Jönsson, M. R. Andersson, M. Fahlman and M. Berggren, Synth. Met., 2004, 146, 233–236 CrossRef CAS PubMed.
- C.-Y. Yu, C.-P. Chen, S.-H. Chan, G.-W. Hwang and C. Ting, Chem. Mater., 2009, 21, 3262–3269 CrossRef CAS.
- A. P. Zoombelt, M. Fonrodona, M. M. Wienk, A. B. Sieval, J. C. Hummelen and R. A. J. Janssen, Org. Lett., 2009, 11, 903–906 CrossRef CAS PubMed.
- C. An, S. R. Puniredd, X. Guo, T. Stelzig, Y. Zhao, W. Pisula and M. Baumgarten, Macromolecules, 2014, 47, 979–986 CrossRef CAS.
- T. Dallos, D. Beckmann, G. Brunklaus and M. Baumgarten, J. Am. Chem. Soc., 2011, 133, 13898–13901 CrossRef CAS PubMed.
- T. T. Steckler, P. Henriksson, S. Mollinger, A. Lundin, A. Salleo and M. R. Andersson, J. Am. Chem. Soc., 2014, 136, 1190–1193 CrossRef CAS PubMed.
- M. E. Foster, B. A. Zhang, D. Murtagh, Y. Liu, M. Y. Sfeir, B. M. Wong and J. D. Azoulay, Macromol. Rapid Commun., 2014, 35, 1516–1521 CrossRef CAS PubMed.
- M. X. Chen, X. Crispin, E. Perzon, M. R. Andersson, T. Pullerits, M. Andersson, O. Inganas and M. Berggren, Appl. Phys. Lett., 2005, 87, 252105 CrossRef PubMed.
- T. T. Steckler, O. Fenwick, T. Lockwood, M. R. Andersson and F. Cacialli, Macromol. Rapid Commun., 2013, 34, 990–996 CrossRef CAS PubMed.
- T. T. Steckler, M. J. Lee, Z. Chen, O. Fenwick, M. R. Andersson, F. Cacialli and H. Sirringhaus, J. Mater. Chem. C, 2014, 2, 5133–5141 RSC.
- Y. Lee, T. P. Russell and W. H. Jo, Org. Electron., 2010, 11, 846–853 CrossRef CAS PubMed.
- H. Yi, R. G. Johnson, A. Iraqi, D. Mohamad, R. Royce and D. G. Lidzey, Macromol. Rapid Commun., 2008, 29, 1804–1809 CrossRef CAS.
- K.-F. Cheng, C.-C. Chueh, C.-H. Lin and W.-C. Chen, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6305–6316 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- M. M. Durban, P. D. Kazarinoff and C. K. Luscombe, Macromolecules, 2010, 43, 6348–6352 CrossRef CAS.
- X. Guo, S. R. Puniredd, M. Baumgarten, W. Pisula and K. Müllen, Adv. Mater., 2013, 25, 5467–5472 CrossRef CAS PubMed.
- J.-L. Bredas, Mater. Horiz., 2014, 1, 17–19 RSC.
- H. Li, F. S. Kim, G. Ren and S. A. Jenekhe, J. Am. Chem. Soc., 2013, 135, 14920–14923 CrossRef CAS PubMed.
- W. Zhou, Z.-G. Zhang, L. Ma, Y. Li and X. Zhan, Sol. Energy Mater. Sol. Cells, 2013, 112, 13–19 CrossRef CAS PubMed.
- S. Wang, A. Kiersnowski, W. Pisula and K. Müllen, J. Am. Chem. Soc., 2012, 134, 4015–4018 CrossRef CAS PubMed.
- X. Zhang, T. T. Steckler, R. R. Dasari, S. Ohira, W. J. Potscavage, S. P. Tiwari, S. Coppee, S. Ellinger, S. Barlow, J.-L. Bredas, B. Kippelen, J. R. Reynolds and S. R. Marder, J. Mater. Chem., 2010, 20, 123–134 RSC.
- J. Liu, R. Zhang, G. v. Sauvé, T. Kowalewski and R. D. McCullough, J. Am. Chem. Soc., 2008, 130, 13167–13176 CrossRef CAS PubMed.
- Cerius2 calculation, Accelrys, S. D., CA, USA.
- D. Balzar, N. Audebrand, M. R. Daymond, A. Fitch, A. Hewat, J. I. Langford, A. Le Bail, D. Louer, O. Masson, C. N. McCowan, N. C. Popa, P. W. Stephens and B. H. Toby, J. Appl. Crystallogr., 2004, 37, 911–924 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The experimental part, CV, TGA, DFT calculations, and NMR and HRMS spectra. See DOI: 10.1039/c5tc00155b |
| This journal is © The Royal Society of Chemistry 2015 |