
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular hierarchy among halogen and hydrogen bond donors in light-induced surface patterning†
Marco
Saccone
a,
Valentina
Dichiarante
b,
Alessandra
Forni
*c,
Alexis
Goulet-Hanssens‡
d,
Gabriella
Cavallo
b,
Jaana
Vapaavuori§
a,
Giancarlo
Terraneo
b,
Christopher J.
Barrett
d,
Giuseppe
Resnati
*b,
Pierangelo
Metrangolo
*be and
Arri
Priimagi
*abf
aDepartment of Applied Physics, Aalto University, P.O. Box 13500, FI-00076 Aalto, Finland
bNFMLab-DCMIC “Giulio Natta”, Politecnico di Milano, Via L. Mancinelli 7, IT-20131 Milano, Italy. E-mail: pierangelo.metrangolo@polimi.it; giuseppe.resnati@polimi.it
cISTM-CNR, Institute of Molecular Sciences and Technologies of CNR, Università degli Studi di Milano, Via Golgi 33, IT-20133 Milano, Italy. E-mail: alessandra.forni@istm.cnr.it
dDepartment of Chemistry, McGill University, 801 Sherbrooke Street West, Montreal, Quebec H3A 0B8, Canada
eVTT-Technical Research Centre of Finland, P.O. Box 1000, FI-02044 VTT, Finland
fDepartment of Chemistry and Bioengineering, Tampere University of Technology, P. O. Box 541, FI-33101 Tampere, Finland. E-mail: arri.priimagi@tut.fi
First published on 25th November 2014
Abstract
Halogen bonding, a noncovalent interaction possessing several unique features compared to the more familiar hydrogen bonding, is emerging as a powerful tool in functional materials design. Herein, we unambiguously show that one of these characteristic features, namely high directionality, renders halogen bonding the interaction of choice when developing azobenzene-containing supramolecular polymers for light-induced surface patterning. The study is conducted by using an extensive library of azobenzene molecules that differ only in terms of the bond-donor unit. We introduce a new tetrafluorophenol-containing azobenzene photoswitch capable of forming strong hydrogen bonds, and show that an iodoethynyl-containing azobenzene comes out on top of the supramolecular hierarchy to provide unprecedented photoinduced surface patterning efficiency. Specifically, the iodoethynyl motif seems highly promising in future development of polymeric optical and photoactive materials driven by halogen bonding.
Introduction
The azobenzene moiety is an important tool in present-day research of molecular-level photoswitching and is extensively used for the development of macroscopic photonic and photomechanical materials.1 The interest in azobenzene lies in its clean, efficient, and reversible photoisomerization between a rod-like trans-form and a bent cis-form,2 which can give rise to a cascade of motions beyond the size scale of an individual molecule. When the azobenzene is incorporated into a liquid-crystalline or a polymeric medium, the photoisomerization can accomplish various tasks, allowing for induction of order–disorder transitions, control over the molecular alignment, photoactuation of the shape and dimensions of the material system, and inscription of surface-relief patterns onto thin azobenzene-containing films.3 In particular, when an azobenzene-containing thin polymer film is irradiated with a polarization/intensity interference pattern of light, the film surface deforms and creates a replica of the incident interference pattern in the form of a surface topography pattern, often denoted as a surface-relief grating (SRG).4 The modulation depth of the photoinduced surface patterns can be hundreds of nanometers, creating efficient diffractive optical elements with a first-order diffraction efficiency of up to 30–40%. SRGs have been used as nanostructuring tools, liquid-crystal alignment layers, and periodic light trapping/light outcoupling layers.5 Despite their application potential, however, the surface pattern formation mechanism is not yet well understood. Therefore, a full understanding of structure–function relationships that govern the SRG process is needed in order to design better performing materials for future advanced applications.Supramolecular side-chain polymers, in which the photoactive azobenzene units are attached to a passive polymer backbone via noncovalent interactions, have in the past few years emerged as a new class of SRG-forming materials.6 The importance of these materials lies in their extreme ease of preparation and modular tunability: the material composition can be easily optimized to meet specific requirements. As a result, high-quality resonance filters, and patterns with record-high modulation depth have been fabricated from azobenzene-containing supramolecular side-chain polymers.6a,7 We have recently shown that halogen bonding8 appears particularly promising for the design of supramolecular SRG-forming materials.9 Halogen bonding is the noncovalent interaction in which a region of positive electrostatic potential on top of the surface of the halogen atom, the σ-hole,10 interacts with a nucleophilic site. High-level ab initio quantum-chemical calculations suggest that dispersion forces may also play an important role in this interaction.11 The features that distinguish halogen bonding from hydrogen bonding are of particular interest, which include higher directionality, hydrophobicity, size of the interacting atom (halogen vs. hydrogen) and the possibility to tune interaction strength by a simple change of the halogen atom.12 These characteristic properties have allowed for the fine tuning of geometrical features relevant to crystal engineering13 and to the design of functional materials with unique light-emissive14 and liquid-crystalline15 properties.
Recently several examples of halogen-bonded supramolecular structures involving azobenzenes have been reported.9,16 For example, we showed that the efficiency of SRG formation increases with the strength of halogen bonding, and that the high performance of halogen-bonded complexes seems to be connected more to the directionality of halogen bonding than to its intrinsic interaction strength. To further develop the practical applications of halogen bonding in the field of photocontrollable materials, we have undertaken a detailed study on the relative surface patterning efficiency that can be imparted by different halogen-bond donors as a result of the presence of electron-withdrawing substituents versus the hybridization of the carbon bearing the heavy halogen. Moreover, we aimed to provide a ranking between various halogen and hydrogen bonds in driving SRG formation, since these two interactions show similar chemical behaviour and a detailed knowledge of the performance of competing noncovalent interactions is crucial for the preparation of materials with designed structures and properties.
Herein, we present a comprehensive study on the surface patterning properties of an extensive library of azobenzene derivatives as presented in Fig. 1. We show that halogen-bonded polymer–azobenzene complexes unambiguously surpass their hydrogen-bonded counterparts in terms of SRG formation efficiency, confirming that it is the directionality rather than the strength of the noncovalent interactions that dictates the macroscopic light-induced movements. Most importantly, we introduce in this field a new halogen-bond donor moiety – iodoacetylene – that further boosts the macroscopic light-induced movements and the surface pattern formation. The optical studies are complemented with theoretical calculations and crystallographic structure determinations that further elucidate the different performance of halogen- and hydrogen-bonded systems.
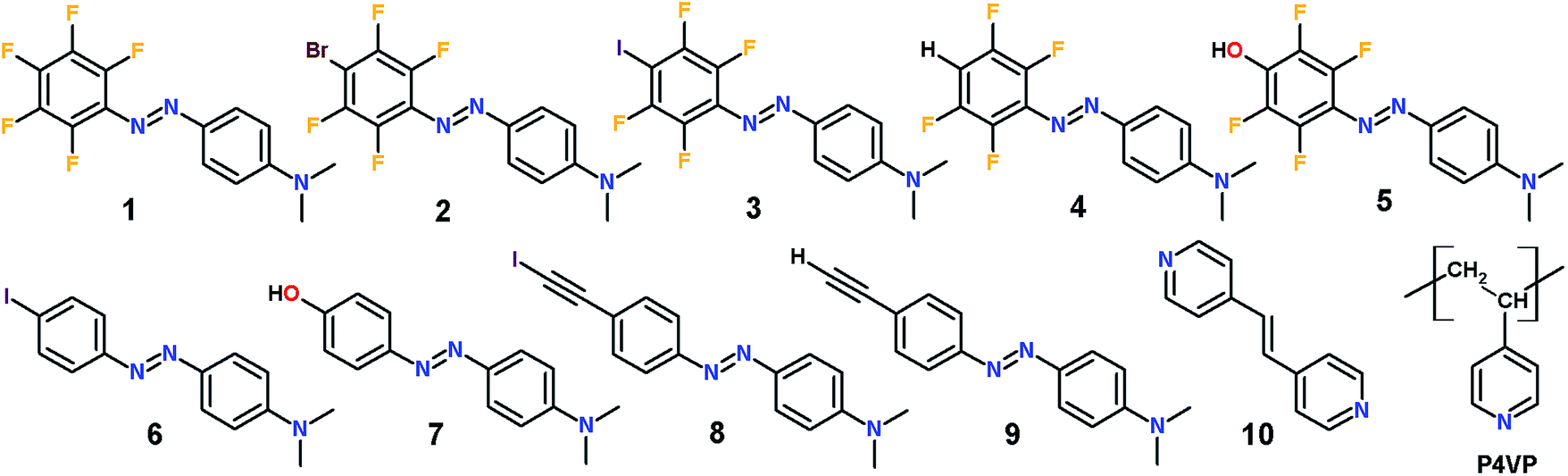 | ||
| Fig. 1 Chemical structures of fluorinated azobenzene modules 1–5, hydrogenated azobenzene modules 6–9, model dipyridyl compound 10 used in the co-crystallization studies, and poly(4-vinyl pyridine) (P4VP) that was used as the polymer matrix for the azobenzene dyes. | ||
Results and discussion
Materials design
The library of azobenzene compounds shown in Fig. 1 allows for (i) establishing an optimal halogen-bond donor in terms of SRG formation capability, and (ii) comparing structurally similar azobenzenes substituted with halogen-bond versus hydrogen-bond donor groups. The series of perfluorinated azobenzenes 1–5 comprises a reference molecule incapable of forming halogen bonds (1), two halogen-bond donors (2, 3) with similar spectral and photochemical properties but different interaction strengths (Br < I), and two hydrogen-bond donors, one weak (4) and one strong perfluorophenol-containing dye (5). Compound 5 serves a dual purpose. Firstly, a close structural relationship with 3 affords an ideal comparison for halogen bonding versus hydrogen bonding in driving the SRG formation process. Secondly, the perfluorinated phenol presumably acts as a very powerful hydrogen-bond donor and could therefore give rise to a very strong noncovalent interaction,17,18 which is interesting in its own right in the field of functional supramolecular materials. This is the rationale for the comparison between 5 and its nonfluorinated counterpart 7, the SRG performance of which is known.9The non-fluorinated dyes 6–9 include the already mentioned phenol-substituted compound 7, an iodo-substituted azobenzene 6 that is expected to act as a weak halogen-bond donor, and ethynyl-substituted dyes 8 and 9. The latter two compounds are particularly interesting, since very recently iodoethynyl-benzenes were investigated both theoretically and experimentally with the purpose of establishing a hierarchy among halogen-bond donors in crystal engineering.19 Based on ATR-FTIR and single crystal X-ray diffraction data, Aakeröy et al. proved that the iodoethynyl benzenes form co-crystals with a larger class of halogen-bond acceptors compared to perfluoroiodobenzenes suggesting that this motif might be employed to enhance the performance of photoresponsive polymers. Each of the studied molecules is substituted with an electron-donating dimethylamino group, which red-shifts the absorption spectrum of the dyes, increases the rate of trans–cis–trans cycling, and boosts the SRG formation efficiency.
Poly(4-vinyl pyridine) (P4VP) was used as a polymeric bond acceptor as it is a popular choice in constructing supramolecular polymeric complexes based on hydrogen bonding or proton transfer,20 and it equally functions as a halogen-bond acceptor.21 P4VP has been used by several groups to prepare light-responsive polymers,6b,22 making it the polymer of choice for the present study. Low-molecular-weight P4VP (Mw: 1200 g mol−1) was selected since long polymer chains tend to disrupt the photoinduced surface patterning. SRG formation was studied in polymer–azobenzene complexes, denoted as P4VP(n)y where n is the azobenzene unit chosen among 1–9 and y is the n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P4VP molar ratio. Typically, y was set to be 0.1, or, in other words, the studied systems contained one azobenzene moiety per ten polymer repeat units. This low degree of complexation minimizes crystallization and macroscopic phase separation of the azobenzene units, which would compromise comparisons between the molecules. Assuming that (E)-1,2-di(4-pyridyl)ethylene (10) is a good mimic of the minimal binding motif of P4VP, the co-crystals formed by 10 with various azobenzene derivatives were used as models for single-crystal X-ray diffraction analysis of the P4VP–azobenzene systems employed in light-induced surface patterning. 4-Methyl pyridine was used as a model of the minimal binding motif of P4VP in computational studies.
:P4VP molar ratio. Typically, y was set to be 0.1, or, in other words, the studied systems contained one azobenzene moiety per ten polymer repeat units. This low degree of complexation minimizes crystallization and macroscopic phase separation of the azobenzene units, which would compromise comparisons between the molecules. Assuming that (E)-1,2-di(4-pyridyl)ethylene (10) is a good mimic of the minimal binding motif of P4VP, the co-crystals formed by 10 with various azobenzene derivatives were used as models for single-crystal X-ray diffraction analysis of the P4VP–azobenzene systems employed in light-induced surface patterning. 4-Methyl pyridine was used as a model of the minimal binding motif of P4VP in computational studies.
Computational studies
As mentioned above, 4-methyl pyridine was used as the smallest pyridine model compound mimicking P4VP to compute the binding energy with azobenzenes 1–9. Table 1 presents the PBE0/6-311++G(d,p) interaction energies, both non-corrected (ΔE) and corrected (ΔEBSSE) for the basis set superposition error, showing a notable energy overestimation (from 6 to 14% depending on the dimer) when neglecting such an error. The ΔEBSSE values computed for the perfluorinated dyes 2 and 3 were −3.501 and −5.135 kcal mol−1,9 respectively, in agreement with the consolidated trend of Br < I in halogen-bonded systems,10 while molecule 1, lacking the σ-hole on the fluorine atom in the para position to the azo group, was confirmed to be unable to give rise to a stable halogen-bonded complex. For the complex of 4-methyl pyridine with the perfluorinated hydrogen-bond donor 5, a stabilization energy of −11.765 kcal mol−1 was obtained, corresponding to a medium-to-strong hydrogen bond.23 The corresponding iodinated and phenolic nonfluorinated donors, 6 and 7, provided stabilization energy values of −2.546 and −10.053 kcal mol−1, respectively, upon binding to the pyridine unit.| Compound | ΔE | ΔEBSSE | V S,max | μ calc |
|---|---|---|---|---|
| 1 | — | — | −0.018 | 10.67 |
| 2 | −3.712 | −3.501 | 0.032 | 10.47 |
| 3 | −5.565 | −5.135 | 0.035 | 10.22 |
| 4 | −4.477 | −3.930 | 0.039 | 8.94 |
| 5 | −12.644 | −11.765 | 0.086 | 7.63 |
| 6 | −2.828 | −2.546 | 0.018 | 9.28 |
| 7 | −10.796 | −10.053 | 0.083 | 5.65 |
| 8 | −5.533 | −5.170 | 0.041 | 8.36 |
| 9 | −3.832 | −3.528 | 0.043 | 8.30 |
The key observation here is that perfluorination of the benzene ring is significantly more effective in strengthening halogen bonding than hydrogen bonding: the interaction strength, in fact, doubles in the first case (6vs.3), whereas a minor <20% enhancement takes place for the latter (7vs.5). This difference can be rationalized by examining the changes in charge density distribution around iodine and the phenolic hydrogen in the isolated dyes upon fluorination. While the molecular dipole moment (Table 1, μcalc) undergoes a greater increase for the phenolic with respect to the iodinated derivative, the electrostatic potential (Fig. 2) is practically unaffected by fluorination near the phenolic hydrogens, though it is strongly positive in both cases (maximum values of VS,max are 0.083 and 0.086 au for 7 and 5, respectively, on the 0.001 a.u. isosurface of electron density). Conversely, the maximum value of the electrostatic potential around the iodine atom, while less positive, doubles upon ring fluorination, from 0.018 a.u. (6) to 0.035 au (3). This can be explained by polarizability arguments: it is well-known that halogen atoms – apart from fluorine – are highly polarizable, and more so for the heavier halogens. As a result, the strength of halogen bonding significantly increases by introduction of electron-withdrawing groups such as fluorine atoms on the molecular moiety bonded to the halogen-bond donor site.24
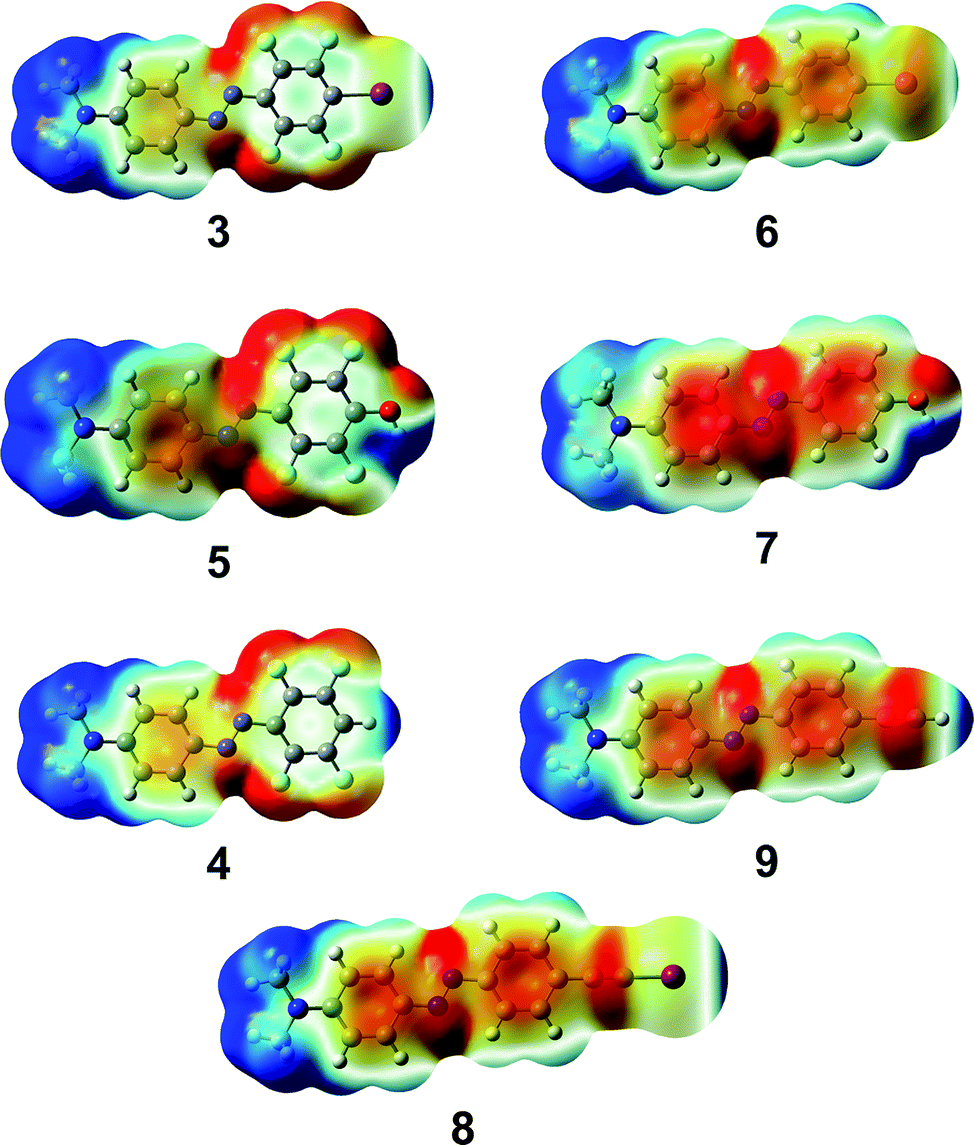 | ||
| Fig. 2 Plots of the electrostatic potential of compounds 3, 6 (first row), 5, 7 (second row), 4, 9 (third row), and 8 (bottom), computed at the PBE0/6-311++G(d,p) level in vacuo. Potentials are mapped on the respective isosurfaces (0.001 a.u.) of electron density. Values of electrostatic potential range from −0.03 (red) to 0.03 (blue) a.u. Atom color scheme: C, gray; H, light gray; N, dark blue; O, red; F, sky blue; and I, magenta. | ||
As recently shown by a systematic investigation of a series of halogen-bond donors,19 the hybridization of the carbon atom adjacent to the halogen bonded atom heavily influences the strength of the formed halogen bonds. In particular, iodoethynyl-containing moieties have recently emerged as remarkably powerful halogen-bond donor moieties.19 In agreement with such findings, compound 8 shows, upon binding to the model pyridine unit, a slightly higher stabilization energy (−5.170 kcal mol−1) than compound 3. It is also noteworthy that the maximum value of the electrostatic potential around the iodine atom is 0.041 a.u., notably larger than the corresponding value obtained for 3 (0.035 a.u.), suggesting that the iodoethynyl moiety should give a more directional halogen bonding.
Such observation prompted us to check the different directionality features of the corresponding hydrogenated derivatives, 9vs.4. While the hydrogen bond of the former with 4-methyl pyridine is slightly weaker than that of the latter (stabilization energies are −3.528 and −3.930 kcal mol−1, respectively), the ethynyl derivative is characterized by a highly sharpened positive region in the electrostatic potential (Fig. 2), with a more positive VS,max value with respect to compound 4 (0.043 and 0.039 a.u., respectively). This suggests a potentially much greater directionality for the hydrogen bonding of the ethynyl derivative with respect to the perfluorinated compound, somewhat resembling halogen bonding.
It is thus clear that 4-methyl pyridine is more tightly bound to the phenolic and hydrogen-bonding donor dyes 5 and 7 than to the halogen-bonding donor dyes 3 and 6, but it is also evident from Fig. 2 that halogen bonds are expected to be significantly more directional,25 as the positive region responsible for the noncovalent interaction is narrowly confined along the extension of the C–I bonds and it is spread out hemispherically around the phenolic hydrogens. On the other hand, the dimeric complexes comprising 4-methyl pyridine and ethynyl derivatives 8 and 9 show quite similar directionality features but the interaction strength of the iodine derivative is greater. In our hypothesis, a more directional noncovalent interaction between the azobenzene dye and the P4VP provides a more efficient junction in transferring the light-induced motions of the photoactive units to the polymer backbone and enhanced SRG formation results.9 Molecules 3 and 5 comprise an ideal pair to gain further insight into this hypothesis.
We also computed the UV-visible spectra for 1–9 by time-dependent DFT calculations (Table S1†), which showed that perfluorination of the benzene ring red-shifts the absorption maxima of both iodine and phenolic derivatives, as was also verified experimentally by measuring the absorption spectra in DMF solutions. Moreover, the spectroscopic properties of the tetrafluoroiodo and iodoethynyl derivatives 3 and 8, respectively, are quite similar indicating that the two halogen-bond donors share many parallel features. Such findings are extremely helpful for the design of new functional materials based on halogenated ethynyl moieties. Details on computations are given in the ESI.†
Structural characterization
(E)-1,2-Di(4-pyridyl)ethylene (10) was used as a co-crystal forming component that mimics P4VP. The aim was to get specific insight into the binding between the pyridine moiety and the azobenzene derivatives 5 and 8 which are new dyes introduced here. These two molecules were co-crystallized with the bis-pyridine derivative 10 in a 2:1 ratio.
Good-quality single crystals of the complex 10·(5)2 were obtained by slow diffusion of diethyl ether into a methanol solution containing the two starting compounds at room temperature. X-ray diffraction analysis shows that each molecule of 10 binds two different phenol dyes 5via short N⋯H hydrogen bonds (Fig. 3a). The O⋯N distance is 2.557(2) Å, the C–O⋯N angle is 125.68(3)° and a sigmoidal trimeric unit is formed. The observed hydrogen bond is among the shortest found in the Cambridge Structural Database (CSD) and is comparable to that found in the pentachlorophenol/4-methyl-pyridine co-crystal wherein the O⋯N distance is 2.515 Å.26 The O–H⋯N hydrogen bond present in the co-crystal 10·(5)2 is shorter than that found in a similar co-crystal between the nonfluorinated analogue 7 and 1,2-di-pyridylethane (O⋯N distance 2.703 Å).9 This difference in hydrogen bond distances of co-crystals formed by the fluorinated and hydrogenated azophenols 5 and 7 parallels very well the difference in interaction energies from theoretical calculations (Table 1), i.e., the enhanced electron density acceptor capability of the phenolic hydrogen in 5, leading to a shorter and stronger hydrogen bond than in 7, is consistently proved by experimental and computational studies.
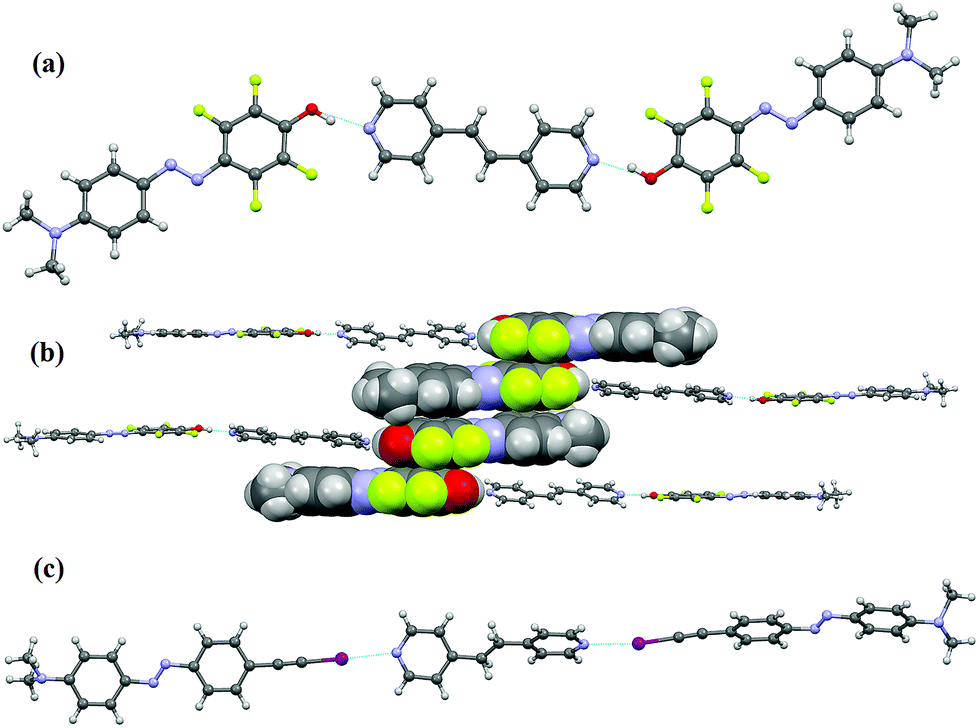 | ||
| Fig. 3 (a) Ball-and-stick representation of a 10·(5)2 hydrogen-bonded trimer. The diazo-molecule is disordered by rotation of 180° around the O⋯N (amino) axis and only one of the two models is shown. (b) View along the crystallographic b-axis of four 10·(5)2 trimers. Four molecules of compound 5, giving strong quadrupolar interactions, are shown in a spacefill style. (c) Ball-and-stick representation of a 10·(8)2 halogen-bonded trimer. Both halogen-bond donor and acceptor are heavily disordered over two positions and here only one of the two models is represented. Hydrogen and halogen bonds are pictured as blue dotted lines. Colour code: carbon, grey; hydrogen, white; oxygen, red; nitrogen, blue; fluorine, green and iodine, purple. | ||
In the three-dimensional crystal packing, the 10·(5)2 trimers form well-defined anti-parallel dimers due to an off-set stacking of the tetrafluorobenzene rings (the centroid–centroid distance is 3.445 Å) (Fig. 3b).27 Further π⋯π stacking involves the tetrafluorobenzene rings and the dimethylaminobenzene rings of adjacent dimers or trimers (the centroid–centroid distance is 3.684 Å).28 Such stacking interactions, while useful in crystal engineering29 as well as in liquid-crystal self-assembly,30 may cause dye aggregation within polymer matrices, thus preventing high doping concentrations that would be beneficial in optoelectronic applications.
The co-crystal formed between 8 and 10 is a strong example of the high directionality of halogen bonding. Single crystals of 10·(8)2 were obtained by slow evaporation at room temperature of a THF solution containing the two starting modules. The single crystal analysis revealed the formation of rod-like halogen-bonded trimers in which a 1,2-di(4-pyridyl)ethylene molecule bridges two iodoethynyl azo-dyes (Fig. 3c) by short and linear halogen bonds with N⋯I distance of 2.754(3) Å and C–I⋯N angle of 175.29(10)°. The disorder present in the structure prevents the possible further aggregation of these trimers to be analysed. The structural characterization of the model co-crystals highlights that both of the new dyes 5 and 8 interact strongly with pyridine-bearing entities; therefore their incorporation into the P4VP matrix may lead to the formation of new supramolecular polymeric materials with efficient photoswitching functionalities.
Photochemical studies
Table 2 shows the absorption maxima, relative decreases in absorbance upon irradiation (488 nm, 50 mW cm−2, circular polarization), and time constant estimates of the cis–trans thermal isomerization of thin films of the supramolecular complexes between P4VP and dyes 1–9. The absorbance decrease relates to the amount of cis-isomers in the photostationary state. The thermal cis–trans relaxation is a known non-monoexponential process in heterogeneous polymer environments and fitting the relaxation curves as a sum of first-order processes lacks physical justification.31 Hence the relaxation time constants, which are obtained using reported procedures,32 are only approximate and are given to facilitate the comparison among compounds of the molecular library of Fig. 1.| P4VP(1) | P4VP(2) | P4VP(3) | P4VP(4) | P4VP(5) | P4VP(6) | P4VP(7) | P4VP(8) | P4VP(9) | |
|---|---|---|---|---|---|---|---|---|---|
| a Obtained from drop-cast films of P4VP(n)0.01 with thickness of several microns. b Obtained from spin-coated films of P4VP(n)0.1, thickness: 150 ± 10 nm. | |||||||||
| λ max (nm) | 456 | 467 | 467 | 454 | 463 | 442 | 415 | 454 | 453 |
| A pss/A0a | 0.79 | 0.70 | 0.69 | 0.70 | 0.73 | 0.69 | 0.78 | 0.52 | 0.53 |
| τcis–transa (S) | 910 | 350 | 620 | 620 | 250 | 370 | 440 | 1250 | 1250 |
| DEb (%) | 0.6 | 2.9 | 6.5 | 1.8 | 6.0 | 2.4 | 5.0 | 7.5 | 3.4 |
| d (nm) | 30 | 105 | 175 | 80 | 170 | 95 | 130 | 180 | 115 |
Apart from compounds 6 and 7, the absorption maxima of the molecules studied are very similar. Fluorination of the bond-donor ring red-shifts the λmax by 25 nm for the iodo-substituted azobenzenes (6vs.3) and 48 nm upon phenol-substitution (7vs.5). The ethynyl group plays a similar, though not as pronounced, role (6vs.8), due to an increase in the Hückel conjugation length. The absorbance decrease upon illumination is very similar, around 30% for compounds 2–6, and around 20% for 1 and 7. Due to similar trans–cis isomerization efficiencies, the photoinduced surface patterning may be compared well with each other within our set of molecules. Interestingly, the photostationary state is much more cis-rich for the ethynyl-substituted molecules 8 and 9, and the lifetime of the cis-isomer is longer, possibly because of the larger aspect ratio of these molecules as compared to those of 1–7.
To summarize, the compounds shown in Fig. 1 cover the range from weak to strong halogen and hydrogen bond donors and the fact that a central core providing similar absorption properties can be maintained allows for facile and valid comparisons between these dyes in the same polymer matrix.
Supramolecular hierarchy in SRG formation
The molecular library presented in Fig. 1 allows us to explore various aspects of the surface patterning process, particularly to address the role of (i) the interaction strength, (ii) halogen versus hydrogen bonding, and (iii) the ethynyl group in the efficiency of the SRG inscription. Such a comprehensive comparison is only valid when using a polymer host of low polydispersity (we used P4VP with Mw = 1200 g mol−1; Mw/Mn = 1.2) containing equal amounts of noncovalently attached azobenzene units (10% mol mol−1 in this case), and films of equal thickness (150 ± 10 nm).The best way to study the efficiency of SRG formation is to monitor the first-order diffraction generated by periodic modulation of the sample surface due to interference irradiation. Several simultaneously formed and coupled gratings contribute to the overall diffraction efficiency,33 but in thin amorphous azo-polymer films, by far the largest contribution arises from the SRG formation. Hence, we can relate the efficiency of the SRG formation to the growth dynamics of the diffracted signal. All samples provided high-quality sinusoidal gratings with a well-defined modulation depth ranging from 30 to 180 nm (Table 2).
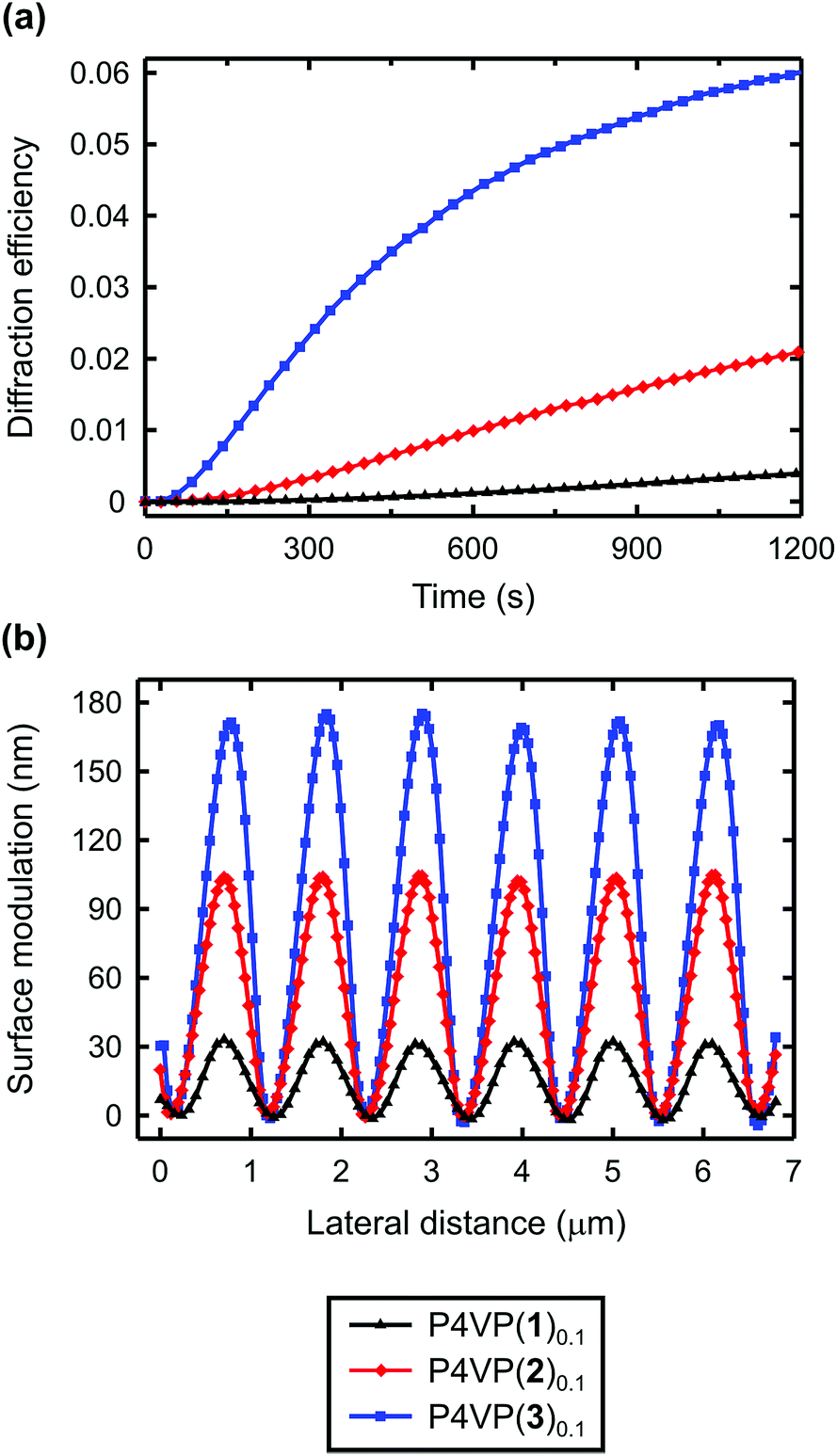 | ||
| Fig. 4 (a) Kinetics of the first-order diffraction efficiency upon the grating inscription process, and (b) AFM surface profiles of the gratings for P4VP(n)0.1, where n = 1 (F is para to the azo group), 2 (Br is para to the azo group), and 3 (I is para to the azo group). | ||
The diffraction efficiency curves for the complexes between P4VP and 3, 5–7 are presented in Fig. 5. As expected, based on Fig. 4, for the halogen-bonded complexes the diffraction efficiency increases with increasing interaction strength; hence fluorination of the iodobenzene ring of 6 to afford 3 greatly enhances the SRG formation. In the systems based on azophenols 5 and 7, the diffraction kinetics is rather similar but the diffraction efficiency is systematically higher for the fluorinated one P4VP(5)0.1. The surface-modulation depths after 30 min inscription are ca. 170 nm and ca. 130 nm for P4VP(5)0.1 and P4VP(7)0.1, respectively. The difference is likely due to slightly higher interaction strength and more confined electropositive surface of the former.
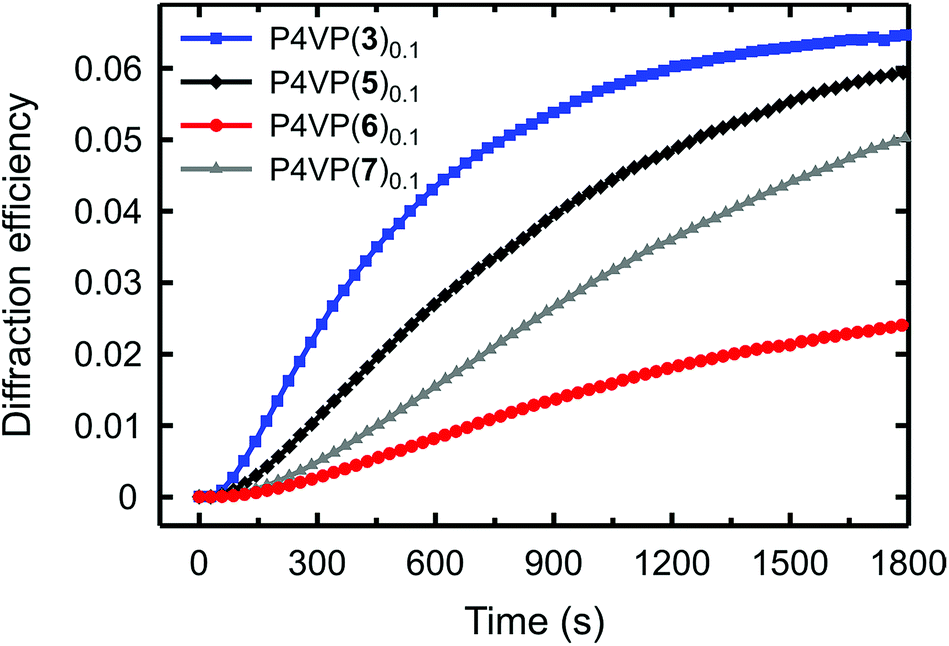 | ||
| Fig. 5 Kinetics of the first-order diffraction efficiency upon the grating inscription process for P4VP(n)0.1, where n = 3 (I is para to the azo group), 5 (OH is in the para position), 6 (I is in the para position on a non-fluorinated ring), and 7 (OH is in the para position on a non-fluorinated ring). | ||
The most interesting comparison is that between P4VP(3)0.1 and P4VP(5)0.1, that is, between similar halogen-bonded and hydrogen-bonded complexes. Even if the eventual diffraction efficiency and the modulation depth are practically the same, the dynamics of the diffraction growth is significantly faster for the halogen-bonded complex, i.e., the SRG formation efficiency is higher for the iodinated dye despite its much weaker interaction with the polymer. As their chemical structures are identical apart from the bond-donor group, and the spectral and photochemical properties are comparable, this comparison unambiguously shows that, when sufficiently strong, halogen bonding surpasses hydrogen bonding in driving the light-induced mass transport in polymer–azobenzene complexes, due to its higher directionality. The order of the efficiency is reversed only when halogen bonding is significantly weaker than hydrogen bonding.
The diffraction curves are shown in Fig. 6, and based on them we can draw two important conclusions. First, among the nine supramolecular systems studied, the iodoethynyl-based P4VP(8)0.1 complex is the most efficient for SRG formation. The difference with P4VP(3)0.1 is not large, but it is systematic and repeatable. We believe that such enhancement results from more favourable photochemical properties, and possibly larger free-volume sweep upon isomerization, which may enhance the SRG formation.35 Note also that the longer cis-lifetime does not seem to hinder the mass transport process as we suggested earlier. The fact that we can maintain efficient optical performance while removing the fluorine atoms from the bond-donor ring may be a great advantage when designing polymeric optical materials based on halogen bonding. The reduced quadrupolar interactions between the chromophores probably allow for using higher chromophore loadings without crystallization and phase separation of the dye, which may be beneficial in applications that require, e.g., high optical anisotropy or nonlinear optical response.
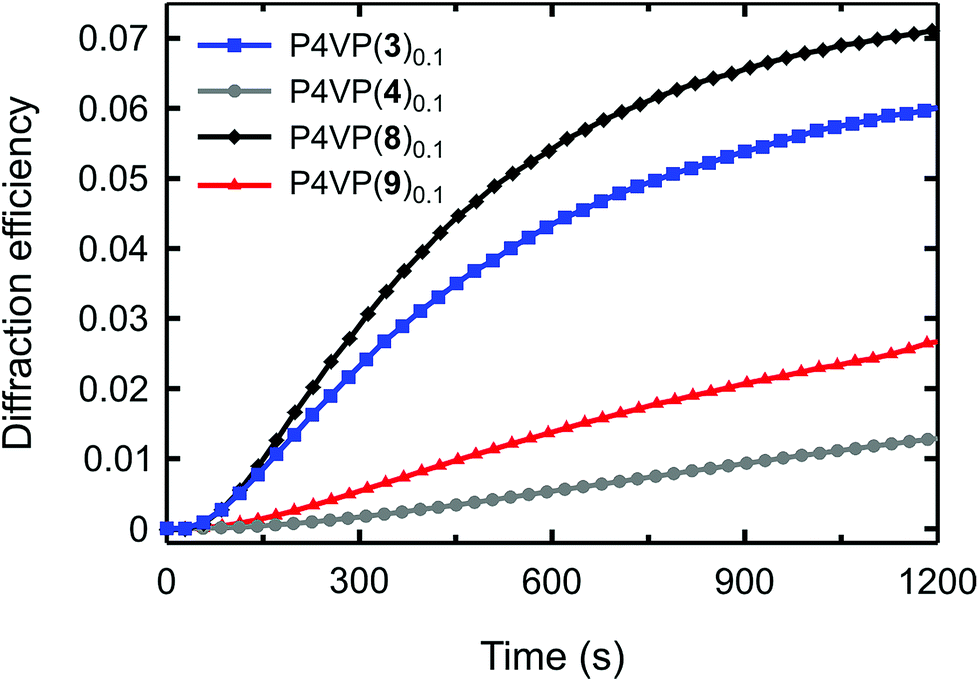 | ||
| Fig. 6 Time evolution of the first-order diffraction efficiency upon the SRG inscription process for P4VP(n)0.1, where n = 3 (I is in the para position), 4 (H is in the para position), 8 (iodoacetylene is in the para position), and 9 (acetylene is in the para position). | ||
The second important conclusion relates to the role of directionality of the polymer–azobenzene noncovalent interaction in the SRG formation. The P4VP(9)0.1 complex performs worse than the halogen-bonded complexes (Fig. 6), but better than P4VP(4)0.1. This relative efficiency supports the data of Fig. 5 and suggests that more directional interactions of comparable strength lead to more efficient mass transport. We attribute this trend to formation of electrostatics-controlled rigid polymer–azobenzene junctions that “force” the passive polymer chains to follow the movements of the azobenzene molecules upon isomerization. The role of directionality explains the potential of halogen bonding in this application, and also calls for further investigation into the SRG formation in, e.g., complexes driven by strong and directional multiple hydrogen bonds.36
Experimental
Compounds 2, 3 and 4 were synthesized as previously reported.9 Dye 7 was purchased from TCI Europe. Synthesis of azobenzenes 1, 5, 6, 8 and 9 is described in detail in the ESI.†The molecular structures of dyes 1–9 were optimized in DMF within the DFT approach, using the PBE0 functional,37 which has been judged to be well suited for describing the electronic and optical features of a series of organic dyes,38 and treating the energetic and geometric features of halogen bonding, both in vacuo39 and in a solvent.40 The 6-311++G(d,p) basis set was used for all atoms. The solvation effects have been included by means of the Polarizable Continuum Model (PCM).41 to determine the absorption wavelengths, standard vertical time dependent42 PBE0/6-311++G(d,p) calculations have been carried out. The molecular dimers of compounds 1–9 with 4-methyl pyridine have been optimized at the PBE0/6-311++G** level of theory in vacuo. Interaction energies ΔEBSSE have been computed by optimization on the BSSE-free potential energy surface as the difference between the energy of the dimer and the sum of the energies of the single monomers. BSSE correction was made by the standard counterpoise method.43 All calculations have been performed with the Gaussian suite of programs.44
The single-crystal X-ray structures were determined using a Bruker Kappa Apex II CCD diffractometer with Mo Kα radiation (λ = 0.71073 Å) and a Bruker Kryoflex low-temperature device. Crystals were mounted in inert oil on glass fibers. Data collection and reduction were performed by SMART and SAINT and absorption correction, based on the multi-scan procedure, by SADABS. The structures were solved by SIR92 and refined on all independent reflections by full-matrix least-squares based on Fo2 by using SHELX-97. Crystallographic data are reported in Table S1.† CIFs containing full crystallographic data can be obtained free of charge from the ESI.†
Surface-relief gratings were inscribed on thin films (150 ± 10 nm as measured with a DEKTAK 6M surface profiler) spin-cast from freshly prepared, filtered DMF solutions of the azobenzene–polymer mixtures on quartz substrates. The inscription was performed using a spatially filtered and collimated Ar+ laser beam (Coherent Innova 70) with circular polarization at a wavelength of 488 nm with an irradiation intensity of 150 mW cm−2. Lloyd's mirror interferometer, with an incidence angle of 15° (spatial period of 1 μm) was used to create the light-interference pattern. The dynamics of the gratings was monitored by measuring the transmitted first-order diffracted beam from a normally incident 633 nm He–Ne laser. The diffraction efficiency of the gratings was defined as η = I1/I0, where I1 and I0 are the intensities of the first-order diffracted beam and the transmitted beam prior to irradiation, respectively. The surface profiles of the inscribed gratings were characterized using a Veeco Dimension 5000 atomic-force microscope.
The UV-Vis spectra were collected from both thin films and dilute (10−5 M) DMF solutions with an Ocean Optics USB2000+ fiber-optic spectrometer and a DH-2000-BAL light source, both in the dark and under irradiation (488 nm, 50 mW cm−2). Thermal cis–trans isomerization was studied by exciting the chromophores to the cis-state with a circularly polarized pump beam (488 nm, 50 mW cm−2) and monitoring the transmittance changes after blocking the pump. The probe was a fiber-coupled xenon lamp equipped with proper bandpass filters. The signal was detected with a photodiode and a lock-in amplifier.
Conclusions
We have investigated the formation of photoinduced surface patterns in halogen-bonded and hydrogen-bonded polymer–azobenzene supramolecular complexes. Using an extensive molecular library we were able to draw several important conclusions on the light-induced mass transport process. First of all, halogen-bonded complexes outperform the corresponding hydrogen-bonded complexes (cf.Fig. 1, compounds 3 and 5) in terms of surface patterning efficiency, as a result of the higher directionality of halogen bonding. Greater directionality promotes more efficient mass transport also when hydrogen bonding is responsible for the dye–polymer binding. Furthermore, within the halogen-bonded series (cf. molecules 1, 2, 3, and 6) the efficiency increases with the interaction strength. The most efficient halogen-bond donor motif that comes on top of the supramolecular hierarchy in driving SRG formation is the iodoacetylene dye 8. This efficiency is due to its favourable photochemical properties as compared to the tetrafluoroiodobenzene motif. We believe this motif to widen the future perspectives of the design of halogen-bonded optical and photoresponsive materials by allowing for high chromophore loadings to be used in polymer matrices. Research in this direction is currently ongoing in our laboratories and will be reported elsewhere.Acknowledgements
PM acknowledges the MIUR for the PRIN 2010–2011 InfoChem project. GR acknowledges the MIUR for the PRIN 2010–2011 project no. 2010ERFKXL. AF acknowledges the CINECA-LISA Award N. HPL13P3CCB, 2013, for the availability of high-performance computing resources and support. AGH wishes to acknowledge FQRNT for a B3 post-doctoral fellowship. JV wishes to acknowledge Emil Aaltonen Foundation for a postdoctoral grant. AP acknowledges the Politecnico di Milano International Fellowship Program, the Academy of Finland, and the Emil Aaltonen Foundation for financial support.Notes and references
- M. M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348 CrossRef CAS PubMed; H. Yu and T. Ikeda, Adv. Mater., 2011, 23, 2149 CrossRef PubMed; Z. Mahimwalla, K. G. Yager, J. Mamiya, A. Shishido, A. Priimagi and C. J. Barrett, Polym. Bull., 2012, 69, 967 CrossRef; L. De Sio, N. Tabiryan, T. Bunning, B. R. Kimball and C. Umeton, Prog. Opt., 2013, 58, 1 Search PubMed; T. Seki, S. Nagano and M. Hara, Polymer, 2013, 54, 6053 CrossRef PubMed.
- M. H. Dhammika Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809 RSC.
- T. Ikeda, J. Mater. Chem., 2003, 13, 2037 RSC; A. Priimagi, C. J. Barrett and A. Shishido, J. Mater. Chem. C, 2014, 2, 7155 RSC.
- N. K. Viswanathan, D. Y. Kim, S. Bian, J. Williams, W. Liu, L. Li, L. Samuelson, J. Kumar and S. K. Tripathy, J. Mater. Chem., 1999, 9, 1941 RSC; A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139 CrossRef CAS PubMed.
- S. I. Na, S. S. Kim, J. Jo, S. H. Oh, J. Kim and D. Y. Kim, Adv. Funct. Mater., 2008, 18, 3956 CrossRef CAS; S. Lee, H. S. Kang and J. K. Park, Adv. Mater., 2012, 24, 2069 CrossRef PubMed; L. M. Goldenberg, V. Lisinetskii, Y. Gritsai, J. Stumpe and S. Schrader, Adv. Mater., 2012, 24, 3339 CrossRef PubMed; A. Priimagi and A. Shevchenko, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 163 CrossRef.
- (a) O. Kulikovska, L. M. Goldenberg and J. Stumpe, Chem. Mater., 2007, 19, 3343 CrossRef CAS; (b) J. Gao, Y. N. He, F. Liu, X. Zhang, Q. Z. Wang and X. G. Wang, Chem. Mater., 2007, 19, 3877 CrossRef CAS; (c) N. Zettsu, T. Ogasawara, N. Mizoshita, S. Nagano and T. Seki, Adv. Mater., 2008, 20, 516 CrossRef CAS; (d) Q. Zhang, X. Wang, C. J. Barrett and C. G. Bazuin, Chem. Mater., 2009, 21, 3216 CrossRef CAS; (e) A. Priimagi, K. Lindfors, M. Kaivola and P. Rochon, ACS Appl. Mater. Interfaces, 2009, 1, 1183 CrossRef CAS PubMed.
- T. Alasaarela, D. Zheng, L. Huang, A. Priimagi, B. Bai, A. Tervonen, S. Honkanen, M. Kuittinen and J. Turunen, Opt. Lett., 2011, 36, 2411 CrossRef PubMed.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711 CrossRef CAS.
- A. Priimagi, G. Cavallo, A. Forni, M. Gorynsztejn-Leben, M. Kaivola, P. Metrangolo, R. Milani, A. Shishido, T. Pilati, G. Resnati and G. Terraneo, Adv. Funct. Mater., 2012, 22, 2572 CrossRef CAS.
- T. Clark, M. Henneman, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291 CrossRef CAS PubMed; T. Clark, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 13 CrossRef.
- K. E. Riley and P. Hobza, Phys. Chem. Chem. Phys., 2013, 15, 17742 RSC; A. Forni, S. Pieraccini, S. Rendine and M. Sironi, J. Comput. Chem., 2014, 35, 386 CrossRef CAS PubMed.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686 CrossRef CAS PubMed.
- X. Ding, M. Tuikka and M. Haukka, Halogen Bonding in Crystal Engineering, in Recent Advances in Crystallography, ed. J. B. Benedict, InTech, New York, 2012, pp. 143–168 RSC; M. Saccone, G. Cavallo, P. Metrangolo, A. Pace, I. Pibiri, T. Pilati, G. Resnati and G. Terraneo, CrystEngComm, 2013, 15, 3102 RSC; R. W. Troff, T. Mäkelä, F. Topić, A. Valkonen, K. Raatikainen and K. Rissanen, Eur. J. Org. Chem., 2013, 9, 1617 CrossRef CAS.
- D. Yan, A. Delori, G. O. Lloyd, T. Friščić, G. M. Dayet, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duanal, Angew. Chem., Int. Ed., 2011, 50, 12483 CrossRef CAS PubMed; O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205 CrossRef PubMed.
- D. W. Bruce, in Supramolecular Chemistry: From concept to Nanomaterials, ed. Steed J. W. and Gale P. A., John Wiley & Sons, Ltd, 2012, p. 3493 Search PubMed.
- A. Priimagi, M. Saccone, G. Cavallo, A. Shishido, T. Pilati, P. Metrangolo and G. Resnati, Adv. Mater., 2012, 24, OP345 Search PubMed; M. Saccone, G. Terraneo, T. Pilati, G. Cavallo, A. Priimagi, P. Metrangolo and G. Resnati, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 149 Search PubMed; O. S. Bushuyev, T. C. Corkery, C. J. Barrett and T. Friščić, Chem. Sci., 2014, 5, 3158 RSC; Y. Chen, H. Yu, L. Zhang, H. Yang and Y. Lu, Chem. Commun., 2014, 50, 9647 RSC.
- J. P. W. Wong, A. C. Whitwood and D. W. Bruce, Chem.–Eur. J., 2012, 18, 16073 CrossRef CAS PubMed; A. Takemura, L. J. McAllister, S. Hart, N. E. Pridmore, P. B. Karadakov, A. C. Whitwood and D. W. Bruce, Chem.–Eur. J., 2014, 20, 6721 CrossRef PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- C. B. Aakeröy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem.–Eur. J., 2013, 19, 16240 CrossRef PubMed.
- O. Ikkala and G. ten Brinke, Chem. Commun., 2004, 2131 RSC.
- R. Bertani, P. Metrangolo, A. Moiana, E. Perez, T. Pilati, G. Resnati, I. Rico-Lattes and A. Sassi, Adv. Mater., 2002, 14, 1197 CrossRef CAS.
- S. Xiao, X. Lu and Q. Lu, Macromolecules, 2007, 40, 7944 CrossRef CAS; A. Priimagi, J. Vapaavuori, F. J. Rodriguez, C. F. J. Faul, M. T. Heino, O. Ikkala, M. Kauranen and M. Kaivola, Chem. Mater., 2008, 20, 6358 CrossRef; Q. Zhang, C. G. Bazuin and C. J. Barrett, Chem. Mater., 2008, 20, 29 CrossRef; J. Vapaavuori, V. Valtavirta, T. Alasaarela, J. Mamiya, A. Priimagi, A. Shishido and M. Kaivola, J. Mater. Chem., 2011, 21, 15437 RSC.
- G. A. Jeffrey, An Introduction to Hydrogen Bond, Oxford University Press, New York, 1997 Search PubMed.
- K. E. Riley, J. S. Murray, J. Fanfrlik, J. Rezac, R. J. Sola, M. C. Concha, F. M. Ramos and P. Politzer, J. Mol. Model., 2011, 17, 3309 CrossRef CAS PubMed.
- A. C. Legon, Angew. Chem., Int. Ed., 1999, 38, 2686 CrossRef CAS; Z. P. Shields, J. S. Murray and P. Politzer, Int. J. Quantum Chem., 2010, 110, 2823 CrossRef; P. Politzer and J. S. Murray, ChemPhysChem, 2013, 14, 278 CrossRef PubMed.
- (a) I. Majerz, Z. Malarski and L. Sobczyk, Chem. Phys. Lett., 1997, 274, 361 CrossRef CAS; (b) T. Steiner, I. Majerz and C. C. Wilson, Angew. Chem., Int. Ed., 2001, 40, 2651 CrossRef CAS.
- J. H. Williams, J. K. Cockcroft and N. Fitch, Angew. Chem., Int. Ed., 1992, 31, 1655 CrossRef.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210 CrossRef CAS PubMed.
- M. H. Yoon, A. Facchetti, E. S. Charlotte and T. J. Marks, J. Am. Chem. Soc., 2006, 128, 5792 CrossRef CAS PubMed.
- K. Kishikawa, Isr. J. Chem., 2012, 52, 800 CrossRef CAS.
- C. S. Paik and H. Morawetz, Macromolecules, 1972, 5, 171 CrossRef CAS.
- C. Barrett, A. Natansohn and P. Rochon, Macromolecules, 1994, 27, 4781 CrossRef CAS; C. Barrett, A. Natansohn and P. Rochon, Chem. Mater., 1995, 7, 899 CrossRef.
- F. Lagugné Labarthet, P. Rochon and A. Natansohn, Appl. Phys. Lett., 1999, 75, 1377A CrossRef CAS PubMed; A. Sobolewska and S. Bartkiewicz, Appl. Phys. Lett., 2008, 92, 253305 CrossRef PubMed; H. Audorff, R. Walker, L. Kador and H. W. Schmidt, J. Phys. Chem. B, 2009, 113, 3379 CrossRef PubMed; A. Sobolewska, S. Bartkiewicz, A. Miniewicz and E. Schab-Balcerzak, J. Phys. Chem. B, 2010, 114, 9751 CrossRef PubMed.
- J. W. Lauher, F. W. Fowler and N. S. Goroff, Acc. Chem. Res., 2008, 41, 1215 CrossRef CAS PubMed; C. Perkins, S. Libri, H. Adams and L. Brammer, CrystEngComm, 2012, 14, 3033 RSC; O. Dumele, D. Wu, N. Trapp, N. Goroff and F. Diederich, Org. Lett., 2014, 16, 4722 CrossRef PubMed.
- A. Goulet-Hanssens, T. C. Corkery, A. Priimagi and C. J. Barrett, J. Mater. Chem. C, 2014, 2, 7505 RSC.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382 CrossRef CAS; J. V. Barth, Annu. Rev. Phys. Chem., 2007, 58, 375 CrossRef PubMed.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029 CrossRef CAS PubMed; C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef PubMed.
- D. Jacquemin, V. Wathelet, E. A. Perpète and C. Adamo, J. Chem. Theory Comput., 2009, 5, 2420 CrossRef CAS; D. Jacquemin, A. Planchat, C. Adamo and B. Mennucci, J. Chem. Theory Comput., 2012, 8, 2359 CrossRef.
- S. Kozuch and J. M. L. Martin, J. Chem. Theory Comput., 2013, 9, 1918 CrossRef CAS.
- A. Forni, S. Rendine, S. Pieraccini and M. Sironi, J. Mol. Graphics Modell., 2012, 38, 31 CrossRef CAS PubMed.
- M. Cossi and V. Barone, J. Chem. Phys., 2001, 115, 4708 CrossRef CAS PubMed.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997 CrossRef CAS; R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218 CrossRef PubMed; M. E. Casida, J. Mol. Struct.: THEOCHEM, 2009, 914, 3 CrossRef PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- M. Frisch, et al., Gaussian 09, Revision D.01., Gaussian Inc., Wallingford CT, 2013 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis procedures and compound characterization, computational details, description of photochemical experiments, and crystal data. CCDC 1025655 and 1025656. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4tc02315c |
| ‡ Present address: Department of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany. |
| § Present address: Département de Chimie, Université de Montréal, Montréal, QC, Canada H3C 3J7. |
| This journal is © The Royal Society of Chemistry 2015 |