
Non-aqueous sol–gel synthesis of hybrid rare-earth-doped γ-Ga2O3 nanoparticles with multiple organic–inorganic-ionic light-emission features†
Roberto
Lorenzi
*a,
Alberto
Paleari
ab,
Nikita V.
Golubev
b,
Elena S.
Ignat'eva
b,
Vladimir N.
Sigaev
b,
Markus
Niederberger
c and
Alessandro
Lauria
*c
aDepartment of Materials Science, University of Milano-Bicocca, via R. Cozzi 55, I-20125 Milano, Italy. E-mail: roberto.lorenzi@mater.unimib.it
bP. D. Sarkisov International Laboratory of Glass-Based Functional Materials, Mendeleyev University of Chemical Technology of Russia, Miusskaya Square 9, 125047 Moscow, Russia
cDepartment of Materials, ETH Zürich, Vladimir-Prelog-Weg 5, 8093 Zurich, Switzerland. E-mail: alessandro.lauria@mat.ethz.ch
First published on 28th October 2014
Abstract
We present a novel strategy for the synthesis of pure and Eu-doped γ-Ga2O3 nanoparticles with an in situ organic capping resulting from a non-aqueous solution-based benzyl alcohol synthesis route. Photoluminescence spectroscopy highlights the concomitant benzoate-related and γ-Ga2O3 exciton-like Eu3+ excitations in the UV, and a blue emission superimposed onto γ-Ga2O3 donor–acceptor recombination, ascribable to organic moieties different from benzoate.
Gallium sesquioxide (Ga2O3) is a wide band gap material (∼4.9 eV) currently used in the semiconductor industry, mainly as a target for the deposition of thin film transistors for next-generation flat-panel displays. Bulky and nanostructured Ga2O3 have also been widely investigated as luminescent materials,1 insulating barriers in magnetic tunnel junctions,2 and as active substrates for sensors and photocatalysis.3 The majority of these studies consider only the most stable form of Ga2O3, that is the monoclinic β phase, although Ga2O3 exists in five different polymorphs: α, β, γ, δ, and ε.4 Indeed, the metastable cubic γ phase presents several fascinating properties that may be crucial for further applications. The synthesis routes and the preparation of γ-Ga2O3 can be chemical, physical, or biochemical reactions. Moreover, it has been produced in the form of colloids or nanopowders through sol–gel derived synthesis,5 thin films through pulsed laser deposition,6 elongated platelets through enzymatic synthesis,7 and as nanoparticles (NPs) embedded in glass through a secondary phase transformation.8 All the reported syntheses result in nano- or microsized phases with very large surface areas and a large number of crystal defects. The large surface area is expected to improve catalytic and sensor performances, while the presence of localized hole and electron traps (i.e., cation and/or oxygen vacancies) is crucial in photonic and optoelectronic applications. In fact, the intense blue emission at around 460 nm, excited by the band-to-band transitions and quite ubiquitous in gallium oxide compounds, is the result of the radiative recombination of donor and acceptor pairs (DAPs). In these oxides, DAPs are formed by an oxygen vacancy
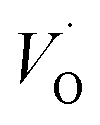 – the donor species and a pair of oxygen and gallium vacancies (VO,VGa)′, located at the same site – the acceptor species.9 Recently, this strong blue emission has been demonstrated to show a tuneable peak wavelength as a function of colloidal nanocrystal size5c and has been successfully coupled, via resonant energy transfer, to the emission of a suitable organic dye bonded onto the nanocrystal surface, thus realizing the first UV-pumped white light-emitting diode based on γ-Ga2O3.5b
– the donor species and a pair of oxygen and gallium vacancies (VO,VGa)′, located at the same site – the acceptor species.9 Recently, this strong blue emission has been demonstrated to show a tuneable peak wavelength as a function of colloidal nanocrystal size5c and has been successfully coupled, via resonant energy transfer, to the emission of a suitable organic dye bonded onto the nanocrystal surface, thus realizing the first UV-pumped white light-emitting diode based on γ-Ga2O3.5b
Herein, we demonstrate the feasibility of a one-pot non-aqueous solution-based synthesis of rare-earth-doped γ-Ga2O3 nanosystems with an organic shell. Unlike other syntheses, this approach enriches the spectroscopic features of the nanosystem, showing that the crystalline core, the embedded rare-earth dopant ions, and the organic capping resulting from this solvothermal reaction can all contribute to the light-emission response of the hybrid doped compound. The analysis of the excitation pattern points to the occurrence of mutual excitation mechanisms involving core, shell, and dopant species interactions, highlighting the versatility of this material as a phosphor with multiple excitation/emission channels.
Nanopowders were prepared by non-aqueous sol–gel chemistry. In a typical synthesis, 1 g of gallium(III) acetylacetonate was dissolved in 20 mL of anhydrous benzyl alcohol in a glovebox. For rare-earth-doped samples, the proper amount of europium acetate was added to the mixture so as to obtain a final doping content of 1% molEu/molGa. The solution was then poured in a 45 mL PTFE liner and transferred to a steel autoclave (Parr Instrument Company) and accurately sealed. The autoclave was removed from the glovebox and heated in a furnace at 200 °C for 2 days. The resulting milky suspension was washed with diethyl ether, and the obtained precipitate was dried in air at 60 °C for 12 hours.
Representative transmission electron microscopy (TEM) images of nanopowder are reported in Fig. 1a and b. The nanoparticles show an elongated shape with preferential growth along the (111) crystal axis (Fig. 1b), with a crystal size about 12 nm long and 5 nm wide. The Bragg reflections observed in the X-ray diffraction (XRD) patterns of Fig. 1c confirm the formation of γ-Ga2O3 (JCPDS 20-0426) and exclude the presence of other crystalline by-products. The Scherrer analysis of the (440) diffraction peak at 2θ = 64° indicates an average crystal size of ∼10 nm, in accordance with the TEM results. Importantly, the TEM and XRD data are significantly different from other non-aqueous sol–gel synthesis of γ-Ga2O3 based on benzylamine,5f suggesting that the benzyl alcohol route leads to a product with higher crystallinity.
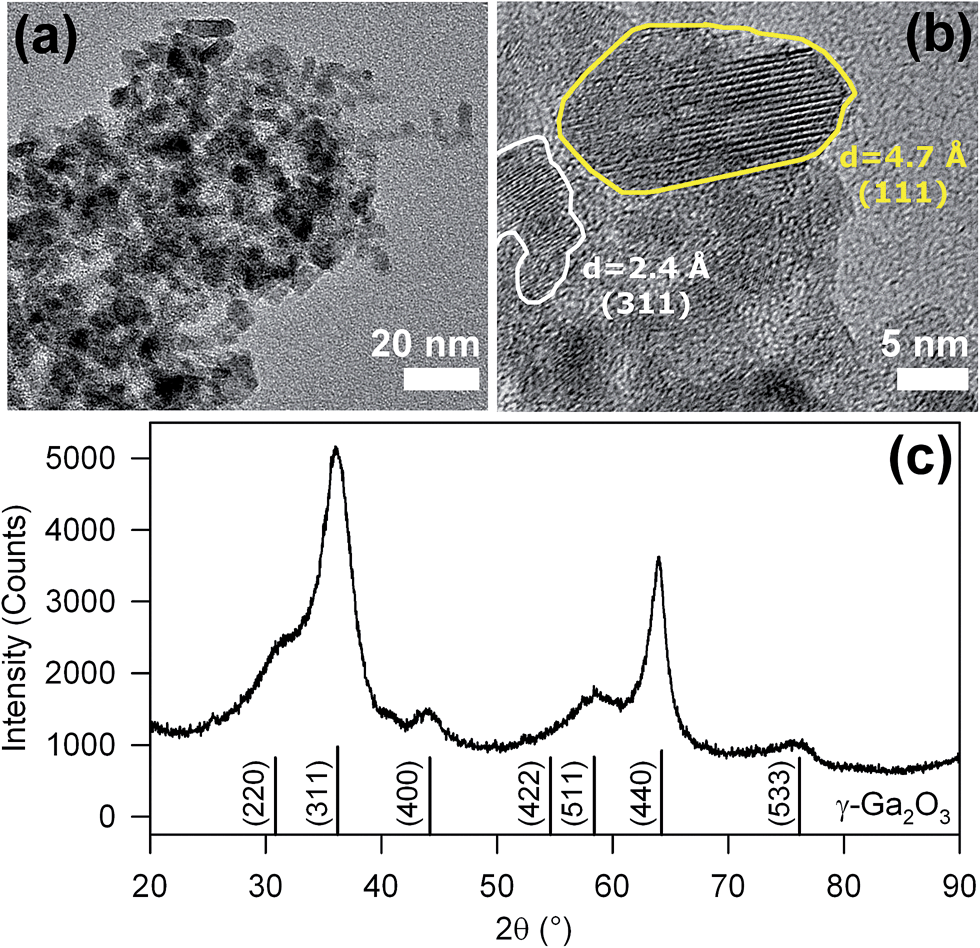 | ||
| Fig. 1 (a) TEM image of the as-synthetized NPs. (b) HR-TEM image of one NP showing crystalline features corresponding to the lattice parameters of γ-Ga2O3 and the typically elongated shape parallel to the (111) direction. (c) XRD pattern of the as-synthetized NPs; the calculated pattern of γ-Ga2O3 is reported for comparison. | ||
Thermal analysis combined with vibrational spectroscopy reveals the stability and structural features of both the core nanocrystals and the organic shell. Differential scanning calorimetry (DSC) and thermo gravimetric analysis (TGA) (Fig. 2a) evidence a weight loss of about 8% between 200 °C and 600 °C, concurrent with several exothermal peaks related to the decomposition of organic residuals. Raman analyses before and after heat treatment in air at 420 °C for 1 h confirm the removal of the organic component. In fact, the as-prepared powders exhibit narrow Raman peaks (Fig. 2b) at 650, 845, 1010, and 1030 cm−1 together with broader modes at 545, 620, 800, 820, and 935 cm−1, which are all consistent with the presence of benzoate groups coordinated to the NPs surface.10 The heated powders instead show only very broad and weak bands, already ascribed to the GaO stretching modes in γ-Ga2O3.3d,11 The removal of organics seems to be the only effect of the annealing, since the heated NPs show no significant changes in the XRD relative intensities and in the full width at half maximum (FWHM). This confirms that the metastable γ-phase is preserved after thermal treatment (see ESI†).
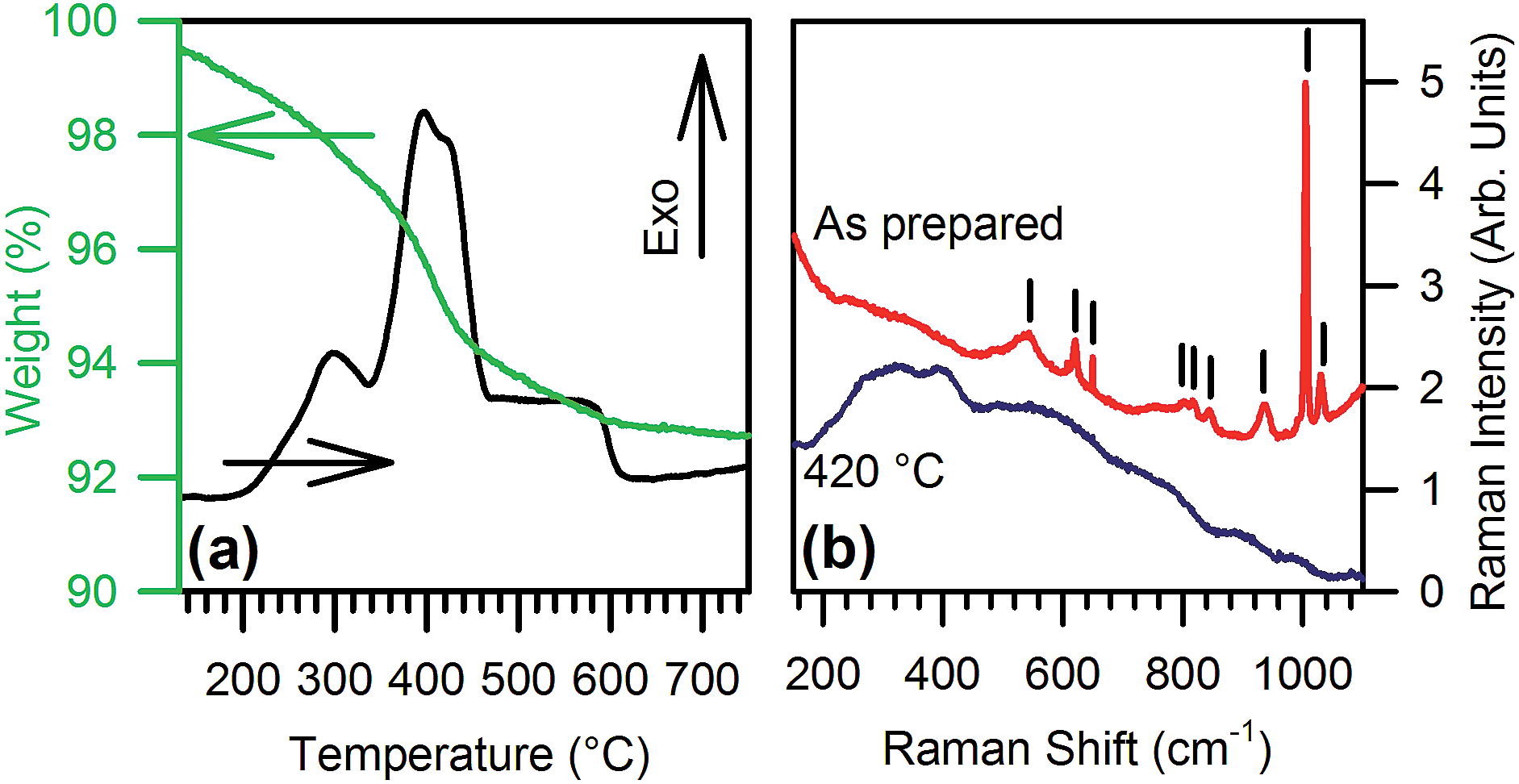 | ||
| Fig. 2 (a) TGA (green) and DSC (black) curves of the as-synthetized NPs. (b) Raman spectra of the as-synthetized NPs (red) and after treatment at 420 °C for 1 h (blue); the vertical bars indicate the peaks ascribed to benzoate. | ||
The photoluminescence (PL) and PL excitation (PLE) spectra of undoped and Eu-doped nanopowders without organic capping are reported in Fig. 3a. The emission and excitation patterns of the undoped sample show two distinct emission paths. The most pronounced band is centred in the blue region at 460 nm and arises from DAP recombination excited at 265 nm. A second, less intense contribution, centred at 330 nm, is due to an exciton-like emission with a specific excitation at 300 nm, as already observed in single-crystal gallium oxide,12 as well as in glass/γ-Ga2O3 nanocomposites.13 The spectra taken in the same conditions on Eu-doped samples (Fig. 3a, black line) do not show any emission from DAP recombination. The Eu-related emission is instead dominated by 5D0 → 7F0,1,2 red emission lines of Eu3+ at 570–670 nm (Fig. 3a, red line), excited by the sharp 4f–4f transitions and are within a broad UV excitation at around 290 nm (Fig. 3a). The latter excitation cannot be assigned to intra-centre Eu3+ transitions, suggesting an energy transfer from the γ-Ga2O3 host. The resonance between the NPs and Eu3+ ions may arise from the overlap between the exciton-like emission of γ-Ga2O3 and the 4f–4f transitions of Eu3+. The broad UV excitation of Eu3+ PL is in fact quite similar to the 300 nm exciton-like band of the undoped sample, rather than the DAP band at 265 nm. The occurrence of energy transfer is further confirmed by the time resolved PL data: the PL decays of the as-synthesized nanopowders (Fig. 3b) feature similar stretched exponential kinetics, irrespective of doping, except for a faster decay in the presence of Eu3+ ions. By assuming a Förster resonance energy transfer (FRET) mechanism governing the emission process, it is possible to evaluate the efficiency EFRET of the NP-Eu3+ system from the collected lifetimes.14 In fact, from the fluorescence lifetime of the donor species, i.e. the lifetime of the exciton-like emission of γ-Ga2O3 NPs, in the presence (τD,A) and in the absence (τD) of the acceptor species, i.e. the europium ions, the efficiency can be calculated by EFRET = 1 − τD,A/τD. Since the kinetics is not a pure single exponential, EFRET is evaluated through the excited state half-life τ1/2. By using experimental values of τ1/2D,A = 2.9 ns and τ1/2D = 4.0 ns, we calculated the effeciency of FRET from the γ-Ga2O3 exciton-like emission to Eu3+ to be EFRET = 27%.
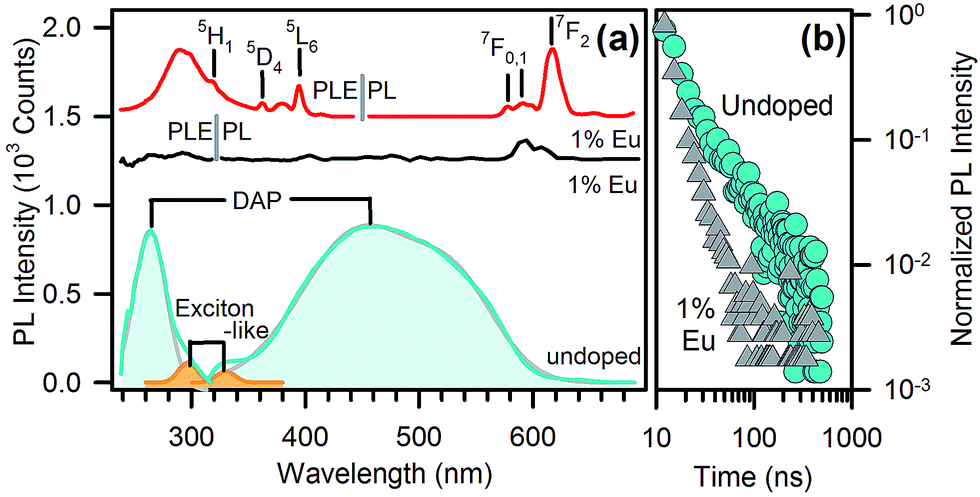 | ||
| Fig. 3 (a) Photoluminescence (PL) and photoluminescence excitation (PLE) spectra (λEXC = 250 nm, λEMI = 430 nm) of intrinsic DAPs and the exciton-like emission of undoped (blue line) and Eu-doped (black line, shifted up by 1200 counts for clarity) γ-Ga2O3 nanoparticles, and the PL/PLE spectra (λEXC = 290 nm, λEMI = 620 nm) of the ionic emission in Eu-doped γ-Ga2O3 NPs (red line, shifted up by 1500 counts for clarity). (b) Lifetime measurement of doped (grey triangles) and undoped (blue circles) γ-Ga2O3 nanoparticles, excited at 250 nm and monitored at 430 nm. | ||
On the other hand, when the organic capping is present, i.e. before thermal treatment, the full hybrid system shows a considerable enrichment of the PL and PLE pattern, with additional emission and excitation channels with respect to the treated samples. The PL and PLE of the as-synthesized NPs, irrespective of doping, are dominated by a mirror-like emission with emission and excitation maxima at 430 nm and 360 nm, respectively (Fig. 4a). This emission is clearly related to the organic shell because of its absence after thermal treatment. However, at least two other spectral features may excite the light emission at 430 nm in the full hybrid system, with maxima centred at 295 nm and 250 nm. Because the PLE spectra in Fig. 4a probe the same emission wavelength of the experiments in Fig. 3a, we expect possible contributions from the nanocrystal excitation spectrum. The shoulder at 295 nm in Fig. 4a can be attributed to an exciton-like contribution, which is observed also in the treated sample (Fig. 3a). The broad excitation band at 250 nm deserves some further comments and will be discussed in more detail later.
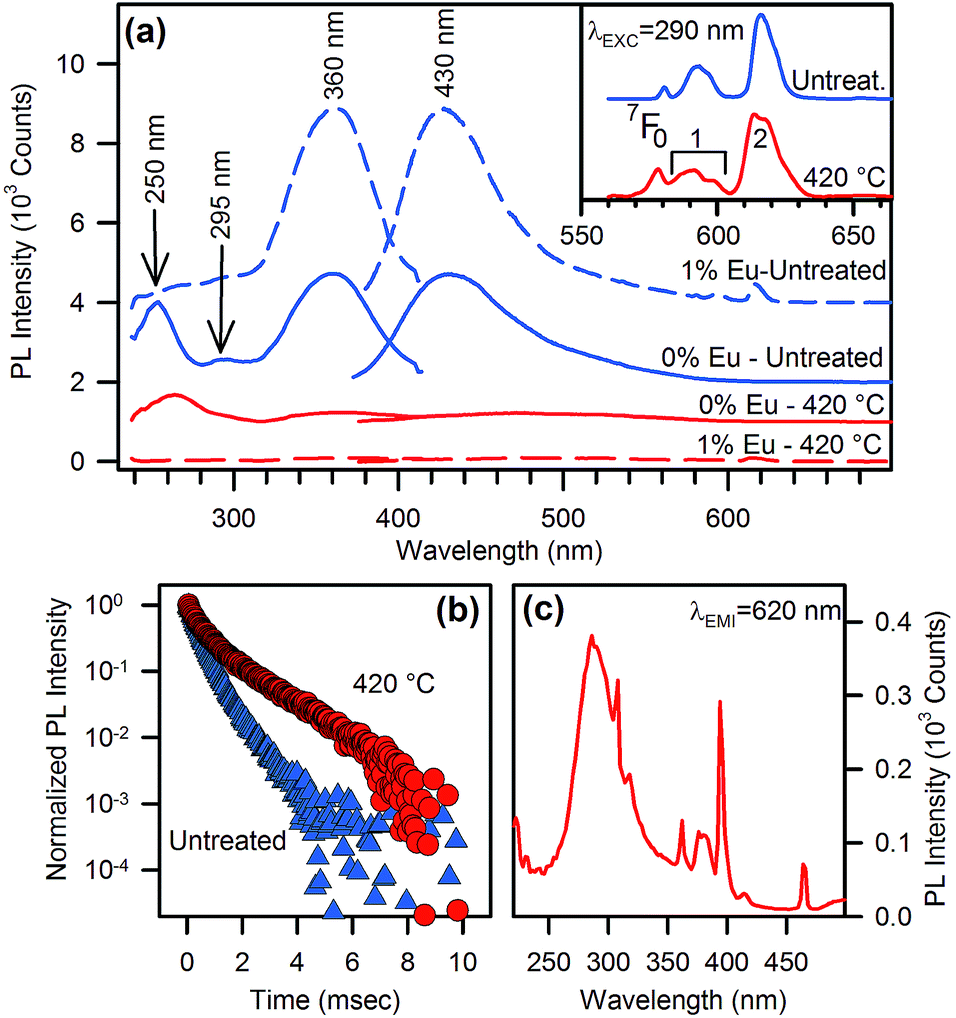 | ||
| Fig. 4 (a) PL/PLE (λEXC = 340 nm, λEMI = 430 nm) of undoped (full lines) and Eu-doped (dashed lines) γ-Ga2O3 NPs before (blue lines) and after (red lines) organics removal; the lines are upshifted by 1000 counts for clarity. Inset: PL (λEXC = 290 nm) of the Eu transition of untreated (blue) and treated NPs (red). (b) Lifetime measurements of the ionic emission in Eu-doped NPs before (triangles) and after (circles) thermal treatment excited at 250 nm and monitored at 620 nm. (c) Excitation spectra of Eu emissions of as-synthetized Eu-doped NPs monitored at 620 nm. | ||
Besides the organics and intrinsic related emissions, Eu3+ ion photophysics gives important evidence of the modifications induced by the thermal treatment. As regards Eu3+ PL (inset in Fig. 4a), the main changes after thermal treatment at 420 °C are: (i) intensity enhancement of the transition 5D0 → 7F0 compared with 5D0 → 7F1, (ii) broadened emissions, and (iii) the structuring of the spectrum in the 5D0 → 7F1,2 transition region. These changes are caused by: (i) a lowering of the Eu–O bond distance,15 (ii) a broader distribution of bond length and angles, and (iii) the occurrence of Eu ions at more than one preferred site, respectively. The latter conclusion is also supported by the lifetime measurements (Fig. 4b). After thermal treatment we observe a considerably slower decay, ascribable to a partial suppression of the non-radiative decay channels related to the organic residuals interacting with Eu3+ ions. Furthermore, the curves fitting unveils a biexponential behaviour, both in the pristine and thermally treated samples (see ESI†), but the thermal treatment leads to a smaller relative weight of the fast component and a drastic increase in the slow component lifetime (from 700 μs to 1.5 ms). As evidenced by the presence of extra peaks in the Eu3+ emission spectrum, these outcomes confirm that a distinct Eu3+ site configuration is introduced by a thermally activated structural rearrangement of the host lattice. Additionally, the relative intensity of the 5D0 → 7F2 and 5D0 → 7F1 transitions is a well-known optical probe of the local symmetry of the Eu3+ ion.16 Specifically, the absence of inversion symmetry and the lowering of local symmetry cause an enhancement of the first transition with respect to the latter one. The measured ratio (∼2.9 both in the as-prepared and thermally treated samples) indicates that the local symmetry of the Eu3+ sites is unaffected by treatment and is similar to the Eu3+ sites in the β-phase (∼2.7).17 Interestingly, this ratio is instead significantly lower in the α- phase (∼1.9), whose pure octahedral structure lacks the simultaneous occurrence of tetrahedral and octahedral sites, different from the β- and γ-phases.18
Eu-doping also shows clear-cut effects on the excitation pattern of the organic capping, providing a useful tool for clarifying the nature of the organic component. Specifically, we observe that the excitation spectrum of Eu3+ monitored at 620 nm shows a strong band corresponding to the exciton-like band at 290 nm, with some minor contributions of comparable intensity in the short- and long-wavelength tails at around 250 nm and 360 nm (Fig. 4c), which disappear after thermal treatment (Fig. 3a). As regards the organic-related blue emission at 430 nm, the presence of Eu ions does not affect the excitation channel at 360 nm, and depresses only the 250 nm excitation band. This band was ascribed to benzoate species attached to the oxide NPs surface.19 A similar attribution was suggested for the excitation band at 360 nm.19a,19b,20 Following these interpretations, the spectral bands at 250 nm and 360 nm should both belong to the same organic species responsible for the emission at 430 nm. Nevertheless, Eu-doping depresses only one of these two excitation channels. This outcome indirectly highlights that the excitation of the Eu-related emission does not occur via a FRET mechanism. In fact, Kasha's rule states that light emission from molecules can only occur from the lowest excited state.14 Accordingly, if the resonant transition responsible for the benzoate-to-Eu3+ energy transfer is the 430 nm emission with two PLE structures at 250 nm and 360 nm belonging to transitions of the same molecular species, then the presence of Eu3+ is necessarily expected to depress both the excitation channels with equal efficiency, contrary to the experimental data of Fig. 4a. Indeed, the main excitations of benzyl rings lie in the 200–250 nm region with emissions in the 300–350 nm and 360–450 nm regions for fluorescence and phosphorescence, respectively, even in presence of the carbonyl group.21 Accordingly, benzoate is unlikely to be responsible for the main excitation channel registered at 360 nm. Moreover, other recent studies on benzoate-enhanced emissions of yttrium oxides, in which the occurrence of other contaminants can be excluded,19,22 show that PL can be only excited in the 250 nm spectral region, as expected for benzoate species, leading only to a near UV fluorescence. Therefore, the presence of an additional organic component other than benzoate must be taken into account in the photophysics of the system, even if it is the main constituent of the organic capping layer, as confirmed by Raman, IR, and 14C-NMR identification.10a,20,23
As a consequence, our data in Fig. 4 evidence a more complex system, in which benzoate ligands occur, together with an additional organic species responsible for the strong luminescence peak at 430 nm and excited at 360 nm. As a matter of fact, the formation of several minor side products resulting from the benzyl alcohol route, including ketones,23 has already been reported; hence, the presence of molecules like aromatic alpha ketones cannot be excluded. On the other hand, aromatic alpha ketones like benzophenone, acetophenone, and propiophenone, typically possess spectroscopic signatures compatible with our experimental results and a PL quantum yield close to unity.21a Furthermore, the Raman spectrum of benzophenone,10b for instance, does not present relevant differences with respect to the species found in the capping layer. Consequently, the observed spectroscopy could be tentatively explained by the presence of such aromatic ketones, even in small quantities.
As discussed previously, the energy transfer to the Eu3+ ions occurs through resonant γ-Ga2O3 NP luminescence (Fig. 3) in the UV rather than through resonance with the DAP blue emission. In a similar way, the organic sensitized emission in untreated NPs involves the preferential coupling of the Eu3+ transitions with the UV π → π* emission of the benzoate species, whereas the energy transfer from the ketone-related n → π* blue emission is much less efficient.
In conclusion, we presented the synthesis of Eu-doped and undoped γ-Ga2O3 hybrid nanoparticles with an organic capping resulting from the nonaqueous process. The photophysics of the NPs demonstrates the role of the intrinsic and organic-related activation of Eu3+ PL. Importantly, the results highlight that the presence of a negligible amount of organic residues other than benzoate can be responsible for important optical features, due to their high PL efficiency.
The authors acknowledge the financial support by ETH Zürich, the Russian Federation under grant no. 11.G34.31.0027 and by grant MK-1398.2014.3, and Cariplo Foundation, Italy, under project no. 2012-0920. M. J. Süess and ScopeM – ETH Zürich are gratefully acknowledged for TEM imaging.
Notes and references
- (a) E. Nogales, B. Méndez, J. Piqueras and J. A. García, Nanotechnology, 2009, 20, 115201 CrossRef CAS PubMed; (b) M. L. Pang, W. Y. Shen and J. Lin, J. Appl. Phys., 2005, 97, 033511 CrossRef PubMed.
- J. C. Le Breton, H. Saito, S. Yuasa and K. Ando, Appl. Phys. Lett., 2009, 94, 152101 CrossRef PubMed.
- (a) X. Wang, Q. Xu, M. Li, S. Shen, X. Wang, Y. Wang, Z. Feng, J. Shi, H. Han and C. Li, Angew. Chem., Int. Ed., 2012, 51, 13089 CrossRef CAS PubMed; (b) S. Arnold, S. Prokes, F. Perkins and M. Zaghloul, Appl. Phys. Lett., 2009, 95, 103102 CrossRef PubMed; (c) Z. Liu, T. Yamazaki, Y. Shen, T. Kikuta, N. Nakatani and Y. Li, Sens. Actuators, B, 2008, 129, 666 CrossRef CAS PubMed; (d) Y. Hou, L. Wu, X. Wang, Z. Ding, Z. Li and X. Fu, J. Catal., 2007, 250, 12 CrossRef CAS PubMed; (e) Y. Hou, X. Wang, L. Wu, Z. Ding and X. Fu, Environ. Sci. Technol., 2006, 40, 5799 CrossRef CAS.
- (a) S. Yoshioka, H. Hayashi, A. Kuwabara, F. Oba, K. Matsunaga and I. Tanaka, J. Phys.: Condens. Matter, 2007, 19, 346211 CrossRef; (b) R. Roy, V. Hill and E. Osborn, J. Am. Chem. Soc., 1952, 74, 719 CrossRef CAS.
- (a) T. Wang, A. Layek, I. D. Hosein, V. Chirmanov and P. V. Radovanovic, J. Mater. Chem. C, 2014, 2, 3212 RSC; (b) T. Wang, V. Chirmanov, W. H. M. Chiu and P. V. Radovanovic, J. Am. Chem. Soc., 2013, 135, 14520 CrossRef CAS PubMed; (c) M. Hegde, T. Wang, Z. L. Miskovic and P. V. Radovanovic, Appl. Phys. Lett., 2012, 100, 141903 CrossRef PubMed; (d) T. Wang and P. V. Radovanovic, J. Phys. Chem. C, 2011, 115, 18473 CrossRef CAS; (e) T. Wang, S. S. Farvid, M. Abulikemu and P. V. Radovanovic, J. Am. Chem. Soc., 2010, 132, 9250 CrossRef CAS PubMed; (f) N. Pinna, G. Garnweitner, M. Antonietti and M. Niederberger, J. Am. Chem. Soc., 2005, 127, 5608 CrossRef CAS PubMed.
- H. Hayashi, R. Huang, F. Oba, T. Hirayama and I. Tanaka, J. Mater. Res., 2011, 26, 578 CrossRef CAS.
- D. Kisailus, Q. Truong, Y. Amemiya, J. C. Weaver and D. E. Morse, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5652 CrossRef CAS PubMed.
- V. N. Sigaev, N. V. Golubev, E. S. Ignat'eva, B. Champagnon, D. Vouagner, E. Nardou, R. Lorenzi and A. Paleari, Nanoscale, 2013, 5, 299 RSC.
- L. Binet and D. Gourier, J. Phys. Chem. Solids, 1998, 59, 1241 CrossRef CAS.
- (a) A. Lauria, I. Villa, M. Fasoli, M. Niederberger and A. Vedda, ACS Nano, 2013, 7, 7041 CrossRef CAS PubMed; (b) B. Schrader, Raman/infrared atlas of organic compounds, VCH, Weinheim, 1989 Search PubMed.
- H. Seshadri, P. Sasidhar and P. Sinha, Int. J. Environ. Waste Manage., 2013, 11, 244 CAS.
- K. Shimamura, E. G. Víllora, T. Ujiie and K. Aoki, Appl. Phys. Lett., 2008, 92, 201914 CrossRef PubMed.
- (a) V. N. Sigaev, N. V. Golubev, E. S. Ignat'eva, A. Paleari and R. Lorenzi, Nanoscale, 2014, 6, 1763 RSC; (b) Z. Liu, X. Jing and L. Wang, J. Electrochem. Soc., 2007, 154, H440 CrossRef CAS PubMed; (c) G. Blasse and A. Bril, J. Phys. Chem. Solids, 1970, 31, 707 CrossRef CAS.
- J. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- H. You and M. Nogami, J. Phys. Chem. B, 2004, 108, 12003 CrossRef CAS.
- E. Zych, J. Phys.: Condens. Matter, 2002, 14, 5637 CrossRef CAS.
- H. Xie, L. Chen, Y. Liu and K. Huang, Solid State Commun., 2007, 141, 12 CrossRef CAS PubMed.
- (a) J. Ahman, G. Svensson and J. Albertsson, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 1336 CrossRef; (b) H. Y. Playford, A. C. Hannon, M. G. Tucker, D. M. Dawson, S. E. Ashbrook, R. J. Kastiban, J. Sloan and R. I. Walton, J. Phys. Chem. C, 2014, 118, 16188 CrossRef CAS.
- (a) M. Karmaoui, R. A. Sá Ferreira, A. T. Mane, L. D. Carlos and N. Pinna, Chem. Mater., 2006, 18, 4493 CrossRef CAS; (b) R. A. Sá Ferreira, M. Karmaoui, S. S. Nobre, L. D. Carlos and N. Pinna, ChemPhysChem, 2006, 7, 2215 CrossRef PubMed; (c) N. Pinna, G. Garnweitner, P. Beato, M. Niederberger and M. Antonietti, Small, 2005, 1, 112 CrossRef CAS PubMed.
- X. Bai, A. Pucci, V. T. Freitas, R. A. Ferreira and N. Pinna, Adv. Funct. Mater., 2012, 22, 4275 CrossRef CAS.
- (a) M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of photochemistry, CRC press, 2006 Search PubMed; (b) S. P. McGlynn, T. Azumi and D. Kumar, Chem. Rev., 1981, 81, 475 CrossRef CAS; (c) R. Martin and G. A. Clarke, J. Phys. Chem., 1978, 82, 81 CrossRef CAS; (d) H. Baba and M. Kitamura, J. Mol. Spectrosc., 1972, 41, 302 CrossRef CAS; (e) H. J. Maria and S. P. McGlynn, J. Chem. Phys., 1970, 52, 3399 CrossRef CAS PubMed.
- X.-L. Liu, P.-X. Zhu, Y.-F. Gao and R.-H. Jin, J. Mater. Chem. C, 2013, 1, 477 RSC.
- M. Niederberger, G. Garnweitner, N. Pinna and M. Antonietti, J. Am. Chem. Soc., 2004, 126, 9120 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials & method, XRD patterns and fitting results on lifetime measurement. See DOI: 10.1039/c4tc02118e |
| This journal is © The Royal Society of Chemistry 2015 |