
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly sensitive fluorescent light-up probe for real-time detection of the endogenous protein target and its antagonism in live cells†
Junlong
Geng‡
a,
Walter L.
Goh‡
b,
Chongjing
Zhang
c,
David P.
Lane
b,
Bin
Liu
*ac,
Farid
Ghadessy
*b and
Yen Nee
Tan
*a
aInstitute of Materials Research Engineering, A*STAR, 3 Research Link, Singapore 117602. E-mail: tanyn@imre.a-star.edu.sg
bp53 Laboratory, A*STAR, 8A Biomedical Grove, #06-06 Immunos, Singapore 138648. E-mail: fghadessy@p53Lab.a-star.edu.sg
cDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117576. E-mail: cheliub@nus.edu.sg
First published on 19th June 2015
Abstract
Real-time detection and monitoring of cancer-related biomolecular interactions in live cells are of paramount importance for disease diagnostics and drug screening. Herein, we developed a target-specific fluorescent light-up probe for cellular detection of Mdm2, the key negative regulator of the p53 tumour suppressor protein. Conjugation of a uniquely designed fluorogen (TPECM) with aggregation induced-emission properties, to a specific p53-derived peptide (12.1Pep) targeting Mdm2, yielded a cell-permeable probe (TPECM–12.1Pep) with turn-on fluorescence properties for real-time live cell imaging of Mdm2. This specific light-up probe is almost non-fluorescent in its isolated state but is highly emissive upon binding to Mdm2, enabling quantitative detection of both Mdm2 and its antagonism. Using a model compound (Nutlin-3a), we demonstrate that the as-developed probes can be used to screen p53–Mdm2 inhibiting drug candidates, both in vitro and in cells. Furthermore, the probe activity can be accurately monitored in cells using a fluorescently activated cell sorting machine. These features will expedite research in the areas of drug discovery, clinical diagnostics and fundamental cell biology.
Introduction, experimental
The Mdm2 protein is the key negative regulator of p53, dubbed “the guardian of the Genome” and master regulator of cellular fate.1 Mdm2 binding leads to ubiquitination of p53, and this modification targets p53 for rapid proteasome-dependent degradation in cells.2 The finely tuned relationship between p53 and Mdm2 levels is often corrupted in cancer cells, either through disabling point mutations in p53 or overproduction of Mdm2 due to gene amplification.3 Both events abrogate the pivotal cellular responses to stress (cell cycle arrest, apoptosis) mediated by p53, thus engendering permissive conditions for tumour growth and differentiation.4 Mdm2 has therefore become a therapeutic target of interest, with numerous small molecules and peptidic antagonists in clinical development.5 Reagent tools facilitating real-time Mdm2 detection, particularly in live cells, will therefore have significant use in diagnostic, drug-screening and basic biological research applications.Currently, the most widely utilized approaches for Mdm2 detection are fluorescence in situ hybridization, fluorescence correlation spectroscopy, immunohistochemistry, and immunoblotting.6 These strategies typically involve complex molecular labelling and/or tedious sample preparation, which are not readily amenable to real-time monitoring in live cells. Recently described approaches have utilized fluorescent fusion proteins to study p53–Mdm2 interactions in live cells.7 In addition to the convoluted technical procedures, the large size of the GFP tag may affect the function or interactions of the target proteins, leading to specious observations. In this regard, small molecule modified fluorescent probes show promising advantages in studying biomolecular interactions in view of their size and easy modification.8 More recently, a molecular rotor was conjugated to a specific Mdm2-interacting peptide, enabling fluorescence turn-on upon Mdm2 binding in vitro.9 However, such probes show high fluorescence background with limited response (∼2-fold) to Mdm2 protein, and are not readily suited for real-time cellular studies. Thus it is highly desirable to develop a new class of fluorescent probes with an improved signal-to-noise ratio and cell-penetrance that could facilitate real-time Mdm2 detection and imaging in the native cellular environment.
In this study, a target-specific fluorescent light-up probe with the unique characteristic of aggregation-induced emission (AIE) has been developed for detection of Mdm2 and drug screening applications (Scheme 1). AIE fluorogens (e.g. tetraphenylethene (TPE)) usually show weak fluorescence in the isolated states, but exhibit strong fluorescence in the aggregated state as a result of the restriction of intramolecular rotation and prohibition of energy dissipation via non-radiative channels.10 Typically, AIE fluorogens with longer emission wavelength are well suited for biodetection and bioimaging due to the low auto-fluorescence background and phototoxicity.11 Therefore, for cellular imaging, TPECM dye (Scheme 1) with an excitation wavelength compatible to a 405 nm laser was designed as it displays an orange-red emission color upon aggregation.12 The peptide, MPRFMDYWEGLS (12.1Pep), a p53-derived sequence engineered for high affinity binding to Mdm2 protein through phage display,9,13 was chosen as a response ligand for Mdm2 detection. The conjugation of 12.1Pep with the TPECM fluorogen yielded the target-specific light-up probe, TPECM–12.1Pep, which enables live-cell detection of Mdm2 and screening of Mdm2 antagonisms.
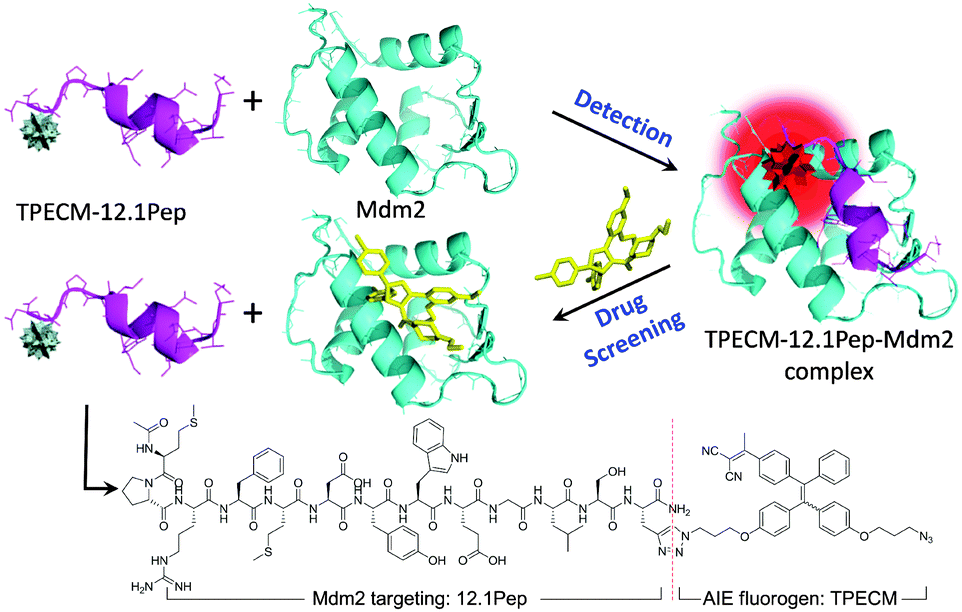 | ||
| Scheme 1 Schematic illustration of Mdm2 detection and drug screening. | ||
Results and discussion
TPECM was synthesized through conjugation of the dicyanovinyl group with TPE molecules.12 The incorporation of dicyanovinyl groups elongated the conjugation system of TPE, thus yielding fluorogens with both red-shifted absorption and emission as compared with those of TPE. In addition, attachment of a methyl group onto the dicyanovinyl group helps improve the stability of TPECM in the presence of biothiol agents present in the culture medium.12 TPECM shows an absorption peak at 385 nm and almost no emission in THF (Fig. S1, ESI†). The fluorescence intensity evolution of TPECM was studied by monitoring the emission intensity from equal molar concentrations of TPECM in THF/water mixtures with various water fractions (fw). TPECM exhibits low fluorescence emission at lower fw values (0–70%) but increases exponentially when fw increased from 80% to 99% (Fig. 1). The corresponding fluorescence intensity of TPECM at 99% fw is ∼90-fold higher than that in pure THF, demonstrating the unique AIE feature of the TPECM fluorogen.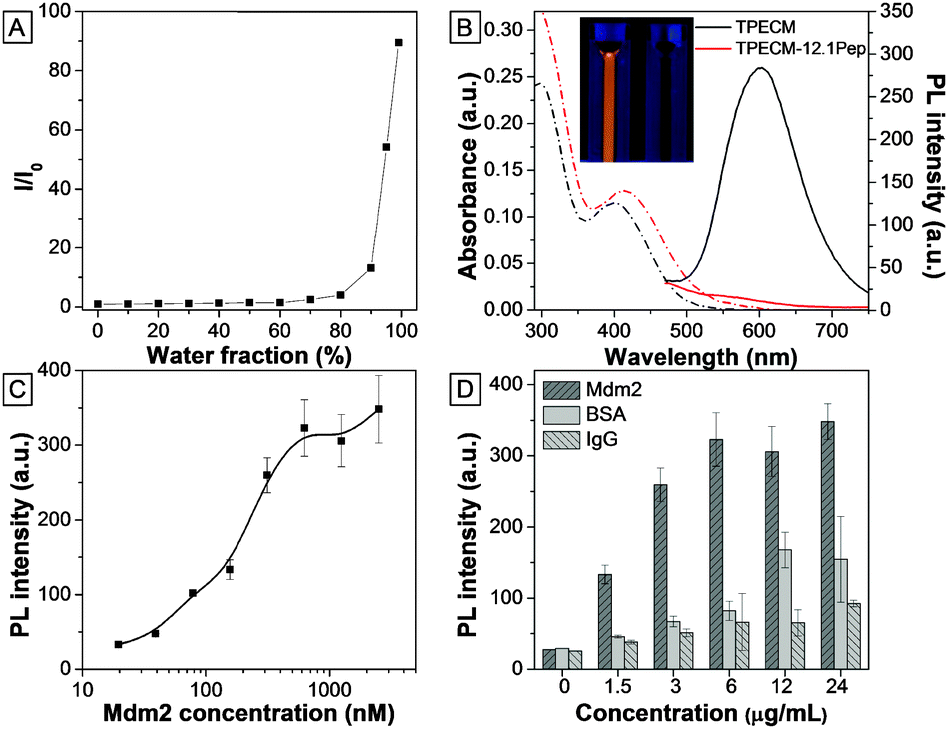 | ||
| Fig. 1 (A) Variations in I/I0 of TPECM with the change in the water fraction in the THF/water mixture. (B) UV-vis (dashed-dotted line) and PL (solid line) spectra of TPECM and TPECM–12.1Pep at 2 μM in water. The inset shows the image of the TPECM and TPECM–12.1Pep (right) solutions under UV-lamp excitation at 365 nm. (C) Fluorescence intensity evolution of 100 nM TPECM–12.1Pep at different concentrations of Mdm2 protein (N-terminal domain) and (D) its comparison with other control proteins. | ||
The TPECM–12.1Pep probe was subsequently prepared through a copper(I) catalyzed click-reaction between azide-functionalized TPECM and alkyne-functionalized 12.1Pep (Scheme S1, ESI†), followed by HPLC purification. The integrity and purity of the probe were characterized using MALDI-TOF MS spectra (Fig. S2, ESI†). UV-vis absorption and fluorescence spectra of the TPECM dye and TPECM–12.1Pep in water mixture are shown in Fig. 1B. Although they exhibited similar absorption profiles in the 350 to 500 nm range, their emission spectra were very different. A strong orange-red emission was observed for TPECM with a maxima at 603 nm while TPECM–12.1Pep is almost non-fluorescent under the same conditions (inset in Fig. 1B). The high fluorescence of TPECM is due to the aggregation of hydrophobic TPECM molecules in aqueous media, determined by laser light scattering (LLS) measurement with an average size of 80 nm (Fig. S3, ESI†). The conjugation of TPECM with 12.1Pep yields TPECM–12.1Pep with improved water solubility over the hydrophobic TPECM fluorogen as confirmed by the undetectable LLS signal. In addition, the probe remains nearly non-fluorescent in DMEM medium and PBS buffer, facilitating its potential application as an effective and specific light-up probe with a low background signal for biological detection and imaging in a cellular context.
To investigate the fluorescence response from binding of the TPECM–12.1Pep probe to Mdm2 protein, increasing concentrations of purified recombinant Mdm2 were titrated against a fixed 100 nM TPECM–12.1Pep. Fluorescence intensities of TPECM–12.1Pep/Mdm2 mixtures increased gradually with increasing Mdm2 concentrations (Fig. 1C). An enhancement factor of 13 is observed for the TPECM–12.1Pep in the presence of 2500 nM Mdm2. This significant fluorescence enhancement is due to the restriction of freely rotating TPECM molecules when the 12.1Pep docks into Mdm2's hydrophobic binding pocket (Scheme 1).14 The calculated apparent Kd (149.6 ± 52.07 nM) for TPECM–12.1Pep–Mdm2 binding is consistent with the previous reports of 12.1Pep–Mdm2 interaction.9,13 This turn-on signal is profoundly weaker in the presence of non-specific proteins (IgG and BSA) at the same mass concentrations (Fig. 1D), suggesting high binding specificity.
The demonstration of Mdm2 specific activity in vitro inspired us to next explore the utilization of TPECM–12.1Pep in live cells. The H1299 Ecdysone-Inducible (H1299-EI) system allows controllable and constitutive expression of p53 in H1299 cells using the inducer ponasterone A (Pon-A).15 As cellular Mdm2 levels are directly up-regulated by p53, induction of H1299-EI cells carrying wild-type p53 (H1299-EI-p53WT) leads to an increase in endogenous Mdm2. We first evaluated cytotoxicity of TPECM–12.1Pep on live cells using the MTT assay. No significant cytotoxicity was observed in H1299-E1 cells after 24 h and 48 h incubation with TPECM–12.1Pep at different concentrations (2 to 20 μM). Cell viabilities remain >90% (Fig. S4, ESI†), suggesting little to no probe-induced cytotoxicity, making TPECM–12.1Pep highly suitable for cellular imaging. Next, T22 reporter cells, carrying a p53 driven β-galactosidase reporter gene, were used to assess the cell permeability and activity of TPECM–12.1Pep.16 The extent of TPECM–12.1Pep cellular uptake can be assessed when benchmarked against Nutlin-3a, a prototypical antagonist which abrogates p53–Mdm2 interaction by binding the Mdm2 N-terminal hydrophobic pocket. A concentration-dependent increase in β-galactosidase activity is observed upon TPECM–12.1Pep treatment, similar to the Nutlin-3a induced response (Fig. S5, ESI†). This strongly suggests cellular uptake and binding of TPECM–12.1Pep to endogenous Mdm2, resulting in the observed increase in p53 activity.
To visualize cellular Mdm2 levels, TPECM–12.1Pep was added to either induced, or uninduced, wild type p53 expressing H1299-EI cells, and imaged using confocal laser scanning microscopy (CLSM). Mdm2-dependent fluorescence is strikingly higher in induced H1299-EI-p53WT (Fig. 2A) compared to p53-null control cells not induced for p53 expression (Fig. 2B). Auto-fluorescence was also not observed in H1299-EI-p53WT cells without TPECM–12.1Pep treatment (Fig. S6, ESI†), suggesting a turn-on fluorescence resulting from the specific interaction of endogenous Mdm2 with TPECM–12.1Pep. The anti-Mdm2 antibody (2A9) was next used to visualize Mdm2 localization in H1299-EI-p53WT live-cells that were p53-induced and TPECM–12.1Pep treated. Fluorescence images of the probe and 2A9 antibody labeled with FITC show a high degree of co-localization, further implying specific interaction in cells (Fig. S7, ESI†). To obtain a quantitative analysis of this interaction, flow cytometry was used to evaluate the intracellular fluorescence of H1299-EI-p53WT with and without Pon-A induction. The mean fluorescence intensity of H1299-EI-p53WT was ∼3-fold over uninduced cells (Fig. 2C), demonstrating the quantifiable measure of Mdm2 protein levels within the complex cellular environment in live cells. As Mdm2 is a direct transcriptional target of p53, TPECM–12.1Pep would be valuable as a live-cell imaging tool for drug discovery and understanding p53 biology in cells.5,17 A key requirement for such a utility would be an Mdm2-induced fluorescence that is critically sensitive to p53's transcriptional state. The fluorescence response of TPECM–12.1Pep was further evaluated using a panel of cells with different p53 and Mdm2 status. Unlike p53-wild type expressing cells, p53–Mdm2 double-knockout cells (DKO), and H1299-EI cells expressing either of the two transcriptionally inactive p53 point mutants (H1299-EI-p53G245S and H1299-EI-p53R248Q) showed significantly lower levels of fluorescence activity when treated with TPECM–12.1Pep (Fig. 2D–F). Notably, western blot analysis using the total cell lysates of the same cells showed a strong correlation between cellular Mdm2 protein levels and the fluorescence intensity within each cell line (Fig. 2G), further exemplifying target specificity and conditional sensitivity of TPECM–12.1Pep as a facile bioimaging probe for real-time detection of Mdm2 in live cells.
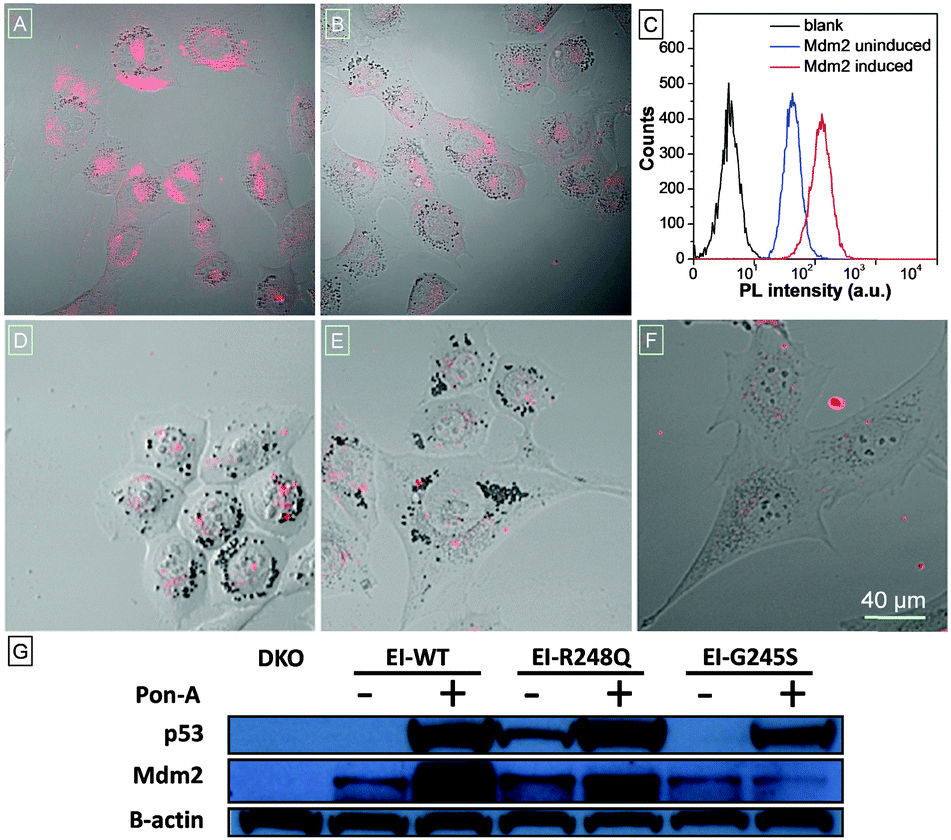 | ||
| Fig. 2 CLSM live-cell images of (A) H1299-EI-WTp53, (B) H1299-EI-p53−/−, (D) H1299-EI-p53G245S, (E) H1299-EI-p53R248Q and (F) p53−/−Mdm2−/−DKO cells after incubation with 2 μM TPECM–12.1Pep. Images have the same scale bar of 40 μm. (C) Flow cytometry histograms of live H1299-EI-WTp53 cells without any treatment (black) and treated with the TPECM–12.1Pep probe after incubation with (red) and without (blue) Pon-A induction, respectively. (G) Whole-cell lysate western blot of DKO and H1299-EI cells showing respective protein levels of p53 and Mdm2 before and after PonA-A induction. | ||
Given the major role Mdm2 plays in regulating p53 levels, significant efforts have gone into developing antagonists of this key interaction, with several small molecules and peptides currently in pre-clinical development.4,17,18 TPECM–12.1Pep acts as a conditionally fluorescent p53 proxy, and is potentially useful to screen competitive antagonists that can displace it from the Mdm2 binding pocket. To explore the utility of the TPECM–12.1Pep probe in small molecule drug screening, we investigated the sensitivity of the probe to Nutlin-3a, a current preclinical compound for p53–Mdm2 inhibition.18 Fluorescence intensity was measured after either Nutlin-3a, or its less active enantiomer, Nutlin-3b was added to pre-formed TPECM–12.1Pep–Mdm2 complexes. A marked decrease in fluorescence intensity was observed with the addition of Nutlin-3a in a concentration dependent manner. In contrast, the weakly binding Nutlin-3b enantiomer showed significantly reduced activity (Fig. 3A). Nutlin-induced displacement could also be seen in a similar fashion when whole-cell lysate from p53-induced H1299-EI-p53WT cells was used in place of purified Mdm2 protein (Fig. 3B), demonstrating potential drug screening or diagnostic applications for TPECM–12.1Pep in complex biological samples. To evaluate this application in live cells, H1299-EI-p53WT cells expressing p53/Mdm2 were pre-treated with Nutlin-3a followed by TPECM–12.1Pep incubation and imaged as before. Nutlin-3a pre-treatment was able to block and ablate TPECM–12.1Pep turn-on fluorescence from cellular Mdm2 binding (Fig. 3C and D), demonstrating the potential for a direct observable approach for drug screening using fluorescence microscopy in live-cells.
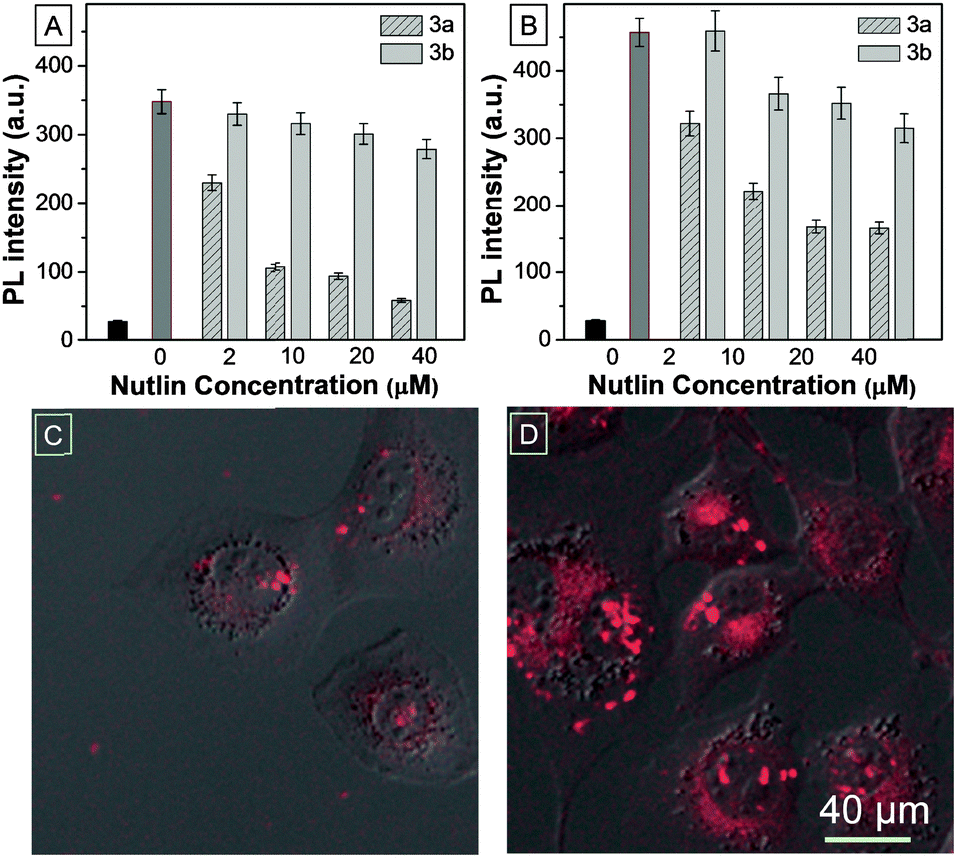 | ||
| Fig. 3 Nutlin competitive chase using (A) purified recombinant N-terminal Mdm2 protein. (B) 24 h induced H1299-EI-p53WT whole cell lysate. The black bar shows background fluorescence from unbound TPECM–12.1Pep while the darker grey bar shows the maximum fluorescence activity (no Nutlin). CLSM images of live Pon-A induced H1299-EI-WTp53 (C) pre-treated or (D) not treated with Nutlin-3a followed by TPECM–12.1Pep incubation. | ||
Conclusions
In conclusion, we report a facile, cell-permeable, conditionally fluorescent probe that is able to sensitively detect changes in endogenous Mdm2 protein levels in live cells. More specifically, the TPECM–12.1Pep probe described in this work facilitates Mdm2 detection in vitro and also enables real-time live cell imaging, which can be used as a target-specific molecular sensing tool for drug screening and cancer diagnostics. To the best of our knowledge, this is the first demonstration of fluorescence light-up response for Mdm2 detection in live cells. In addition, this specially designed imaging probe can not only detect the levels of Mdm2 in live cells, but also the activity of p53 which drives Mdm2 expression. The specific interaction between TPECM–12.1Pep and Mdm2 protein was also demonstrated through the use of a known Mdm2 small molecule antagonist, and hence holds great potential for drug-screening platforms using complex cell lysates or live cells. The capability of TPECM–12.1Pep to sense cellular events through the detection of an endogenous protein target could be harnessed for upstream biological applications. Further development of such target-specific light-up probes with near-infrared emission wavelength and improved target selectivity will undoubtedly be useful in forwarding basic research as well as exploring cancer diagnostics and clinical applications in in vivo systems.Acknowledgements
The authors would like to acknowledge the Agency for Science, Technology and Research (A*STAR), Singapore, for the financial support under the JCO CDA grant 13302FG063 and Singapore National Research Foundation (R-279-444-381).Notes and references
- D. P. Lane, Nature, 1992, 358, 15–16 CrossRef CAS PubMed.
- Y. Haupt, R. Maya, A. Kazaz and M. Oren, Nature, 1997, 387, 296–299 CrossRef CAS PubMed; M. H. G. Kubbutat, S. N. Jones and K. H. Vousden, Nature, 1997, 387, 299–303 CrossRef PubMed; S. Y. Shieh, M. Ikeda, Y. Taya and C. Prives, Cell, 1997, 91, 325–334 CrossRef; M. J. J. Blommers, G. Fendrich, C. García-Echeverría and P. Chêne, J. Am. Chem. Soc., 1997, 119, 3425–3426 CrossRef; C. Prives and P. A. Hall, J. Pathol., 1999, 187, 112–126 CrossRef.
- C. J. Brown, S. Lain, C. S. Verma, A. R. Fersht and D. P. Lane, Nat. Rev. Cancer, 2009, 9, 862–873 CrossRef CAS PubMed; J. Michel, E. A. Harker, J. Tirado-Rives, W. L. Jorgensen and A. Schepartz, J. Am. Chem. Soc., 2009, 131, 6356–6357 CrossRef PubMed; T. Kawamura, J. Suzuki, Y. V. Wang, S. Menendez, L. Batlle Morera, A. Raya, G. M. Wahl and J. C. Izpisua Belmonte, Nature, 2009, 460, 1140–1144 CrossRef PubMed; F. Toledo and G. M. Wahl, Nat. Rev. Cancer, 2006, 6, 909–923 CrossRef PubMed.
- B. Vogelstein, D. Lane and A. J. Levine, Nature, 2000, 408, 307 CrossRef CAS PubMed.
- P. M. Oconnor, J. Jackman, I. Bae, T. G. Myers, S. J. Fan, M. Mutoh, D. A. Scudiero, A. Monks, E. A. Sausville, J. N. Weinstein, S. Friend, A. J. Fornace and K. W. Kohn, Cancer Res., 1997, 57, 4285–4300 Search PubMed; N. Issaeva, P. Bozko, M. Enge, M. Protopopova, L. Verhoef, M. Masucci, A. Pramanik and G. Selivanova, Nat. Med., 2004, 10, 1321–1328 CrossRef CAS PubMed; A. Vazquez, E. E. Bond, A. J. Levine and G. L. Bond, Nat. Rev. Drug Discovery, 2008, 7, 979–987 CrossRef PubMed; A. E. Rabideau, X. L. Liao and B. L. Pentelute, Chem. Sci., 2015, 6, 648–653 RSC; L. Qin, F. Yang, C. Zhou, Y. Chen, H. Zhang and Z. Su, J. Am. Chem. Soc., 2014, 136, 18023–18033 CrossRef PubMed; T. Kamijo, J. D. Weber, G. Zambetti, F. Zindy, M. F. Roussel and C. J. Sherr, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8292–8297 CrossRef; Y. H. Lau, P. de Andrade, S. T. Quan, M. Rossmann, L. Laraia, N. Skold, T. J. Sum, P. J. E. Rowling, T. L. Joseph, C. Verma, M. Hyvonen, L. S. Itzhaki, A. R. Venkitaraman, C. J. Brown, D. P. Lane and D. R. Spring, Chem. Sci., 2014, 5, 1804–1809 RSC; J. Pomerantz, N. Schreiber-Agus, N. J. Liegeois, A. Silverman, L. Alland, L. Chin, J. Potes, K. Chen, I. Orlow, H. W. Lee, C. Cordon-Cardo and R. A. DePinho, Cell, 1998, 92, 713–723 CrossRef.
- N. Sirvent, J. M. Coindre, G. Maire, I. Hostein, F. Keslair, L. Guillou, D. Ranchere-Vince, P. Terrier and F. Pedeutour, Am. J. Surg. Pathol., 2007, 31, 1476–1489 CrossRef CAS PubMed; J. Weaver, P. Rao, J. R. Goldblum, M. J. Joyce, S. L. Turner, A. J. F. Lazar, D. Lopez-Terada, R. R. Tubbs and B. P. Rubin, Mod. Pathol., 2010, 23, 1301–1306 CrossRef PubMed; Y. A. Valentin-Vega, J. A. Barboza, G. P. Chau, A. K. El-Naggar and G. Lozano, Hum. Pathol., 2007, 38, 1553–1562 CrossRef PubMed; L. Sironi, S. Freddi, L. D'Alfonso, M. Collini, T. Gorletta, S. Soddu and G. Chirico, J. Biomed. Nanotechnol., 2009, 5, 683–691 CrossRef PubMed.
- H. D. Herce, W. Deng, J. Helma, H. Leonhardt and M. C. Cardoso, Nat. Commun., 2013, 4, 2660 Search PubMed; Y. C. Li, L. W. Rodewald, C. Hoppmann, E. T. Wong, S. Lebreton, P. Safar, M. Patek, L. Wang, K. F. Wertman and G. M. Wahl, Cell Rep., 2014, 9, 1946–1958 CrossRef CAS PubMed; K. Zolghadr, U. Rothbauer and H. Leonhardt, Methods Mol. Biol., 2012, 812, 275 Search PubMed.
- L. Yuan, W. Lin, K. Zheng, L. He and W. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC; J. O. Escobedo, O. Rusin, S. Lim and R. M. Strongin, Curr. Opin. Chem. Biol., 2010, 14, 64–70 CrossRef CAS PubMed; D. B. P. Thiel, M. Kaiser and C. Ottmann, Angew. Chem., Int. Ed., 2012, 51, 2012–2018 CrossRef PubMed; Y. Guan, X. N. Shan, S. P. Wang, P. M. Zhang and N. J. Tao, Chem. Sci., 2014, 5, 4375–4381 RSC.
- W. L. Goh, M. Y. Lee, T. L. Joseph, S. T. Quah, C. J. Brown, C. Verma, S. Brenner, F. J. Ghadessy and Y. N. Teo, J. Am. Chem. Soc., 2014, 136, 6159–6162 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC; Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed; R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC; C. W. T. Leung, Y. Hong, S. Chen, E. Zhao, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 62–65 CrossRef PubMed; H. Shi, J. Liu, J. Geng, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 9569–9572 CrossRef PubMed; Y. Yuan, R. T. K. Kwok, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2014, 136, 2546–2554 CrossRef PubMed; J. Liang, B. Z. Tang and B. Liu, Chem. Soc. Rev., 2015, 44, 2798–2811 RSC.
- Y. Yuan, C. J. Zhang, M. Gao, R. Zhang, B. Z. Tang and B. Liu, Angew. Chem., Int. Ed., 2015, 54, 1780–1786 CrossRef CAS PubMed; F. Hu, Y. Huang, G. Zhang, R. Zhao, H. Yang and D. Zhang, Anal. Chem., 2014, 86, 7987–7995 CrossRef PubMed.
- C. J. Brown, S. G. Dastidar, S. T. Quah, A. Lim, B. Chia and C. S. Verma, PLoS One, 2011, 6, e24122 CAS.
- G. M. Popowicz, A. Doemling and T. A. Holak, Angew. Chem., Int. Ed., 2011, 50, 2680–2688 CrossRef CAS PubMed.
- M. J. Scian, K. E. R. Stagliano, M. A. E. Anderson, S. Hassan, M. Bowman, M. F. Miles, S. P. Deb and S. Deb, Mol. Cell. Biol., 2005, 25, 10097–10110 CrossRef CAS PubMed.
- X. Lu, S. A. Burbidge, S. Griffin and H. M. Smith, Oncogene, 1996, 13, 413–418 CAS.
- D. P. Lane and P. M. Fischer, Nature, 2004, 427, 789–790 CrossRef CAS PubMed; L. Römer, C. Klein, A. Dehner, H. Kessler and J. Buchner, Angew. Chem., Int. Ed., 2006, 45, 6440–6460 CrossRef PubMed.
- S. G. Dastidar, D. P. Lane and C. S. Verma, J. Am. Chem. Soc., 2008, 130, 13514 CrossRef CAS PubMed; B. Vogelstein, D. Lane and A. J. Levine, Nature, 2000, 408, 307–310 CrossRef PubMed; P. H. Kussie, S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine and N. P. Pavletich, Science, 1996, 274, 948–953 CrossRef; T. A. Davis and J. N. Johnston, Chem. Sci., 2011, 2, 1076–1079 RSC; L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, Science, 2004, 303, 844–848 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental protocols and characterization data. See DOI: 10.1039/c5tb00819k |
| ‡ J. L. Geng and W. L. Goh contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2015 |