
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insulating and semiconducting polymeric free-standing nanomembranes with biomedical applications
Maria M.
Pérez-Madrigal
*ab,
Elaine
Armelin
ab,
Jordi
Puiggalí
ab and
Carlos
Alemán
*ab
aDepartament d'Enginyeria Química, ETSEIB, Universitat Politècnica de Catalunya, Avda. Diagonal 647, Barcelona E-08028, Spain. E-mail: m.mar.perez@upc.edu; carlos.aleman@upc.edu
bCenter for Research in Nano-Engineering, Universitat Politècnica de Catalunya, Campus Sud, Edifici C', C/Pasqual i Vila s/n, Barcelona E-08028, Spain
First published on 28th May 2015
Abstract
In recent decades, polymers have experienced a radical evolution: from being used as inexpensive materials in the manufacturing of simple appliances to be designed as nanostructured devices with important applications in many leading fields, such as biomedicine at the nanoscale. Within this context, polymeric free-standing nanomembranes – self-supported quasi-2D structures with a thickness ranging from ∼10 to a few hundreds of nanometers and an aspect ratio of size and thickness greater than 106 – are emerging as versatile elements for applications as varied as overlapping therapy, burn wound infection treatment, antimicrobial platforms, scaffolds for tissue engineering, drug-loading and delivery systems, biosensors, etc. Although at first, a little over a decade ago, materials for the fabrication of free-standing nanosheets were limited to biopolymers and insulating polymers that were biodegradable, during the last five years the use of electroactive conducting polymers has been attracting much attention because of their extraordinary advantages in the biomedical field. In this context, a systematic review of current research on polymeric free-standing nanomembranes for biomedical applications is presented. Moreover, further discussion on the future developments of some of these exciting areas of study and their principal challenges is presented in the conclusion section.
 Maria M. Pérez-Madrigal | Maria M. Pérez-Madrigal received her BSc degree in Chemical Engineering (2010) and her MSc degree in Polymers and Biopolymers (2011) at the Universitat Politècnica de Catalunya (UPC), Barcelona, Spain. She is currently a PhD student under the supervision of Prof. Carlos Alemán and Dr Elaine Armelin. Her research focuses on combining conducting polymers with conventional polymers to obtain biointerfaces at the nanoscale (nanomembranes and nanofibers) for applications such as tissue engineering scaffolds or drug-delivery systems. |
 Elaine Armelin | Elaine Armelin is Associate Professor in the Chemical Engineering Department and the Center for Research in NanoEngineering (CRnE), Universitat Politècnica de Catalunya (UPC, Spain). She received her Bachelor's degree in Chemistry in 1995 and Master Degree in Organic Chemistry in 1997 at the Universidade de São Paulo (USP-Brazil). In 1997 she moved to Barcelona to perform her PhD studies in Polymer Science, under the supervision of Prof. Puiggalí, at the UPC. She is a co-author of more than 80 peer reviewed publications and patents. Her main interests are characterization of polymeric materials and corrosion protection by using organic–inorganic coatings. |
 Jordi Puiggalí | Jordi Puiggalí is a Full Professor at the Chemical Engineering Department of the Universitat Politècnica de Catalunya. He is the head of the “Macromolecular Chemistry” and “Synthetic Polymers, Structure and Properties” research groups. He also belongs to the Centre of Research on NanoEngineering, being the co-director of the laboratory of Nanochemistry. The main objectives of his research concern the development of new polymers and nanocomposites for biomedical applications, the structural studies of semicrystalline polymers and finally the study of crystallization processes and their influence on materials properties. His work has resulted in more than 200 articles and book chapters. |
 Carlos Alemán | Carlos Alemán earned his PhD in Chemistry at the Universitat Politècnica de Catalunya in 1994. He is currently Full Professor of Physical Chemistry in the same University, leading the “Innovation in Materials and Molecular Engineering” (IMEM) group in Chemical Engineering Department, and co-leading with Prof. J. Puiggalí the “Nanochemistry” group in the Centre of Research on NanoEngineering. His main research interests focus on conducting polymers for biomedical applications, peptides as materials and development of peptide–polymer conjugates and composites. He is the author or co-author of over 450 review articles, research papers and book chapters. |
1. Introduction
Over the past few decades, biomaterials science has been increasingly evolving into a more interdisciplinary field combining elements of medicine, chemistry, biology, engineering and materials science. Recently, nanotechnological concepts and procedures have also been applied to obtain and improve devices with very different applications in biomedicine and biotechnology.1–3 Specifically, there is growing interest in the fabrication of nanomembranes (also known as ultra-thin films or nanosheets) since their distinctive features make them suitable for designing sensors,4,5 nanobiological reactors,6 biomotors,7 biointerfaces for cellular matrices,8 antimicrobial surfaces,9,10 or drug release devices.11,12 In practice, the term nanomembrane refers to quasi-2D structures with macroscopic surface area and thickness values ranging from 10 to a few hundreds of nm. A few years ago, Kunitake et al.,13 pioneers in this field, coined the term “giant nanomembrane” to denote self-supporting (namely, free-standing) membranes with thickness from 1 to 100 nm and an aspect ratio of size and thickness greater than 106. Such a high aspect ratio facilitates the handling of these nanomembranes, while the self-supporting properties, which enable the nanosheet to be removed from its supporting substrate retaining the mechanical integrity, are required to physically separate two spaces. Besides, free-standing nanomembranes (hereafter denoted as FsNMs) are characterized by other special properties, such as low weight, high flexibility, robustness, and, in some cases, transparency.14Research on FsNMs for biomedical applications started less than a decade ago when it was proved that ultra-thin sheets prepared using biodegradable and biocompatible polyesters and polysaccharides exhibited excellent mechanical properties and adhesion strength. Rapidly, this led to considerable interest in these nanosheets as novel substrate/biointerfaces to promote cell adhesion, migration, proliferation and differentiation. More recently, FsNMs have been used for different therapeutic uses, for example treatment of microbial infections, control of cell adhesion and repair of tissue defects. In this review, we discuss the most representative examples of FsNMs made of insulating and electrochemically inactive polymers for advanced biomedical applications. After this, we present the very recent achievements in the application of electroactive conducting polymers (ECPs) what we call the “second generation of FsNMs”, which incorporate electrochemical and electrical functionalities for biomedical applications. Before such a discussion, brief explanations about the different strategies that can be used to fabricate FsNMs and the main concepts of ECPs are provided.
2. Preparation of free-standing nanomembranes
Typically, FsNMs are produced by depositing nanofilms on solid substrates, or via fluid–fluid or liquid–air interfaces. In addition, some other approaches, including layer-by-layer (LbL) assembly,15–17 Langmuir–Blodgett transfer (LB),18 spin-coating,19 electrophoretic deposition20 and cross-linking of self-assembled monolayer (SAM) techniques,21 are also often employed. These methods differ in two general aspects: (1) the degree of control over the final FsNM thickness, composition and stability; and (2) the step in which the nanomembrane is removed, transferred or extracted from the solid surface or the fluid–fluid interface.22In the LbL technique, which was developed by Decher et al.15–17 in the early 1990s, the formation of the FsNM is achieved by depositing on a solid surface alternating layers of oppositely charged materials (Fig. 1a), such as polyions, metals, nanoparticles, ceramics or biological molecules. Accordingly, FsNM stability arises from primary or secondary interactions between those layers. This approach stands out because of its simplicity and the high degree of control over the FsNM thickness. Furthermore, the precise guidance exerted over the chemical composition of the layers results in a tremendous versatility when designing FsNMs.23 Regarding the SAM technique, Stroock et al.21 extensively described the synthesis of polymeric FsNMs with 10–15 nm in thickness and well-defined lateral size and shape.
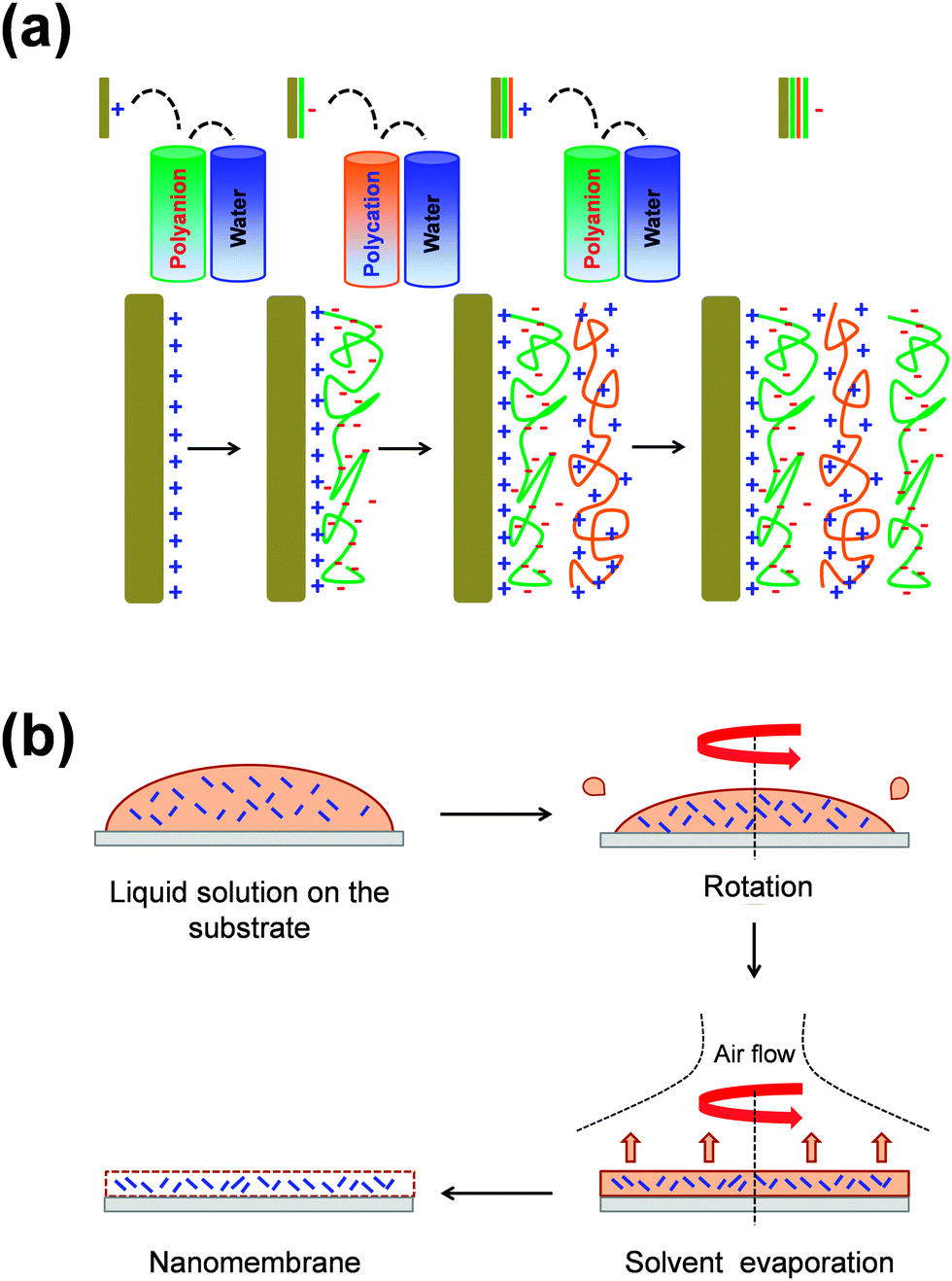 | ||
| Fig. 1 Schematic illustration of the (a) LbL and (b) spin-coating processes. | ||
On the other hand, spin-coating (Fig. 1b) is another interesting approach that allows the preparation of single- or multi-layered FsNMs in a few steps. In this case, the optimization of the spin-coating parameters (e.g. spinning speed and time, and the solution concentration) leads to ultra-thin films with controlled features, thickness and homogeneity. In this procedure, which seems to be the most versatile and easy-going, the liquid polymeric solution is spin-coated onto a solid substrate previously coated with a sacrificial layer. Hence, by dissolving the sacrificial layer in an appropriate solvent, the FsNM is detached from the substrate and released into a liquid environment where it can be then easily handled using syringes or pipettes. Most of the FsNMs described in this review were fabricated using a combination of both the LbL assembly and spin-coating procedures.
3. Electroactive conducting polymers
Electroactive conducting polymers (ECPs) are widely known to be electrochemically active organic semiconducting materials. These polymers are special in that they consist of long conjugated polymeric chains that present alternation of double and single-bonded sp2 hybridized atoms. This conjugation, which is described in terms of electronic wave functions that are delocalized over the entire chain, allows charge mobility along the polymer backbone and between adjacent chains, thus endowing the polymer with semiconductive and electrochemical properties.24–26 Although the band structure of ECPs is similar to that of inorganic semiconductors, they also exhibit some of the attractive properties associated with conventional polymers (i.e. electrically insulating polymers), such as ease of synthesis and flexibility in processing. However, the mechanical stability of ECPs is typically lower than that of both inorganic semiconductors and conventional polymers.The doping process converts neutral ECPs into semiconductive. In this process, the ECP is oxidized (p-doping) or reduced (n-doping) and, as a result, positive or negative charge carriers, respectively, are introduced into the backbone. Backbone charge carriers, which typically consist of charged polarons (i.e. radical cations or anions) or bipolarons (i.e. dications or dianions),27 are accompanied by counter ions with opposite charges that are entrapped into the polymer matrix to maintain electrical neutrality. Simultaneously, the band structure of the ECP is modified to accommodate the formation of polaronic and bipolaronic bands, which facilitate the electrical conduction.
Although Bolto et al.28 reported in 1963 polypyrrole derivatives with a resistivity as low as 1 Ohm cm, the discovery and development of conducting polymers were attributed to Shirakawa, MacDiarmid, and Heeger, who reported the conductivity of polyacetylene doped with iodine,29 when they awarded with the Novel Prize in Chemistry 2000. Generally, ECPs are categorized into three groups: non-cyclic polyenes (e.g. polyacetylene), cyclic polyenes (e.g. poly(p-phenylene) and its derivatives), and polyheterocyclic ECPs (e.g. polypyrrole (PPy), polythiophene (PTh), and polyaniline (PAni)). The last group is actually considered the most versatile due to the excellent properties exhibited by such ECPs, especially in terms of electrochemical and environmental stabilities. Fig. 2 displays the chemical structure of the most representative ECP of each group.
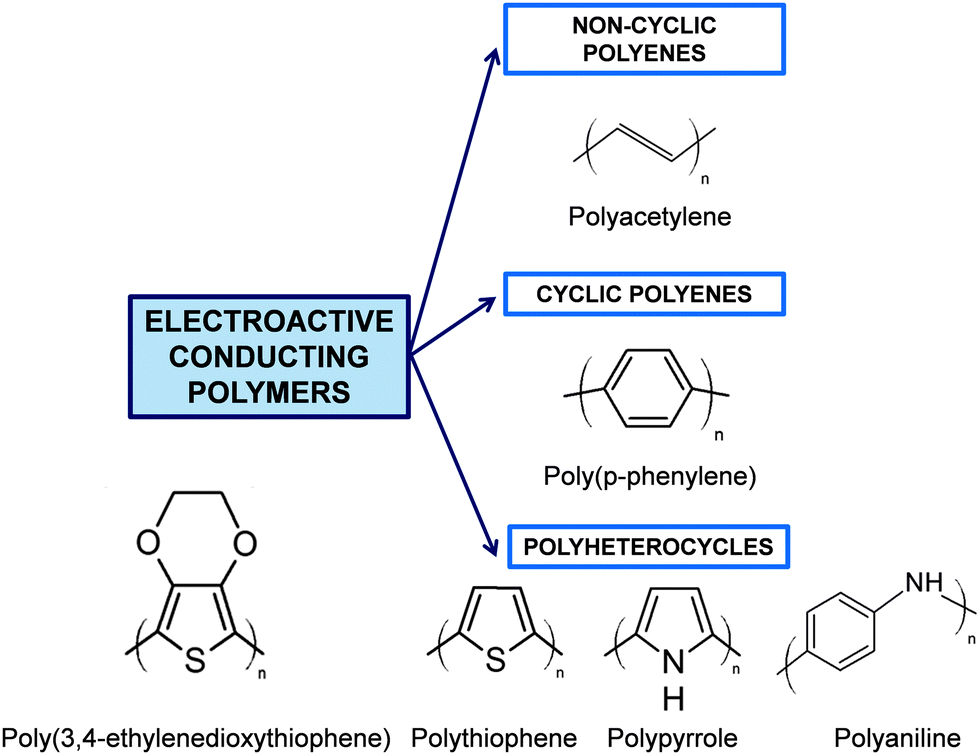 | ||
| Fig. 2 General classification of ECPs and chemical structure of the most representative compounds of each group. | ||
Although chemical synthesis provides many different possible routes to obtain a variety of ECPs, electrochemical synthesis is frequently the preferred alternative since this procedure is relatively straightforward and enables the control over the yield, morphology and electrochemical properties of the resulting material. Some ECPs, such as PPy and poly(3,4-ethylenedioxythiophene) (PEDOT), have been polymerized by both chemical and electrochemical processes, the advantages and disadvantages of both strategies are summarized in Table 1. Both procedures involve the oxidative polymerization of monomers dissolved in the medium by the action of a chemical oxidant or by applying an anodic potential at the electrode, respectively. The oxidative mechanism for polyheterocyclic ECPs, occurring in chemical and electrochemical polymerization processes, is summarized in Fig. 3.
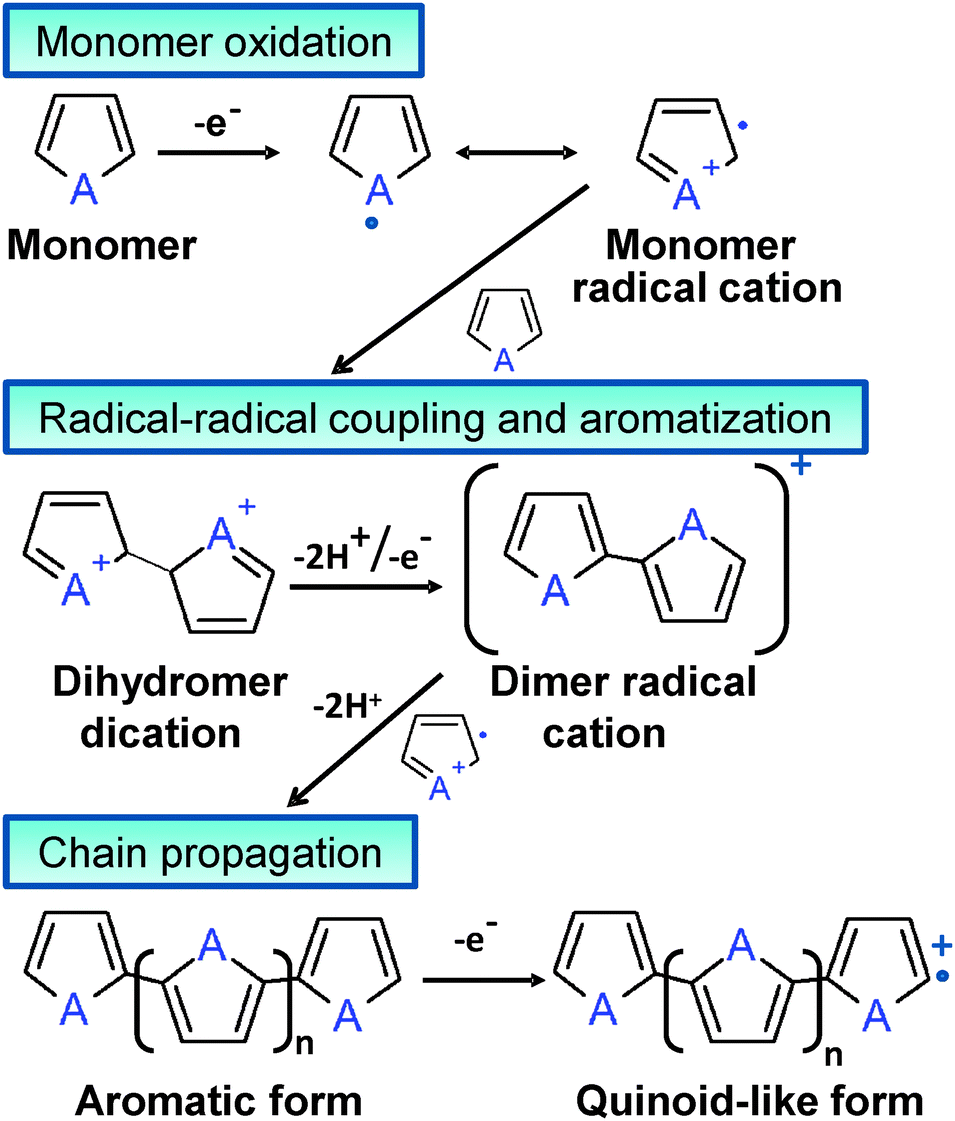 | ||
| Fig. 3 Mechanism for heterocyclic ECPs polymerization, A = N–H and S for PPy and PTh, respectively. | ||
| Polymerization method | Advantages | Disadvantages |
|---|---|---|
| Chemical |
– Large-scale production
– Easy post-incorporation of other molecules to modify the ECP covalently – Many options to modify the chemical structure |
– The synthetic process is more complicated than the electrochemical one – Preparation of ultra-thin films becomes a difficult task |
| Electrochemical |
– Ease of synthesis
– Entrapment of molecules in the polymer network becomes an easy process – The synthetic and doping processes occur simultaneously – Very useful to prepare ultra-thin films of controlled thickness |
– Removal of the films from the electrode surface is frequently a very difficult task. – Post-covalent modification of ECPs is quite complex or, even, impossible |
Because of the outstanding electrical, electrochemical, optical and magnetic properties shown by ECPs,24–26 these materials have been applied in a great number of technological fields that include organic optoelectronics,30 organic photovoltaics,31 energy storage technology,32,33 and the fabrication of electrochromic34 and electroluminescent35 devices. Recently, as ECPs have also extended to biotechnological, bioengineering and biomedical fields,36–39 special attention has been paid to tailor desirable properties (i.e. electrical, chemical and physical properties) to better suit the nature of the specific bioapplication. For example, the ability to entrap and controllably release biological molecules through reversible doping, and the ability to transfer charges from a biochemical reaction are two of the promising inherent functions of ECPs.
Currently, ECPs are frequently used as biosensors, tissue-engineering scaffolds, neural probes, drug-delivery devices and bio-actuators, as has been recently reviewed.36–40 In particular, for tissue engineering applications, ECPs show interesting advantages but also some limitations (i.e. hydrophobicity, the lack of mechanical integrity and the absence of biodegradability).37,40–42 ECP blending with conventional insulating polymers or biopolymers is the most popular approach followed to solve these drawbacks.43–48 An alternative and also effective strategy, which was recently reviewed by Albertsson and co-workers,43 is the synthesis of new biodegradable polymers containing conducting oligomers. Due to the growing interest in this class of new biomaterials, the palette of degradable and electrically conductive polymers is progressively expanding to meet the demands of specific applications within the biomedical field. For example, in the last year Schmidt and co-workers reported biodegradable electroactive copolymers composed of oligoaniline-based blocks linked to polyethylene glycol or polycaprolactone blocks, which deliver anti-inflammatory drugs upon application of electrochemical stimuli.49
In this work we use the concepts discussed in the two previous sections to review the fabrication of FsNMs for biomedical applications. Firstly, we will discuss the design and development of FsNMs made of biopolymers and insulating polymers. After this, we will focus on FsNMs fabricated by blending conventional polymers with ECPs. Finally, several concluding remarks on the achievements and future perspectives of this outstanding research field will be exposed.
4. Biomedical applications of insulating and electrochemically inactive free-standing nanomembranes
Generally, FsNMs for biomedical applications are made using synthetic or naturally derived polymers. Among the synthetic ones, linear aliphatic polyesters, such as poly(lactic acid) (PLA) and poly(lactic-co-glycolic acid) (PLGA), have been extensively chosen since their biodegradation rate and mechanical properties can be easily controlled through variations in their molecular weight.50 Also, poly(ether ester) copolymers have been successfully employed due to their excellent properties (e.g. elasticity, toughness, strength and easy processability), which arise from the combination of both soft and hard segments along their chemical structure.51 In regard to biopolymers, the most frequently used are collagen and polysaccharides, such as alginate and chitosan, their applications being usually focused in tissue engineering.52,53In the biomedical context, FsNMs need specific requirements such as biocompatibility and, in some cases, biodegradability and/or bioresorbability also need to be met. Furthermore, by successfully controlling the chemical composition and the fabrication process, it is possible to tune and adjust the physicochemical, mechanical, chemical and morphological properties of FsNMs to promote cell–substrate interaction, which is particularly relevant in regenerative medicine (i.e. cellular organization can be directly regulated through the cellular microenvironment).54,55 Their structure and flexibility allow FsNMs to coat the surface of devices that interact with biological systems or, alternatively, FsNMs can be introduced into a needle by aspiration, and then be moved and released/injected into liquid environments (e.g. finely positioned on surgical incisions or directly used for wound treatments).
Next sub-sections review the most representative examples of FsNMs made of insulating polymers for advanced biomedical applications. Table 2 summarizes their main characteristics, which are described below organized by polymeric families (namely, polyesters, polysaccharides and other polymeric sources).
| Material | Preparation | Thickness (nm) | Propertiesa | Biomedical applications | Ref. |
|---|---|---|---|---|---|
| a E: elastic modulus. Lc: critical load related to the adhesion. σmax: ultimate tensile strength. εmax: ultimate tensile elongation. | |||||
| PLA | Spin-coating | 23 ± 5 | E: 1.7 ± 0.1 GPa, Lc = (1.7 ± 0.3–1.8 ± 0.2) × 10−5 N m−1. Transparency | Wound dressing, and atraumatic fixation tools | 56–58 |
| PLA | Spin-coating assisted multilayering | 60 ± 6 | Transparency | Barrier against burn wound infection | 59 |
| PLA | Spin-coating | 320 ± 27 | E: 136 ± 44 MPa. Transparency | Patch for bone or tendon repair and healing, and adhesive bioactive matrices for cell anchoring, spreading and proliferation | 60 and 61 |
| PLA-meshes | Spin-coating with the nanosheet collected on a stainless steel mesh | From 29 ± 1 to 703 ± 4.4 | E: from 3.5 ± 1.3 to 7–10 GPa. Transparency. Weak adhesiveness for higher thickness nanosheets | Tailorable environment for anisotropic cell proliferation and differentiation without using conventional photolithographic approaches | 62 |
| PLA coated with collagen at one side | Spin-coating | 59.5 ± 9.5 (PLA) and 5–10 (collagen) | Weak adhesiveness | Scaffolds that endow each side of the nanosheet with different discrete functions: anti-adhesive and pro-healing | 63 |
| PLA + SPION | Spin-coating | From 33 ± 3 to 245 ± 9 | E similar to PLA. Magnetically responsive and colored | Magnetically controllable support | 64 |
| PLA–PAH–D–HA | Spin-coating + LbL | ∼500 nm | Robust and flexible | Drug delivery | 65 |
| PLGA | Thermal fusion of adsorbed nanoparticles | — | — | Platelet substitutes and carriers | 66 |
| Chitosan–Na-alginate | Spin-coating assisted LbL | From 30.2 ± 4.3 to ∼75 | E = 1.3/9.6 GPa. Pressure resistance, high flexibility and adhesiveness, transparency, biodegradability and semi-absorbent ability | Nano-adhesive plasters for tissue surfaces. Tissue defect repair without chemical bonding agents. Multi-overlapping therapy for venous hemorrhage. Arachnoid plasters in microsurgery without using chemical bonding agents. | 68–71, 74 and 75 |
| PVA–TC–(chitosan–Na-alginate) | Spin-coating assisted LbL | 177 | High flexibility, adhesive strength and transparency | Overlapping therapy, treatment of burn wound infections, antimicrobial platforms and drug-loading | 72 and 73 |
| HA–collagen | LbL | 42 ± 4 (fibrous collagen) and 62 ± 7 (non-fibrous collagen) | For fibrous collagen: E = 12.5 ± 1.5 GPa, σmax = 289 ± 9 MPa and εmax = 1.0 ± 0.1%. For non-fibrous collagen: E = 4.3 ± 0.6 GPa, σmax = 227 ± 10 MPa and εmax = 1.7 ± 0.3% | Scaffolds for regenerative medicine | 76 |
| Cell–gelatin–(chitosan–Na-alginate)3 | LbL onto cultured cells on PNIPAM-grafted surfaces | — | — | Fabrication of complex artificial soft tissues to substitute some elements of native tissues | 78 |
| PEO–PAA | LbL | 80 nm per bilayer | Elastomeric properties, smooth to touch and transparent | Biosubstrates, drug delivery devices and pH-sensitive sensors | 85 |
| PAA–PEG–PAH–PSS | Spraying polymer solutions | 55 to several hundreds | Components biocompatible, biotolerant or bioinert. Transparent and pH responsive | Therapeutic devices and aids | 86 |
| NBPT–PEG | Spin-coating | ∼5 | Mechanical stability, biocompatibility and high transparency. Protein-repelling behavior | Support in transmission electron microscopy studies in their specific application to sensitive biological targets | 87 |
| PEG | Chemical crosslinking of a hydrogel precursor | 10–350 | Mechanical stability, high flexibility (E = ∼2 MPa), hydrophilicity, and biorepulsion | Highly sensitive support and sensor elements for biological samples | 88 |
| PAH–PAA | LbL dipping + pH treatment for the creation of pores | 253–688 | Pores with diameters ranging from 300 nm to 2 μm (average: 1 μm) | Drug-delivery systems tuned by varying the number of layers and the pore size. | 11 |
| PDDA–P4VPPS | LbL | ∼29 per bilayer | pH-dependence, disintegration in alkali environments | Support (sacrificial layers) for biological templates | 92 |
| PHEMA | Surface-initiated ATRP on a PDA layer | 16–75 | Good chemical stability, low mechanical strength, transparency and colorless | Multi-stimuli responsive sensors after functionalizing the surface of the films | 94 |
| PMPC–(PS, PAH, PAA) | ATRP of MPC and spin-coating assisted LbL | 11 (PMPC) + 85 ± 2 (PS, PAH, PAA) | Physiological stability, surface wettability and anti-biofouling | Biointerface for tissue-engineering scaffolds | 95 |
| PMMA | Spin-coating | 199 ± 20 | High flexibility, physiological stability and expected mechanical behavior similar to those of PLA FsNMs | Fabrication of platforms with space-selective cell culture environments by injet printing protein microarrays at the surface. Cell-directed culture in muscular tissue engineering | 96 |
| PMMA | Spin-coating PMMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PS mixtures :PS mixtures |
From 38.8 ± 1.1 to 110.2 ± 2.0 nm | Porous and perforated structures | Potential applications in cell culture devices, high flux biosensors and drug delivery systems | 97 |
| PS–(CNT–Fn) | Spin-coating (PS) by microcontact printing (CNT–Fn) | From tens (∼40) to hundreds (∼360) | High flexibility, cell adhesiveness and surfaces with tailored morphology | Platforms to direct the cellular organization and engineer the hierarchically assembled tissue structure. Flexible devices for regenerative medicine | 98 |
| (PAH–PSS)nPAH–Au–(PAH–PSS)n–PAH | Spin-coating assisted LbL | 25–70 | E = 30–40 GPa, σmax > 100 MPa and εmax = 2%. Light blue color. Long life, extraordinary sensitivity and unique auto-recovering ability | Although no biomedical application was directly examine, these FsNMs are potential candidates for their use in systems requiring outstanding mechanical and dynamical properties. | 4 and 99 |
| PAA–DMLPEI | LbL | <100 (from 2 to 20 bilayers) | Mechanical robustness and pH responsive | Coatings with microbicide properties for airborne and waterborne bacteria | 101 |
| DGI | Free-radical polymerization under UV radiation | <5 | High mechanical strength and thermal stability. High-density of functional groups exposed to the outer surfaces | Practical applications in the biomedical field depend on the surface post-functionalization | 104 |
4.1. Polyester free-standing nanomembranes
Takeoka and co-workers56 developed PLA FsNMs (Fig. 4a) with a thickness of 23 ± 5 nm by spin-coating a PLA solution (5 mg mL−1) at 4000 rpm for 20 s onto a poly(vinyl alcohol) (PVA) sacrificial film. The mechanical properties and adhesion strength of the resulting transparent films, which exhibited a flat and uniform surface without cracks, were evaluated using the bulging test and the microme-scratch test, respectively. These PLA-based FsNMs showed a low elastic modulus (1.7 ± 0.1 GPa) and high adhesiveness [the critical load for the detachment of the nanosheet adsorbed onto a SiO2 substrate was (1.7 ± 0.3) × 105 N m−1], which encouraged the analysis of their feasibility as a wound dressing in an in vivo experimental gastrostomy model test. The results were extremely positive, showing an excellent sealing efficacy that did not require adhesive agents. Moreover, the incision healed completely without scars and tissue adhesion. In an effort to deeply investigate the possible biomedical application of these PLA FsNMs, their antiadhesive and fixative characteristics were further investigated: an intraperitoneal polypropylene overlaid mesh (IPOM) with PLA nanosheets was placed on an intact peritoneum (Fig. 4b).57 The results evidenced that PLA FsNMs are feasible to induce adhesion prophylaxis in the IPOM, having a beneficial effect as an atraumatic fixation tool. In addition, it was found that nanosheets do not cause inflammatory reaction, which was attributed to the excellent biocompatibility of PLA.57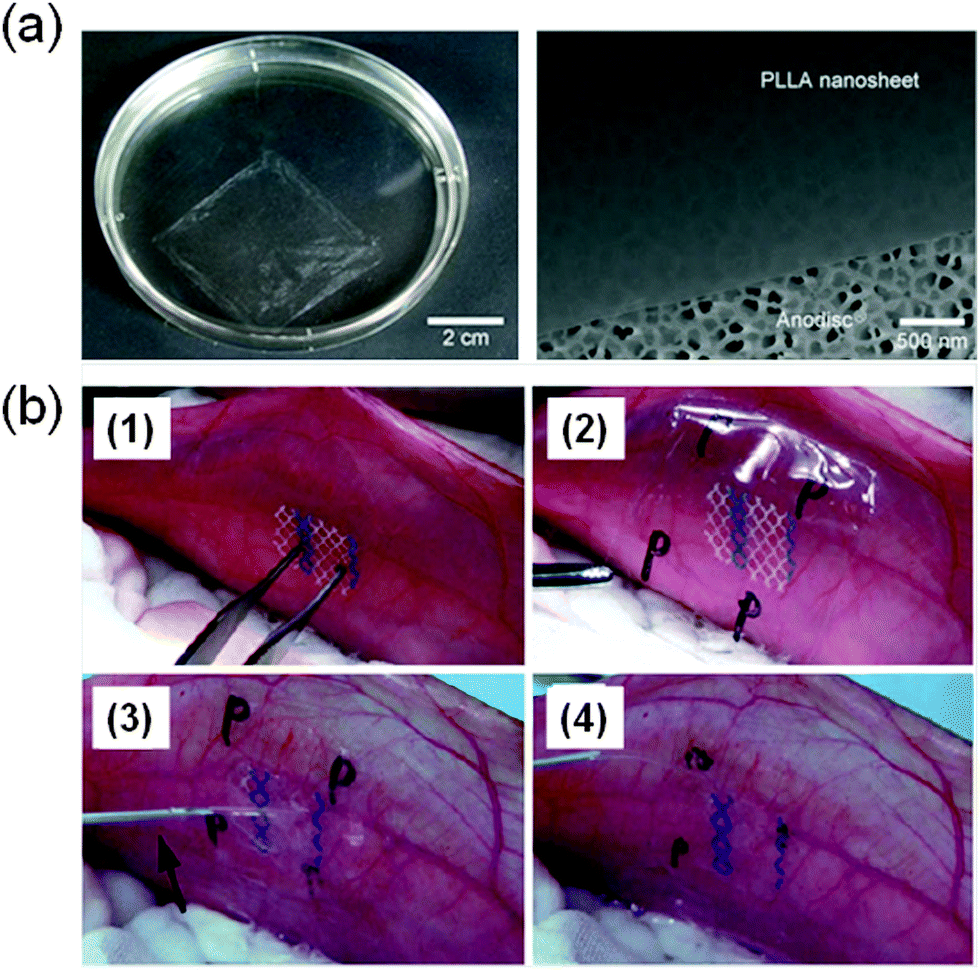 | ||
| Fig. 4 (a) PLA FsNMs with a thickness of 23 ± 5 nm prepared by Takeoka and co-workers:56 the macroscopic image of the nanosheet suspended in water (left) and the SEM image of the nanosheet on an anodisc membrane (right). Reproduced with permission.56 Copyright 2009, Wiley-VCH. (b) Technique of overlaying a PLA FsNM on a large-pore polypropylene mesh for an intraperitoneal onlay mesh (IPOM) without a fixation suture: (1) mesh placed on the peritoneum of a rabbit; (2) overlaying of a PLA FsNM supported by a water soluble PVA sacrificial layer on which the letter “P” is written to distinguish the PLA side; (3) dissolution of the PVA substrate with saline (arrow); and (4) after dissolution of the PVA film, the letter “P” is not visible. Reproduced with permission.57 Copyright 2011, Springer. | ||
Within this research line, in another study, Takeoka and co-workers58 tested the feasibility of PLA FsNMs as a wound dressing against burn infections in an in vivo mouse model partial-thickness injury. PLA FsNMs tightly adhered onto the burn lesion on the mouse back without any adhesive agents, while adhesion to any opposing tissues/organs was not observed. Moreover, its transparency enabled the visual observation of the wound condition, and thus the infection evolution and the healing process were easily monitored. In addition, PLA FsNMs functioned as a barrier against Pseudomonas aeruginosa when it was inoculated on the wound lesion. Therapy with the PLA nanosheet protected against an inflammatory response.59 Hence, the tested system rendered an effective dressing material for the management of partial-thickness burn wounds.
More recently, the same group obtained PLA nanosheets by combining a spin-coating-assisted multi-layering process of PVA and PLA with a peeling step. Therefore, after spin-coating and dissolution of the sacrificial PVA layers between PLA layers in distilled water, a number of PLA FsNMs which corresponded to the number of multi-layering processes of PVA and PLA were obtained.59 The thickness of the resulting FsNMs, which were transparent and extremely flexible, was determined to be 60 ± 6 nm when spin-coating a PLA solution (10 mg mL−1) at 4000 rpm for 20 s. Later, these nanosheets were fragmented to form a stable suspension, and then reconstructed to form a single continuous film that attached to various interfaces without the need of adhesive agents (Fig. 5). The new patched film was applied as a physical barrier against burn wound infection with P. aeruginosa. Both in vitro and in vivo assays evidenced that the patched film exhibited excellent barrier ability to prevent infection during the treatment of burns for 3 days.
 | ||
| Fig. 5 Patched PLA FsNMs coming from several fragmented nanosheets: (a) macroscopic (i) and microscopic (ii and iii) images of fragmented PLA nanosheets adhered on a SiO2 substrate using a digital camera, a digital microscope, and a scanning electron microscope, respectively; (b) detachment of fragmented PLA FsNMs from a poly(tetrafluoroethylene) plate. Reproduced with permission.59 Copyright 2013, Wiley-VCH. | ||
With the aim of designing a novel patch for bone or tendon repair and healing, Pensabene et al.60 studied the biocompatibility, adhesion and proliferation activity of several cell types onto PLA FsNM prepared by spin-coating (2 wt% PLA solution in dichloromethane at 4000 rpm for 20 s using PVA as a sacrificial layer). Their average thickness, which was assessed by AFM, was 320 ± 27 nm, while the elastic modulus, derived from bulge test experiments, was 136 ± 44 MPa. Both immortalized cell lines and primary cell lines cultured on those FsNMs exhibited good morphological and metabolic features and the ability to fully differentiate. In addition to the in vitro assays, the adhesion of such PLA FsNMs to ligament and femur was evidenced under ex vivo conditions using a rabbit model. Moreover, their short- and medium-term biocompatibility were evaluated using C2C12 cultured cells.61 The results evidenced early differentiation with the fusion of cells into firmly adherent myotubes, proving that such flexible nanofilms behaved as good bioactive matrices for cell anchoring, spreading and proliferation.
The effect of an underlying substrate on the interaction between cells and PLA FsNMs was recently investigated.62 For this purpose, polyester-based nanosheets (with thickness values from 29 ± 1 nm to 703 ± 4.4 nm, depending on the PLA concentration during the spin-coating process) were also fabricated by spin-coating and PVA acted as the sacrificial layer. After releasing PLA FsNMs in a liquid environment, they were collected on a stainless steel mesh (PLA-mesh), and subsequently used for cell adhesion studies using rat cardiomyocyte cells (H9c2). The results were compared to the ones obtained on a control interface: SiO2 substrates coated with an ultra-thin layer of PLA (PLA-substrate). Although topological and mechanical properties of PLA FsNMs did not influence the cell viability after 24 h of culturing, cells did geometrically sense the stiffness of the underlying material, thus affecting the adhesion geometry. Briefly, the PLA-mesh induced an anisotropic adhesion of H9c2 cells along the metal wire, while H9c2 cells on the PLA-substrate adhered isotropically, independent of the nanosheet thickness. Accordingly, cells distinguished the increase in the nanosheet stiffness and preferentially adhered onto the rigid interface. Interestingly, cellular anisotropy decreased by increasing the thickness of the PLA FsNM deposited onto the mesh because the nanosheet stiffness was more homogeneous throughout the surface. Considering the huge difference between the Young's modulus of PLA FsNMs (from 3.5 ± 1.3 to 7–10 GPa depending on the thickness) and the metal substrate (hundreds of GPa), it was concluded that cell adhesion was mechanically regulated by the stiffness of the underlying substrate when the thickness of the PLA FsNM was in the order of tens of nanometers.
PLA FsNMs have also been modified and/or functionalized by means of collagen deposition, magnetic particle entrapment and drug loading. In a recent study, Niwa et al.63 prepared PLA nanosheets by modifying only one surface with collagen to endow different discrete functions to each side of the nanosheet: anti-adhesive and pro-healing properties. For this purpose, PLA FsNMs were prepared by spin-coating using conditions similar to those previously described (thickness: 59.5 ± 9.5 nm). A collagen layer was subsequently deposited on the surface following two approaches: solvent casting or spin-coating. The latter strategy resulted in an ultra-thin collagen coating (thickness of 5–10 nm), which is more homogeneous and hydrophilic than that obtained by solvent casting exhibiting a thickness of ∼120 nm. The disorganized collagen fibrils formed on PLA nanosheets when covered using the spin-coated method induced a hydrophilic microenvironment that improved cell adhesion and spreading, in comparison to that obtained by solvent casting. Besides, fine networks of actin filaments were clearly identified in cells cultured on the former biointerface, as opposed to the latter system.
As a first step to develop remotely controlled and manipulated magnetic FsNMs, Taccola et al.64 embedded superparamagnetic iron oxide nanoparticles (SPIONs) into PLA nanosheets by spin-coating (5–20 mg mL−1 of PLA solution in chloroform and 1–15 mg mL−1 of SPION colloidal solution at 3000 rpm for 20 s using a PVA sacrificial layer). Magnetic composites were coloured with different degrees of intensity depending on the nanoparticle load, and the inclusion of SPIONs in the polymeric matrix did not alter their magnetic behaviour, yielding FsNMs with high saturation magnetization and magnetic susceptibility. For nanosheets with a high concentration of SPIONs, the magnetic response increased because of the formation of clusters, albeit their presence did not alter the integrity and stability of the FsNM. The results were good enough to promote further investigation in this area, suggesting that controllable supports with magnetic properties could be fabricated in a near future for biomedical applications.
FsNMs with unidirectional drug delivery ability have the potential to improve therapeutic efficacy, while minimizing undesirable side effects. In this sense, Sun and co-workers65 fabricated robust and flexible FsNMs by sandwiching drug-containing polyelectrolyte multilayer films between a capping and a barrier layer of PLGA. The drug-containing films were prepared by the LbL assembly of chemically cross-linked poly(allylamine hydrochloride)–dextran (PAH–D) microgel and hyaluronic acid (HA), which can load methotrexate (MTX), a negatively charged cancer-inhibiting drug. The PLGA barrier layer, which was obtained by spin-coating, prevents MTX release, and the PLGA capping layer regulates the unidirectional MTX release kinetics in a precisely controlled manner. In vitro cancer treatments evidenced that MTX released from the FsNM preferentially inhibited the proliferation of HeLa cells. The versatility of LbL assembled polymeric films as drug carriers allows the loading of a wide variety of drugs and bioactive agents. Thus, the design of highly efficient unidirectional drug-delivery systems with minimum side effects on normal tissues has great potential for clinical applications.
Finally, Okamura et al.66 prepared disk-shaped PLGA nanosheets and modified their surfaces with dodecapeptide H12 (HLGGAKQAGDV), which is a fibrinogen γ-chain carboxy-terminal sequence (γ400–411) that specifically recognizes the active form of glycoprotein IIb/IIIa on activated platelets. Under flow conditions, H12-PLGA FsNMs were found to interact with activated platelets on a collagen surface faster than H12-PLGA microparticles did. In addition, only FsNMs induced 2D spreading of platelet thrombi on collagen-immobilized plates. These results suggested that PGLA FsNMs might be a suitable candidate as novel platelet substitutes and, also, an alternative to human platelet concentrates infused to treat bleeding in patients with severe thrombocytopenia.66
4.2. Polysaccharide free-standing nanomembranes
Polysaccharides are a class of biological macromolecules that participate in a wide range of biochemical and biomechanical functions. Because of their unique properties, these biopolymers are currently playing an important role in materials science. The structure and function of polysaccharides, and their nanoscale assembly for biomedical materials were recently reviewed by Boddohi and Kipper.67 However, this section will draw the attention to those studies that used polysaccharides as the main polymeric source to develop and design FsNMs. As in the previous section, biomedical applications and promising results will be highlighted.In 2007, Fujie, Okamura and Takeoka68 constructed what they called a “nano-adhesive plaster”, a multilayered FsNM (thickness: 30.2 ± 4.3 nm) made of chitosan and sodium alginate (Na-alginate) that was deposited onto a PVA–silicone rubber substrate using the spin-coating assisted LbL approach. More specifically, the layers of each polyelectrolyte were spin-coated on the silicone rubber (millimetre scale thickness: 1.0 mm) previously covered with a sacrificial PVA layer (microscale thickness: 1.2 μm). As a result, the nano-adhesive plaster was formed by the superposition of three different types of free-standing sheets (Fig. 6a). Chitosan and Na-alginate contain amino and carboxylic groups, respectively, as cationic and anionic polyelectrolytes at ambient pH, which enormously facilitated their assembly in the FsNM. The elastic modulus, which was determined by the bulge test, was found to be 1.3 GPa. The good adhesiveness and flexibility of the nano-adhesive plaster were evidenced by transferring it, after modification with a luminescent pigment for ease of visibility, not only onto the skin of a human arm68 (Fig. 6b–d) but also onto a tissue (rat cecum) surface.69 In a subsequent study, the same group proved the potential biomedical application of this adhesive, which is a robust and flexible nano-adhesive plaster made of chitosan and Na-alginate in a surgical intervention.70 Thus, a FsNM with a thickness of 75 nm repaired an in vivo visceral pleura defect induced on beagle dogs without any loss in the respiratory function and without significant inflammatory response. In addition, the influence of the thickness on the mechanical properties of polysaccharide FsNMs was demonstrated, which enabled an easy modulation of the nanosheet features.
 | ||
| Fig. 6 (a) Elements used to fabricate the nano-adhesive plaster floating in acetone: silicone rubber (substrate), PVA sheets (sacrifial layer) and polyssacharide FsNMs photographed in the dark (the polysaccharide FsNM was modified with the luminescent pigment for easy visibility). Nano-adhesive plasters on the human skin (b) before the polysaccharide FsNM was released from the silicone rubber and (c) after release from such a substrate. (d) Same image as (c) except that it was captured in the dark. Reproduced with permission.68 Copyright 2007, Wiley-VCH. | ||
Besides, the therapeutic effect of the nano-adhesive plaster on the murine cecal puncture was evaluated (Fig. 7).71 The sealing effect of the multilayered chitosan and Na-alginate-based FsNM inhibited effectively the bacterial infection, and increased the survival rate of the individuals. Interestingly, the treatment with the polysaccharide nanosheet provoked less inflammatory response than the suture.71 Despite the promising results, a small percentage of bacteria was able to pass through the FsNM. To overcome this aspect, an antibiotic-loaded polysaccharide nanosheet was developed.72 In this new approach, an antibiotic (tetracycline, abbreviated as TC) was sandwiched between a new PVA layer (named PVAc in Fig. 8), which acted as a protector, and the LbL assembled polysaccharide platform (total thickness: 177 nm). The resulting FsNM, which comprehended three functional layers (Fig. 8), exhibited an important anti-microbial effect and a relatively low inflammatory tissue response.72 As a consequence, in vivo studies using overlapping therapy with these new PVA–TC–polysaccharide FsNMs showed a significant increase in mouse survival after cecal puncture. Later research evidenced that TC-loaded chitosan and Na-alginate multilayered FsNMs exhibited a potential antibacterial activity when covering mice dorsal skin artificially burnt and infected with P. aeruginosa.73 All mice treated with the TC-loaded polysaccharide FsNM survived, whereas mice treated with TC-unloaded nanosheets and mice left untreated displayed increased rates of mortality due to bacterial infection. Moreover, TC-loaded FsNMs prevented not only local inflammation but also systematic inflammation.
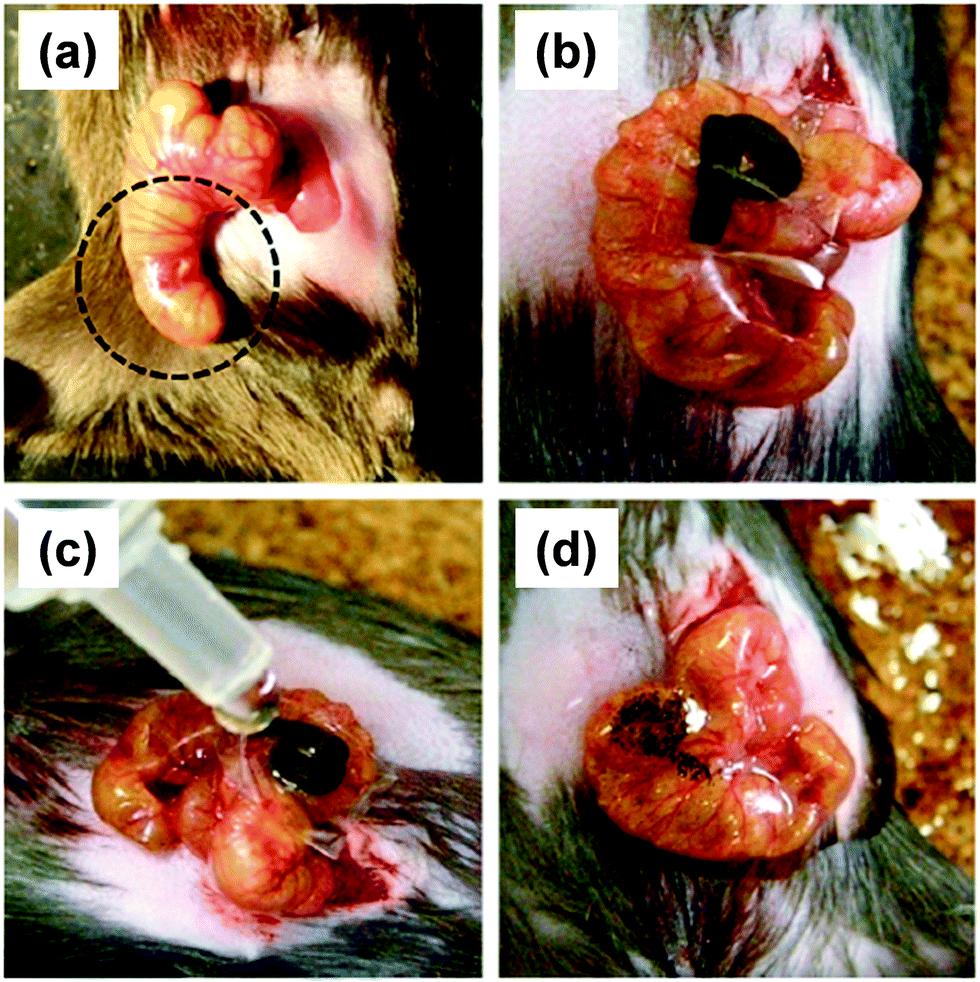 | ||
| Fig. 7 FsNMs made of chytosan and Na-alginate were applied to a murine model of cecal puncture: (a) cecal defect site punctured with a needle (dashed line circle showing an actual puncture site); (b) sealing with the polysaccharide nanosheet supported by a sacrifial PVA film, in which the letter “P” was written; (c) dissolution of the PVA film with saline water solution; and (d) after dissolution of the sacrificial layer, the letter “P” is not visible. Reproduced with permission.71 Copyright 2010, Elsevier. | ||
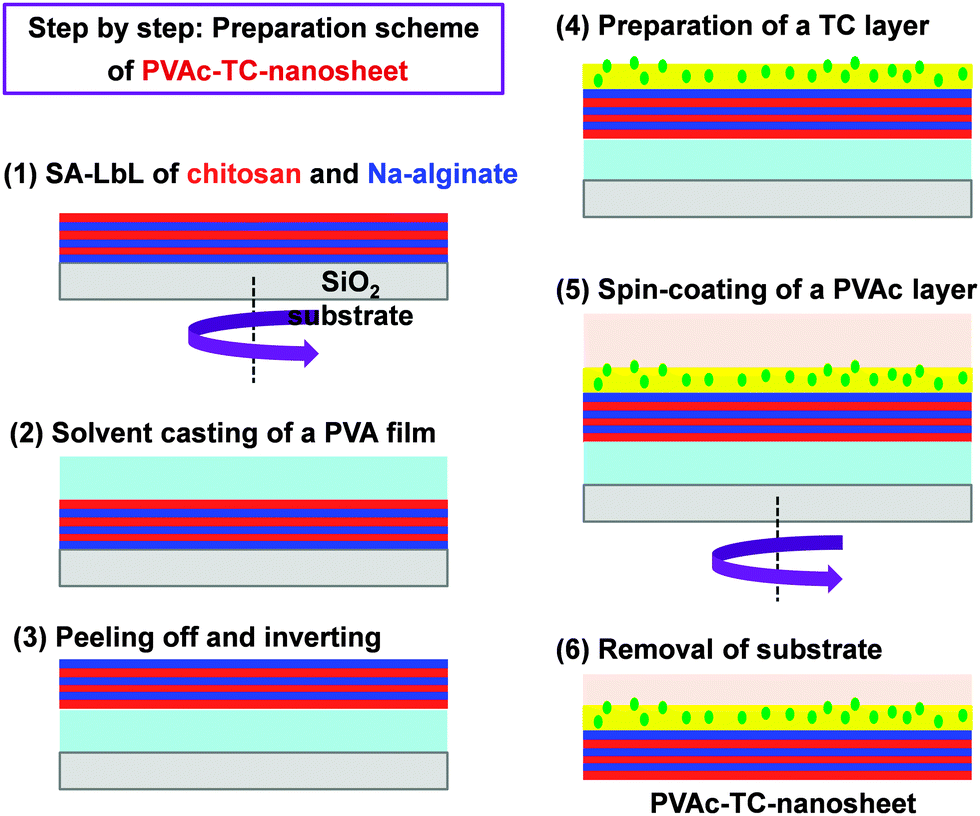 | ||
| Fig. 8 Preparative scheme for tetracycline (TC) loaded polysaccharide nanosheets made of chitosan and Na-alginate using the spin-coating assisted LbL technique (SA-LbL). | ||
Hagisawa et al.74 described a novel therapy in which the overlapping of several chitosan–Na-alginate FsNMs with a thickness of 75 nm (i.e. multioverlapping therapy) sealed and stopped a massive venous hemorrhage in rabbits. Before in vivo tests, the mechanical durability of the nanosheets and their hydrostatic pressure resistance were evaluated. Four pieces of overlapping nanosheets were able to stand 80 ± 6 mm Hg of pressure. In regard to their degradability, polysaccharide nanosheets experience a decrease in their thickness under degradation conditions (phosphate-buffered saline, PBS, at 37 °C). Nevertheless, they retained more than 70% of the thickness after seven days. Positively, after one month, no inflammatory tissue reaction was detected around the FsNM attachment and findings revealed a complete wound healing. The multioverlapping therapy would represent a great advantage for surgical operation, especially in trauma patients with bleeding from large veins.
Otani et al.75 have reported the therapeutic use of bilayered polysaccharide FsNMs made of chitosan and Na alginate as an arachnoid plaster in a microneurosurgery environment because of their semi-absorbent and potent physical adhesive strength features. These authors observed that the application of overlapping FsNMs without using chemical bonding agents completely avoided cerebrospinal fluid leakages in the cerebral cortex. There was evidence of a relationship between the number of overlaid nanosheets and the reinforcement effect: the more the layers displayed, the more they improved. Besides, after six months, no inflammatory infiltration was detected.
In addition to chitosan and Na-alginate, proteoglycan HA has also been used to fabricate polysaccharide FsNMs. For instance, Fujie et al.76 prepared multilayered nanosheets of HA and collagen on a water-soluble sacrificial supporting substrate using the LbL assembly method. The thickness of these FsNMs was found to grow exponentially with the number of HA and collagen layers. This was attributed to the fact that the amount of HA adsorbed in the LbL structure is less than that of collagen.77 Therefore, the polyion pairs mediated by the electrostatic interaction between collagen and HA molecules induced nonlinear growth in the LbL system. The mechanical properties of these FsNMs, as determined by the bulge test, were found to depend on the fibrous or non-fibrous structure of collagen layers. Thus, the elastic modulus of nanosheets made of non-fibrous collagen and a high content of HA (thickness: 62 ± 7 nm) was 4.3 ± 0.6 GPa, which is a value comparably smaller than that of the previously reported chitosan/Na-alginate nanosheet (9.6 GPa for a thickness of 75 nm70). This low elastic modulus was attributed to the hydrophilic and moisture-sensitive nature of HA molecules. In contrast, the elastic modulus of FsNMs made of fibrous collagen (thickness: 42 ± 4), which had a low content of HA, increased to 12.5 ± 1.5 GPa, thus evidencing a greater mechanical durability. Collagen structures in bone and skin have an elastic modulus of 17.2 and 4 GPa, respectively. Therefore, FsNMs made of HA and fibrous or non-fibrous collagen efficiently imitate the mechanical properties of these tissues. Consistently, FsNMs made of non-fibrous collagen exhibited a surface with softer elastic properties than those observed in FsNMs with fibrous collagen. Besides, cell adhesion studies using NIH-3T3 cells showed that HA/non-fibrous collagen FsNMs gave lower cell adhesive elongation in comparison to nanosheets with fibrous collagen and a low content of HA. On the basis of this study, the authors concluded that cell adhesive properties can be tuned by changing the structural components of the nanosheets (i.e. the content of polysaccharide and collagen fibrils),76 thus opening a new door for the production of novel engineered scaffolds for regenerative medicine as well as cell biology.
An innovative and audacious strategy has been recently reported by Chen et al.78 who prepared cell–polymeric nanocomposites for the fabrication of FsNMs lined with cells (Fig. 9). First, poly(N-isopropylacrylamide) (PNIPAM), a temperature-responsive polymer, was grafted onto glass-slides. After this, cells were cultured on the resulting hydrophobic layer. Once 80–90% cell confluence was achieved, the LbL process was conducted on the surface of the cell sheet. However, gelatin, a natural biocompatible polyelectrolyte, was previously deposited as the cell-contacting layer to maintain a high cell viability during the deposition process.79,80 Thus, this gelatin layer is of great importance during the LbL self-assembly step on the cell-sheet surface.79,80 Specifically, three alternating charged polysaccharide bilayers of chitosan and Na-alginate, (chitosan–Na-alginate)3, were assembled onto the gelatin-coated cells cultured on the PNIPAM-grafted surfaces. The deposition of gelatin, chitosan and Na-alginate layers, which are extracellular matrix (ECM) related components, on the cell sheet resulted in a cell adhesive surface that efficiently interacted with cells.81 The free standing cell–gelatin–(chitosan–Na-alginate)3 film was peeled off from the PNIPAM-grafted surface upon temperature changes. By this strategy, the assembly of cell sheets with ultra-thin ECM components to form FsNMs can be potentially used to fabricate complex artificial soft tissues to substitute some elements in native tissues.
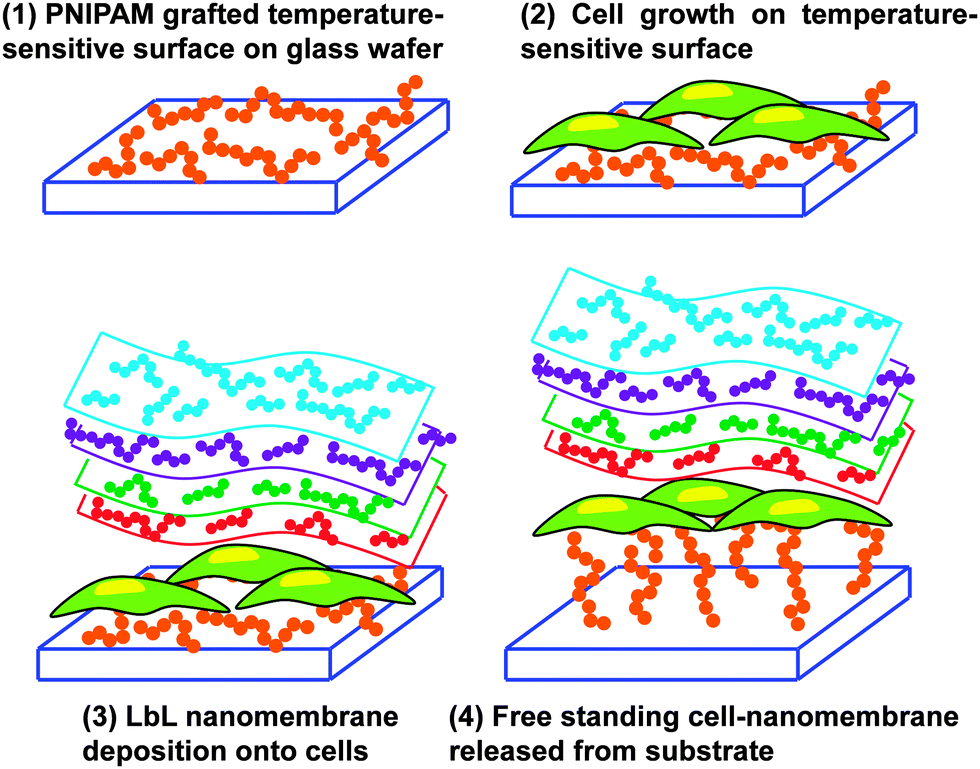 | ||
| Fig. 9 Schematic illustration of fabrication of cell–gelatin–(chitosan–Na-alginate)3 FsNMs. | ||
4.3. Other polymeric sources for free-standing nanomembranes
Poly(ethylene oxide) (PEO) is a thermoplastic biocompatible material widely used in biomedical applications, for example scaffolds, drug delivery systems and sensor devices.82–84 To extend their application list, the LbL technique was used to fabricate FsNMs made of PEO.85 This was achieved by creating solid-state hydrogen bonded assemblies that allowed the incorporation of stable interdigitated layers of PEO and poly(acrylic acid) (PAA) at the nanometer length scale (the thickness of each PEO–PAA bilayer was 80 nm). Free-standing films containing 100 bilayers were transparent, smooth to touch and exhibited elastomeric properties during handling. Although these were not explicitly proved with experiments, Lutkenhaus et al.85 suggested many biomedical applications for these hydrogen bonded assemblies, for example biosubstrates, drug delivery devices and pH-sensitive sensors.Ono and Decher86 reported the use of pH-responsive self-standing polyelectrolyte multilayer membranes, which were fabricated by spraying polymer solutions, for biomedical applications. Concretely, a silicon wafer substrate was covered by a sacrificial multilayered film made of PAA and poly(ethylene glycol) (PEG), which disintegrated by a pH change releasing a target membrane. In that work, the released membrane consisted of a simple polyelectrolyte multilayered film of poly(allylamine hydrochloride) (PAH) and poly(sodium-4-styrenesulfonate) (PSS), which was initially constructed on the top of the PAA–PEG pH-responsive film. The pH response mechanism is based on the alteration of the hydrogen bonds established between PAA and PEG when the carboxylic acid groups present in PAA transform into carboxylate ions. From a biomedical point of view, the advantages of the PAA–PEG pH-responsive film, which was obtained with thickness values ranging from 55 to several hundreds of nanometers and areas of a few square centimetres, rely on the fact that PAA and PEG are biocompatible, biotolerated or bioinert. Accordingly, their release would not lead to adverse effects in the bioenvironment, and thus could be used in therapeutic devices and aids.
In another example, the preparation of PEG-terminal FsNMs was based on the extensive cross-linking of aromatic 4′-nitro-1,1′-biphenol-4-thiol (NBPT) self-assembled monolayers (SAMs) deposited onto a gold support by exposure to low energy electrons, thus resulting in a mechanical and thermally stable monolayer.87 Simultaneously, the terminal nitro groups of the NBPT molecules were converted to reactive amine moieties to which epoxy functionalized PEG chains were subsequently coupled. As a result, these films exhibited protein repelling properties, which in turn ensure the lack of protein denaturation when they are used as a support in transmission electron microscopy studies.87
Meyerbröker and Zharnikov developed highly elastic, hydrophilic and ultra-thin membranes consisting entirely of PEG.88 This was achieved by preparing an ultra-thin stable PEG-hydrogel precursor film composed of a mixture of epoxy- and amine-terminated PEG moieties, which was deposited onto a supported sacrificial layer (i.e. a 100 nm gold film evaporated onto a silicon substrate). Then, the complementary terminal groups underwent chemical crosslinking.89 Afterwards, the PEG–gold bilayer was separated from the silicon support and the sacrificial layer was dissolved releasing the PEG FsNM, which could be transferred onto a grid or any other arbitrary substrate. These nanosheets exhibited sufficient mechanical stability, and also high flexibility (extraordinary low Young's modulus of only ∼2 MPa). Such a behavior is characteristic of elastomers,90 and it was never observed before in nanomembranes. Indeed, the only analogous systems are polyisoprene and polyisobutene nanomembranes, which are not biocompatible, prepared using Langmuir–Blodgett technology.17 On the basis of their properties, PEG nanosheets are potentially useful as a highly sensitive support and a sensor element for biological samples.
In another study, porous multilayered films of PAH and PAA were prepared using the LbL dipping technique, nanopores being subsequently created using a pH treatment.11 The porosity induced by pH adjustment resulted in a significant increase in the thickness. For example, the thickness of films made of 8 PAH–PAA bilayers before and after the creation of nanopores was 101 and 253 nm, respectively. Ketoprofen and cytochalasin D, which represent different types of drugs that can be entrapped in these films, were successfully loaded and showed zero order release kinetics over a long period of time. The amount of drug loaded and released could be tuned by varying the number of bilayers in the porous regions of the films, whereas variations of the pore size controlled the release flux of the given drug. On the other hand, sheet- and tube-like FsNMs of PAA–PAH were prepared by exfoliating PAA–PAH multilayered films from substrates in an aqueous acid solution containing Cu2+.91 Initially, multilayered films were prepared by LbL deposition onto silicon wafers, quartz or glass tube substrates. Their ion-triggered exfoliation was achieved by breaking the electrostatic interactions between the PAA layer and the underlying substrate. However, it should be noted that, although PAA–PAH bilayers were nanometric, ion-triggered exfoliation was applied to films with 15 bilayers of submicrometric thickness (∼0.8 μm) and, therefore, these cannot be considered as nanomembranes. The developed technology was proposed for the fabrication of small-caliber artificial blood vessels.
Gui et al.92 fabricated LbL multilayer films made of poly(diallyldimethylammonium) (PDDA) and poly(4-vinylpyridine propylsulfobetaine) (P4VPPS), a zwitterionic polysulfobetaine, in an acid aqueous solution at pH 2 with 0.5 M NaCl. The average growth rate was estimated to be ∼29.0 nm per PDDA–P4VPPS bilayer. These films were pH-dependent, thus disintegrating in alkali aqueous solution, especially at pH ≥ 12, which suggested a potential application as sacrificial layers for the release of other polymeric films. This was proved by depositing a multilayered film of PDDA and poly(sodium 4-styrenesulfonate) (PSS) on the top of a 10-bilayered PDDA–P4VPPS sacrificial film. After treatment with alkali aqueous solution at pH 12, the PDDA–PSS film was released maintaining its integrity in air. Previously, Dubas et al.93 had reported the fabrication of PDDA–PSS FsNMs using PDDA–PAA LbL multilayer films as sacrificial layers.
Kohri et al.94 presented a new approach for the fabrication of smooth and stable FsNMs composed of a polymer brush, poly(2-hydroxyethyl methacrylate) (PHEMA), supported on an ultra-thin (i.e. ∼6 nm) colourless polydopamine (PDA) layer. This was achieved through the surface-initiated atom radical polymerization (ATRP) of 2-hydroxyethyl methacrylate (HEMA) on the PDA layer, which resulted in optically transparent and colourless free-standing PHEMA brush films with a tailored thickness of 16–75 nm. Prior to this process, the PDA ultra-thin film was placed onto a silicon or glass substrate covered by a cellulose acetate sacrificial layer (thickness: ∼115 nm). The scheme of the synthetic design is displayed in Fig. 10. Although no application was explicitly tested, the authors suggested that PHEMA FsNMs can be used as multi-stimuli responsive sensors after functionalizing their surface.94
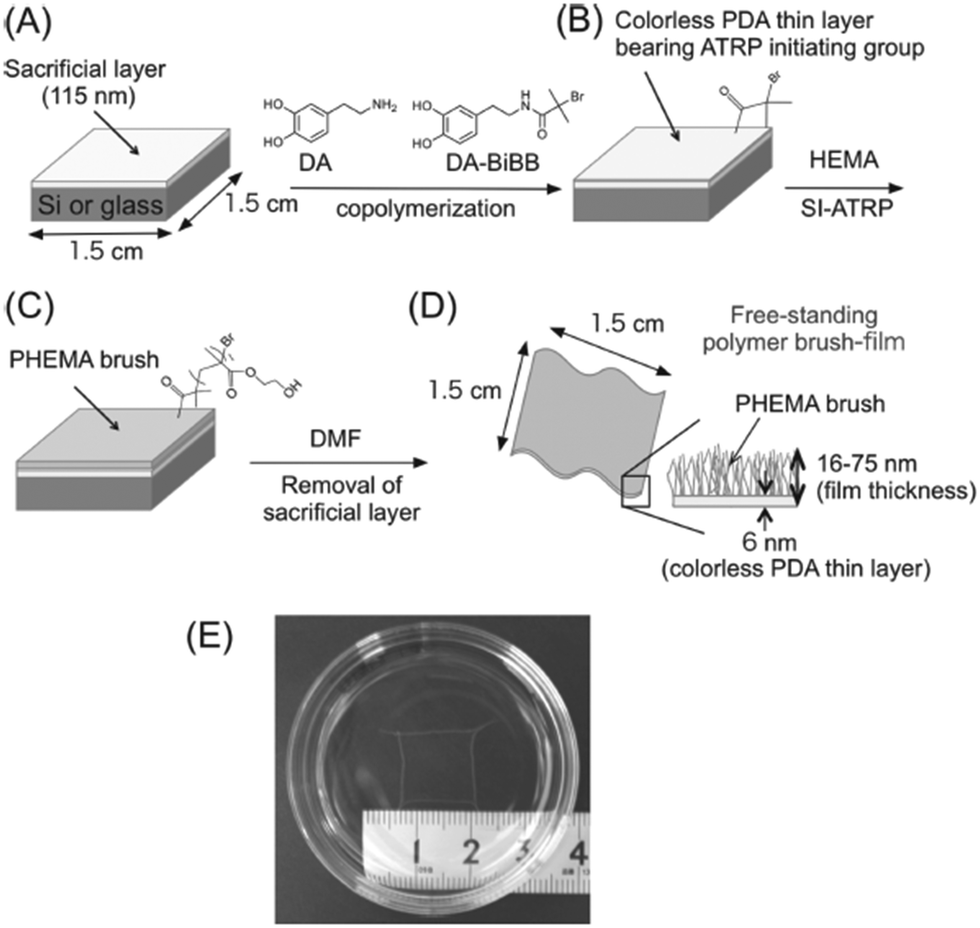 | ||
| Fig. 10 Schematic representation of the preparation of a free-standing polymer brush film based on a colorless PDA thin layer: (A) spin-coating of a sacrificial cellulose acetate layer onto the silicon plate; (B) creation of a colorless PDA thin layer containing an ATRP-initiating group; (C) construction of polymer brushes on the substrate by surface initiating ATRP; (D) recovery of polymer brush film based on a PDA layer by the dissolution of the cellulose acetate layer in DMF; and (E) photographic image of the obtained free-standing PHEMA brush film floating in dimethylformamide. Reproduced with permission.94 Copyright 2013, Wiley-VCH. | ||
Surface-functionalized FsNMs were produced grafting poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC) brushes by the ATRP of 2-methacryloyloxyethyl phosphorylcholine (MPC) on nanosheets made of PAH, PAA and PS, which were prepared by the spin-coating assisted LbL technique (a thickness of 85 ± 2 nm).95 The properties of PMPC-nanosheets (physiological stability, surface wettability and anti-biofouling response) were regulated by several parameters, such as the thermal crosslinking of the FsNM, the grafted amount of MPC and the thickness of PMPC brushes (ca. 11 nm). PMCP-nanosheets were easily peeled, transferred using tweezers with the aid of a water-soluble PVA sacrificial layer, cut into any shape using scissors, and patterned using a needle. Nanosheets patched on cell culture substrates exhibited anti-biofouling properties such as anti-coagulant behaviour of human blood cells as well as the potential to microscopically pattern murine fibroblasts cells. The overall results provided an important physicochemical insight into the remarkable biological response of surface-functionalized nanosheets, which represent not only a powerful tool for biomedical applications but also an alternative to conventional micropatterned techniques used in bionanotechnology.
Fujie et al.96 developed spatio-selective cell culture environments by inkjet printing bio-patterns onto FsNMs composed of poly(methyl methacrylate) (PMMA). The thickness of the nanosheets, which were prepared by spin-coating, was ca. 200 nm. Interestingly, several cell adhesion promoters, such as poly(L-lysine) modified with fluorescein isothiocyanate, were micropatterned on the FsNM surface and, subsequently, functionalized with fibronectin by electrostatic interaction. The high flexibility of PMMA FsNMs made it an ideal substrate for cell adhesion and spreading. In vitro assays showed the selective deposition of C2C12 skeletal muscle myoblasts following these patterns. Accordingly, the protein micropatterned FsNM system was proposed as an interesting tool for cell-directed culture in muscular tissue engineering.
PMMA was also employed for the preparation of FsNMs displaying the phase separation morphology. This was achieved by spin-coating a polymer mixture solution of PMMA and polystyrene (PS) onto a PVA-coated substrate.97 Due to the intrinsic immiscibility of PMMA and PS, rapid quenching during the spin-coating process induced their phase separation. In order to obtain porous nanosheets, PS regions were dissolved by immersing the PMMA–PS FsNM into cyclohexane. The thickness of these films, which ranged from 38.8 ± 1.1 to 110.2 ± 2.0 nm, and the diameter of the pore, which varied between 64.4 ± 9.3 and 187.2 ± 33.9 nm, were controlled through the PMMA:PS ratio and the spin-coating conditions. Furthermore, the authors demonstrated that when the thickness of the ultra-thin films is comparable to the dimensional scale of the phase separation domains, it is possible to prepare perforated FsNMs with nanopores in the range of tens of nanometers.97 The availability of these perforated FsNMs is proposed to be of immense value in many biomedical applications, for example cell culture devices, high flux biosensors and drug delivery systems.
Flexible PS FsNMs for directing cellular organization were prepared by Fujie et al.98 by combining spin-coating and microcontact printing methodologies. The PS nanosheet thickness, which ranged from tens to hundreds of nanometers, was controlled through the concentration of the polymer solution used during the spin-coating process. In this work, the sacrificial layer was made of PNIPAM. After hydrophilization of the PS nanosheet surface, a composite made of multi-walled carbon nanotubes and fibronectin (CNT–Fn) was micropatterned on the FsNM using poly(dimethylsiloxane) (PDMS) molds with microscopic groove-ridge features (width and separation of ∼50 μm). Then, unpatterned regions were rendered cytophobicity by Pluronic F-127 to promote initial cell alignment. As a result, the CNT–Fn nanocomposite was homogeneously distributed onto the flexible PS nanomembrane (Fig. 11a). Nanomechanical mapping revealed the high flexibility of these engineered nanomembranes by simply decreasing the film thickness. This flexibility is illustrated in Fig. 11b and c, which display the bending of a silicone tube wrapped with 40 nm thick PS/CNT–Fn FsNMs. Therefore, the combination of their outstanding features (i.e. high flexibility, cell adhesiveness and surfaces with tailored morphology) suggests that such systems recreate the properties of the extracellular matrix (ECM). Cell-adhesive micropatterns facilitated the alignment of C2C12 skeletal myoblasts, while CNTs enhanced the cellular elongation and differentiation to generate functional myofibers.
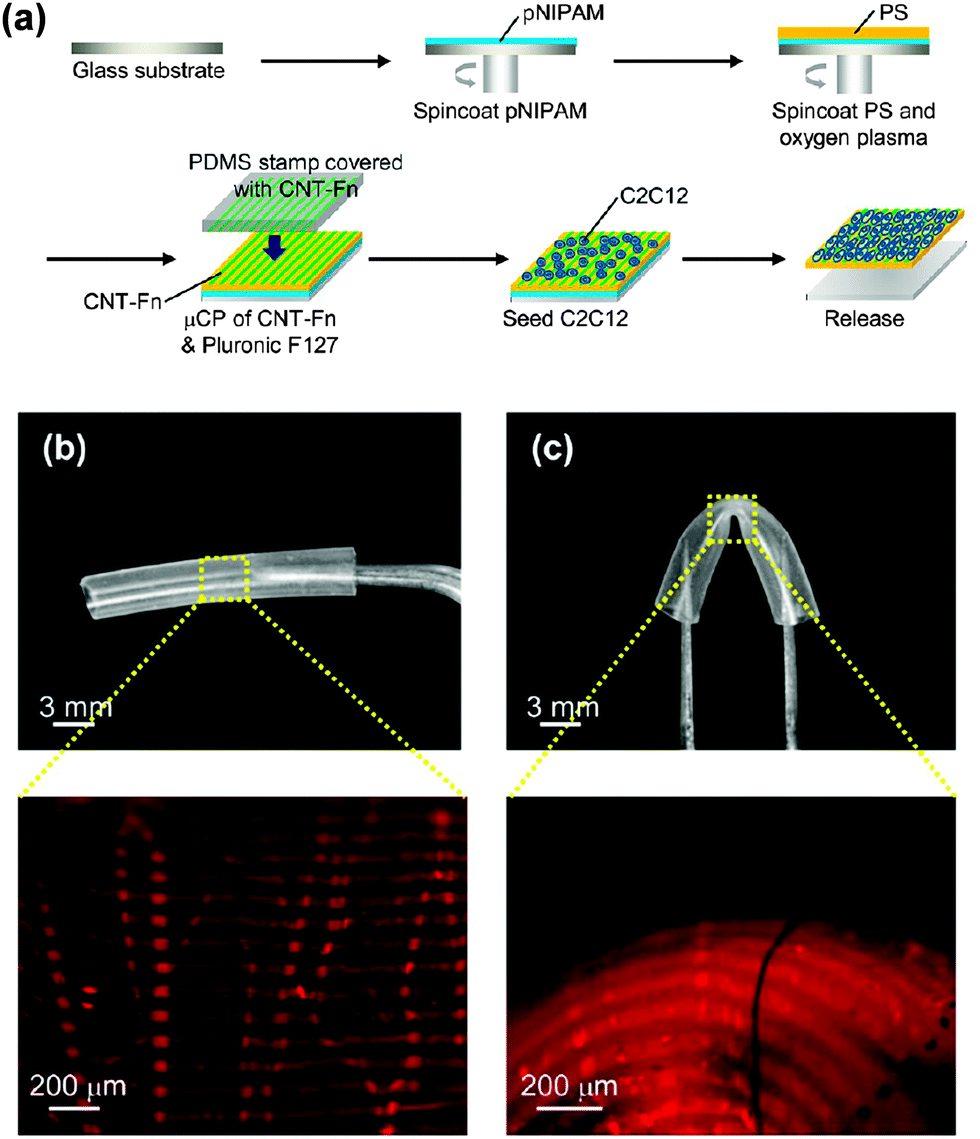 | ||
| Fig. 11 (a) Schematic of the preparation of PS nanomembranes with micropatterned CNT–Fn nanocomposites and aligned C2C12 myoblasts. Macroscopic and magnified fluorescence images of the nanomembranes (40 nm thick) wrapping a silicone tube (3 mm diameter) (b) before and (c) after bending the tube. The micropatterns (rhodamine labeled fibronectin) were flexible and adhered to the curvature of the tube in the bending state. Reproduced with permission.98 Copyright 2013, American Chemical Society. | ||
In an earlier study, Tsukruk and co-workers4,99 prepared hybrid organic–inorganic FsNMs with extraordinary sensitivity and unique auto-recovering ability. These nanosheets (a thickness of 25–70 nm) were prepared by spin-assisted LbL assembly of PAH and PSS on a sacrificial substrate. The most innovative aspect of these FsNMs is the intercalation of a central layer containing gold nanoparticles (a diameter of 12.7 nm) sandwiched between PAH–PSS bilayers. Thus, the general formula of these nanosheets can be described as: (PAH–PSS)nPAH–Au–(PAH–PSS)n–PAH, where n was varied between 3 and 11. The thickness of the system with a gold central layer was ∼20 nm higher than that of films without gold nanoparticles, independent of n. The mechanical properties of these hybrid FsNMs (an elastic modulus of 30–40 GPa, an ultimate strain of about 2% and an ultimate tensile strength higher than 100 MPa) were found to surpass those reported for a much thicker (i.e. submicrometer scale) nanoparticle-containing free-standing LbL films.99 Moreover, the overall examined properties suggested a wide variety of prospective applications,4 those related to the development of chemical and temperature micro-array sensors being particularly relevant in the biomedical field.
Metallic nanoparticles were also used to fabricate a superhydrophobic/hydrophilic asymmetric FsNM using the LbL approach on a Teflon substrate.100 In this case, the layer assembly was achieved between a poly(ethyleneimine)–Ag+ complex (PEI–Ag) at pH 9 and PAA at pH 3.2. The incorporated silver ions were reduced to silver nanoparticles during the thermal treatment applied to promote the cross-linking by imide bond formation. As a result, silver loaded films displayed asymmetric wettability: the top surface was superhydrophobic, while the bottom one was hydrophilic. On the one hand, the superhydrophobic side limited the bacterial adhesion and exhibited self-cleaning properties; on the other hand, the hydrophilic side delivered bactericidal silver ions. These silver-functionalized hybrid films can have great potential in open wound patches or in the barrier, separation, transportation or drug delivery fields, although exceeding the nanoscale (thickness ranging between 2 and 20 μm, depending on the number of layers). This aspect was overcome by Hammond and coworkers,101 who designed contact-killing ionically cross-linked LbL ultra-thin films using N,N-dodecyl, methyl-polyethylenimine (DMLPEI), which has microbicidal activity, layered with PAA (thickness < 100 nm for films with a number of bilayers ranging from 2 to 20). A pH treatment was applied during the assembly process to rearrange the microbicidal polycation chains. Thus, at acid pH, the amount of positive charges on the surface available to interact with the bacterial cell membrane is higher than that at neutral pH. Consistently, films as thin as 10 nm made at pH 3 were more lethal to both airborne and waterborne bacteria than films made at pH 7.
Baxamusa et al.102 proposed the fabrication of FsNMs without using any sacrificial layer for their release from the substrate. More specifically, these authors proposed the direct delamination of ultra-thin films made of poly(vinyl formal) (PVF) resin, PS or PMMA. Delamination of a thin film from its substrate spontaneously occurs when the strain energy (Gv) in the film exceeds the interfacial energy resisting separation (γ). In practice, this occurs when the film thickness (L) satisfies the following condition:103
 | (1) |
Lastly, extraordinarily thin FsNMs (thickness < 5 nm) were obtained by spatially confined polymerization of a unique and elaborate 2D supramolecular system composed of two liquid-crystalline lamellar bilayer membranes made of self-assembled nonionic surfactant, dodecylglyceryl itaconate (DGI), which was sandwiched by a water layer (Fig. 12).104 Nanosheets are achieved using a simple free-radical polymerization under UV radiation with high yield and in large quantity. The covalently bonded two-molecular-thick sheets exhibited high mechanical strength and thermal stability. Moreover, an important characteristic of these ultra-thin sheets is the high-density of functional groups exposed to the outer surfaces. Post-functionalization of the hydroxyl groups at the head of DGI located on the outer surfaces of these nanomembranes opens the door to many practical applications in the biomedical field.
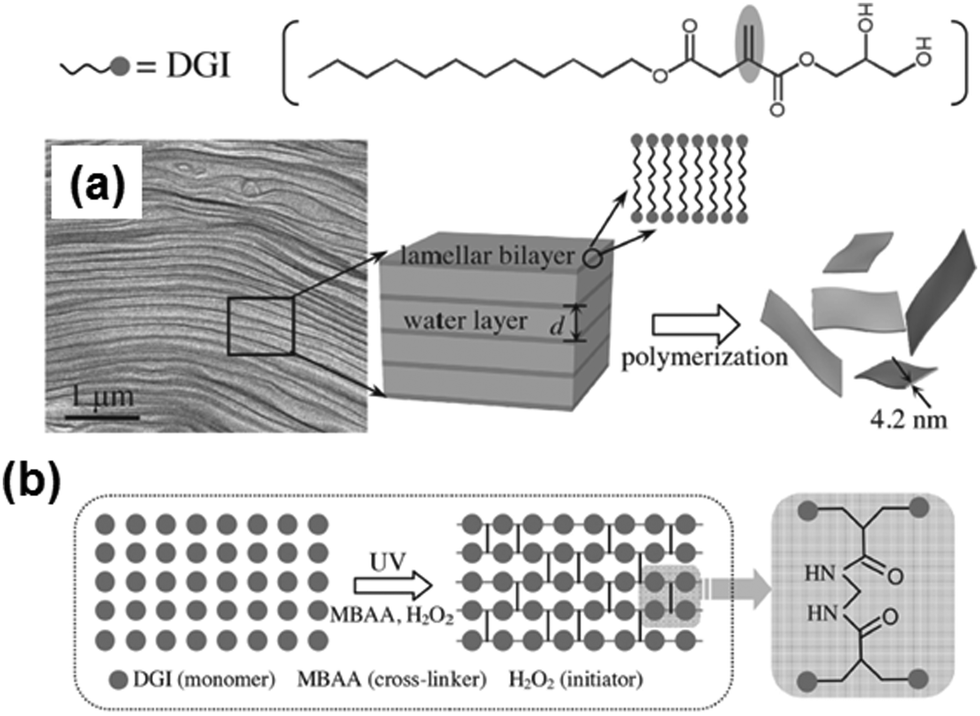 | ||
| Fig. 12 Schematic illustration for the formation of free-standing, single-bilayer-thick polymeric nanosheets derived from DGI: (a) freeze-fracture electron microscopy image of the DGI system, and (b) top-view schematic showing cross-linking between DGI along one lamellar plane. Reproduced with permission.104 Copyright 2014, Wiley-VCH. | ||
5. Biomedical applications of free-standing nanomembranes with electroactive conducting polymers
Human body tissues, such as neural, cardiac or skeletal muscle ones, respond to electrical and electrochemical stimuli, signals being able to regulate cell growth and behaviour. However, conventional biomaterials do not result in appropriate biointerfaces to conduct electrical and/or electrochemical stimulation since they lack both electrical conductivity and electrochemical activity. In order to overcome this drawback, ECPs have been used as biocompatible materials which can support cell adhesion, migration and proliferation.105 Their properties are of great potential since they can be used to electrically stimulate local tissue,106–109 exchange ions reversibly with cells, thus promoting adhesion and proliferations of these living systems,41,110,111 fabricate electronic devices (e.g. biocapacitors and biobatteries) that can be implanted in the organism,112 act as time controlled drug- or bactericidal-release devices,113,114 detect biomolecules,115–117 recognize specific fragments of DNA,118,119etc. Recently, Llorens et al.120 reviewed the advancements in the preparation of structures (i.e. essentially fibrous mats) based on biodegradables and ECPs for biomedical applications. On the other hand, it should be remarked that the electrochemical detection of biomolecules using ECPs has received intense interest within the biomedical field.121–126 In this application, ECPs act as interfaces between bio-substrates and inorganic electrodes favouring lowered impedance between the electrode and electrolyte interface. Accordingly, ECP-based biosensors consist of organic films (or even hydrogels) supported on inorganic electrodes rather than in self-supported organic membranes and, therefore, their discussion has not been included in this review.Nevertheless, one of the major drawbacks in obtaining free-standing nanosheets composed exclusively of ECPs is the lack of mechanical integrity, poor stability, brittleness and the restricted processability of many of these materials. These limitations, combined with their non-degradability, affect the number of biomedical applications of all-ECP FsNMs. Because of this reason, in this section we separate the few studies on FsNMs fabricated only with ECPs and inorganic–ECP hybrids, from those based on insulating polymer–ECP blends.
The biomedical applications of some ECPs are limited by the biocompatibility and cytotoxicity of these materials, which in turn depend on their chemical nature. In a recent study, Humpolicek et al.127 examined the biocompatibility of non-conducting and conducting PAni forms (i.e. PAni emeraldine base and PAni hydrochloride, respectively). Although both forms of PAni did not induce any sensitization and skin irritation, the two materials but especially PAni hydrochloride exhibited considerable cytotoxicity. Polymer purification via re-protonation ↔ de-protonation cycles led to a significant reduction in cytotoxicity, suggesting that the negative effects were provoked by low molecular weight reaction residues or by oligomers rather than by the own PAni molecules. Subcutaneous implantation of PAni emeraldine base films into animal models showed inflammation response and fibrous encapsulation.128,129 In opposition, PPy exhibited good biocompatibility. Wang et al.130 evaluated the biocompatibility of PPy supported membranes with nerve tissue. PPy showed no evidence of acute and subacute toxicity, pyretogen, hemolysis, allergen and mutagenesis, and the cells showed better survival and proliferation rates than with the control saline solution. These results suggested that PPy might be a candidate material for bridging the peripheral nerve gap.130
George et al.131 evaluated the cytotoxicity of PPy in vitro using primary cerebral cortical cells and in vivo surgically implanting PPy substrates in the cerebral cortex of rats. The results evidenced that PPy is at least as biocompatible as Teflon (used as a control) and, in fact, performed better in many cases. Similarly, PTh and its derivatives have shown tremendous potential for interfacing electrically conducting polymers with biological applications due to their low cytotoxicity and high biocompatibility. For example, PEDOT41 and poly(α-terthiophene)132 (PTh3) have been used to fabricate bioactive platforms, even though their monomers and small oligomers are cytotoxic. Cytotoxicity is particularly low for some water-soluble PTh derivatives bearing hydrophilic pendant moieties, which have been proposed as excellent lysosome-specific materials for bio-imaging applications.133
Despite its current importance among conducting materials and its recently developed biomedical applications, in this review we have not included studies based on graphene nanosheets.134,135 Graphene typically presents porous and honeycomb-like structures and, in some cases, it has been considered as an alternative to ECPs in the preparation of single component FsNMs. However, the use of graphene is restricted by the topology of its bidimensional nanosheets, which is not comparable to that of ECPs.136,137
5.1. All-electroactive conducting polymers and inorganic-electroactive conducting polymer free-standing nanomembranes
In spite of the fact that the mechanical properties of ECPs are in general poor, different strategies have been developed to produce free-standing membranes of micrometric thickness made of PPy,138–142 PAni143–145 and PTh146–148 for their application in different fields, such as separation membranes, electrode materials, sensors and catalysts.In 2010, Wang et al.149 reported a novel one-pot procedure to prepare PPy FsNMs which was based on an interfacial polymerization (IP) that occurred at the interface between air and a Cu2+-containing ionic liquid (Fig. 13a). Resulting films were compact and uniform, and exhibited finely controlled thickness, with values from tens to hundreds of nanometers (i.e. as low as 60 nm, Fig. 13b). However, FsNMs with a thickness under 50 nm were brittle and weak, and broke easily. PPy FsNMs doped with p-toluenesulfonic acid (TSA) displayed good electrical properties. Specifically, the electrical conductivity of films with a thickness of ∼230 nm was 1.14 S cm−1, although this value was lower than that reported previously for PPy in other systems. Moreover, asymmetrical films with different smoothness and water wettability on each side of the film were also prepared by altering the concentration of Cu2+ ions in the liquid phase (Fig. 13c). Even though no biomedical application was proposed by the authors, it is highly promising for this field the development of a general methodology for the preparation of ECP films with different hydrophilicity and roughness on each side.
 | ||
| Fig. 13 (a) Schematic illustration of the formation of a film at the air/ionic liquid interface. (b) High magnification of the cross-sectional SEM images of the PPy FsNM. Top-view SEM image of (c) the rough side and (d) the smooth side of the film. (inset) Water contact angle, showing values of 115° and 80° for the rough and smooth sides, respectively. Reproduced with permission.149 Copyright 2010, American Chemical Society. | ||
Jeon et al.150 fabricated PPy FsNMs by organic crystal surface-induced polymerization of the monomer in an aqueous suspension containing hydrated crystals of sodium decylsulfonate below the Krafft temperature (i.e. temperature above which thermodynamic stable micelles are formed), and using FeCl3 as the oxidant (Fig. 14a). The main role of the crystals was to act as a template onto which the polymerization occurred. FsNMs obtained using this procedure were composed of a unique ECP domain with a thickness of ∼21 nm, widths of 2–6 μm, and lengths greater than 10 μm (Fig. 14b and c). The electrical conductivity of these nanosheets was determined to be 30.6 S cm−1, thus one order of magnitude higher than the value displayed by spherical PPy nanoparticles (diameters 30–50 nm) prepared by emulsion polymerization (2.9 S cm−1). Besides, the feasibility of PPy FsNMs to perform as HCl and NH3 vapour detectors was evaluated: the system exhibited high sensitivity and a fast response with respect to PPy nanoparticles, which was attributed to the increased surface area and porosity of the former nanostructure. Despite the fact that the application studied for PPy FsNMs was not biologically related, the methodology used is of relevant importance to successfully obtain FsNMs.
 | ||
| Fig. 14 (a) Schematic illustration of the synthesis procedure for PPy FsNMs using hydrated crystals of sodium decylsulfonate. (b) TEM image of PPy FsNMs. (c) Magnified image of the circle displayed in (b). Reproduced with permission.150 Copyright 2011, Elsevier. | ||
More recently, Qi et al.151 developed PPy FsNMs by in situ freezing interfacial polymerization (FIP). This approach differs from the conventional interfacial polymerization in that the chemical reaction takes place at a solid/liquid interface. In this specific work, water and cyclohexane, which are immiscible, were used as the liquid and solid phase, respectively, solid cyclohexane being achieved upon crystallization. Special attention was paid to the following issues: (1) the reaction temperature, which should be lower than the freezing point of the organic solvent and water, but above that of the monomer; and (2) the minimization of the polymerization of the monomer and the formation of PPy particles before the formation of solvent crystals. FsNMs were very smooth and their thickness was about 100 nm. In this case, the electrical conductivity was determined to be 430 S cm−1, whereas for the optimal synthetic conditions and doping parameters, the electrical conductivity reached peaks as high as 2010 S cm−1, opening the door to potential biomedical applications such as biosensors.
Jha et al.152 reported a novel strategy for the one-pot fabrication of PPy FsNMs by adding dropwise a dichloromethane solution that contained the monomer and a porphyrin derivative to an aqueous FeCl3 solution kept in a beaker. Initially, a porphyrin–PPy bilayer formed spontaneously at the air/FeCl3 interface, which after being washed rendered the PPy nanosheet. Following this method the nanosheet thickness can be tailored by changing the monomer concentration in the dropping solution. Hence, the thickness of the PPy FsNM prepared by Jha et al.152 increased from 50 to 250 nm when the pyrrole concentration augmented from 0.01 to 1 M. In addition, the conductivity of these PPy FsNMs, which was initially only ∼10−5 S cm−1, was enhanced after optimizing the doping parameters (i.e. the conductivity improved by ∼30 and ∼150 times on exposure to hydrochloric acid and iodine, respectively). Most importantly, the conductivity of these nanosheets showed no change when kept in air for more than 6 months, revealing a very noticeable stability in air. Although no specific application was examined, potential applications were mentioned (e.g. biosensors and artificial muscles).
On the other hand, the number of studies devoted to PAni-based nanosheets, which mainly consist of PAni–inorganic hybrid composites, is very scarce and their application in the biomedical field is practically inexistent. For example, very recently, the fabrication of layered PAni–graphene–PAni nanosheets (the thickness of PAni and graphene layers was 3.7 and 8.9 nm, respectively), which exhibited excellent gravimetric capacitance, was reported.153 This sandwiched structure was essentially oriented towards applications in energy storage devices, solar cells, semiconducting devices, etc. Niu et al.154 used a “skeleton/skin” strategy for the preparation of free-standing, thin and flexible single-walled carbon nanotube (SWCNT)–PAni hybrid films by a simple in situ electrochemical polymerization method. In this approach, directly grown SWCNT films with a continuous reticulate structure acted as the template, whereas PAni layers acted as the skin. The resulting hybrid films displayed a much higher conductivity compared to that of SWCNT–PAni composite films based on the post-deposition of the SWCNT film. Flexible, thin and lightweight supercapacitors were fabricated using SWCNT–PAni hybrid films. Although the applications of PAni-based nanomembranes153,154 were not related with biomedicine, the above described properties may be useful for the fabrication of energy storage components for biomedical equipment.
Regarding PTh and its derivatives, Greco et al.155 reported the preparation of FsNMs made of PEDOT and PSS complexes (PEDOT–PSS), where PSS acted as the doping agent. In this study, the supporting layer method, which enables the release and recovering of the free-standing nanosheet, was used (Fig. 15). Firstly, a layer of water-soluble PVA is deposited as the sacrificial layer on a substrate (PDMS) by spin-coating. Then, the desired nanosheet is supported on that sacrificial layer. Later, once the bilayered film is dried, it is peeled off from the substrate. The thickness of those FsNMs, which was controlled through the rotation speed, ranged from ∼100 nm (rotation speed of 1000 rpm) to ∼40 nm (rotation speed ≥ 4000 rpm). Moreover, the mechanical and electrical properties of PEDOT–PSS FsNMs were extensively investigated as a function of their thickness. With decreasing thickness, the elastic modulus values, which were determined by the strain-induced buckling test, varied between 0.81 and 1.02 GPa, while the electrical conductivity decreased from 1.44 to 0.88 S cm−1. Interestingly, solvent cast PEDOT–PSS micrometric films (a thickness of 7.5 μm) showed a conductivity of 1.38 S cm−1. Therefore, a percolation threshold was reached based on the microscopic grain-like structure or the effect of residual water present in the FsNM. This is schematically illustrated in Fig. 16, which shows that the interconnectivity between neighbour conductive particles is not improved in FsNMs with increasing thickness. However, the stacking of multiple conductive grains in micrometric films results in an improved number of interconnections and long-range connectivity. Because of the proven biocompatibility of PEDOT,41,156,157 PEDOT–PSS FsNMs were considered as first proof of concept towards the development of smart conductive substrates for cell growth and stimulation.155 Although no cell culture study was reported in that work, PEDOT–PSS FsNMs have been recently used to fabricate electrochemical microactuators in the form of microfingers with a variety of lengths.158 The reversible actuation of these microactuators, which consists in the bending of such microfingers, has been demonstrated by imposing electrochemical oxidation and reduction cycles on PEDOT–PSS supports. A number of possible applications can be envisaged for these small, soft actuators, such as microrobotics for cell manipulation.
 | ||
| Fig. 15 Schematic representation of the main steps of fabrication and release for obtaining PEDOT–PSS nanofilms by a supporting layer technique. (a) Si substrate; (b) spin-coating deposition of the PDMS substrate layer; (c) spin-coating deposition of the PEDOT–PSS nanofilm; (d) casting of a thick PVA supporting layer; (e) cutting and (f) peeling of the bilayer (PVA supporting layer + PEDOT–PSS nanofilm); (g) freestanding PEDOT–PSS nanofilm floating in water after dissolving PVA. Reproduced with permission.155 Copyright 2011, The Royal Society of Chemistry. | ||
 | ||
| Fig. 16 Schematic representation of the nanofilm structure made up of PEDOT-rich particles (orange) surrounded by the PSS matrix. The suggested percolative mechanism in nanofilms as thickness increases is depicted; the length of conductive pathways between neighbor PEDOT particles (dashed red line) increases with thickness up to a percolation threshold, when multiple parallel pathways become available. | ||
The same group embedded iron oxide superparamagnetic nanoparticles into PEDOT–PSS FsNMs using the supporting layer method described above (Fig. 15).159 More specifically, a stable colloidal dispersion of iron oxide nanoparticles added to the PEDOT:PSS mixture was used to prepare nanofilms through spin-coating. The thickness and surface roughness of the nanosheets depended on the amount of incorporated nanoparticles, ranging from 218 ± 13 to 269 ± 19 nm and from 1.5 to 8.5 nm, respectively. In this investigation, the attention was focused on the characterization of the morphological, electrical, magnetic and magneto-optical properties, which also depended on the amount of entrapped iron oxide nanoparticles. The electrical conductivity was found to decrease from 1.96 ± 0.14 S cm−1 to 0.38 ± 0.06 S cm−1 with increasing concentration of nanoparticles. These FsNMs open up new perspectives in technological fields (electronics, telecommunications and optics) than can be successfully used for the construction of biomedical devices, even though no practical evidence for any application was provided.
PEDOT–PSS was also employed by Greco et al.160 to prepare and characterize robust wrinkled conductive surfaces. This was achieved by following the simple two-step approach (metal deposition and subsequent heating) developed for the fabrication of nanowrinkles on shape-memory polymer sheets.161 Specifically, the ECP nanosheet (thickness ranging from 53.6 ± 1.2 to 120.9 ± 1.5 nm) was deposited onto a thermo-retractable PS sheet by spin-coating an aqueous dispersion of PEDOT:PSS. A subsequent thermal treatment induced the substrate shrinking causing the microbuckling of the upper PEDOT:PSS layer due to compressive stress patterning. Adhesion and proliferation assays of C2C12 murine skeletal cells on uniaxial wrinkled samples indicated that cells preferentially aligned on low and narrow ridges (∼1.5 μm in height) rather than on high and wide ones (∼2.5 μm in height). This observation was corroborated when aligned myotubes in the C2C12 differentiation stage were only formed on the former topology. Furthermore, the co-culturing of C2C12 cells with a fibroblast feeder layer improved the formation of aligned and mature myotubes. The achievement of tuneable conductive nanowrinkled interfaces represents a unique tool for the development of innovative biomedical devices.
PEDOT has also been used to prepare mechanically robust, electrically conductive and transparent hybrid FsNMs. This was achieved by Lee et al.,162 who coated densified carbon nanotube sheets with PEDOT by vapour phase polymerization. For FsNMs with a thickness of ∼66 nm, the main properties were high mechanical strength and modulus (135 MPa and 12.6 GPa, respectively), low resistance (below 200 Ω per square), moderate optical transparency (56% at 550 nm wavelength), high flexibility and minor changes in resistance upon bending. Another interesting characteristic of these FsNMs was their remarkable shape-recovery ability in a liquid and at the liquid/air interface, as opposed to previous carbon nanotube sheets. On the basis of these properties, these hybrid PEDOT-containing FsNMs were proposed to be of potential interest in the design of sensors, actuators, optical devices, and fuel cells, as well as for electrochemical capacitors.
Ultra-thin films (a thickness of 47 nm) of poly(3-thiophene methyl acetate) (P3TMA) have been prepared by spin-coating. For this system, the remarkable influence of the film–air interface on the thermal properties was examined by comparing the response of nanosheets and bulk P3TMA powder.163 Although P3TMA, which is soluble in tetrahydrofuran, chloroform and dimethylformamide, among a few others, does not form free-standing films,164 the understanding of its properties is of great interest because, as it will be discussed in the next sub-section, P3TMA has been combined with conventional biodegradable polymers to fabricate different types of electroactive FsNMs with biomedical applications. Interestingly, the glass transition temperature determined using microcantilevers coated with ultra-thin P3TMA films resulted in 5.2 °C higher than that obtained for bulk powder samples. Moreover, ultra-thin films showed nanospherical aggregates of small (∼40 nm) size, while powder bulk samples presented a micrometric granular morphology (Fig. 17).
 | ||
| Fig. 17 (a) Low (top) and high (bottom) magnification SEM micrographs of P3TMA powder. (b) SEM micrograph of P3TMA nanosheets. | ||
5.2. Insulating polymer-electroactive conducting polymer free-standing nanomembranes
As mentioned above, blending of ECPs with insulating polymers is the most commonly followed approach to overcome the poor mechanical integrity of organic semiconductors. Accordingly, free-standing membranes have been fabricated by solvent casting mixtures of ECPs with conventional insulating polymers, such as PVA144 and nylon 66.165 However, in all cases, such membranes were of micrometric thickness and their potential use (e.g. optical pH sensors144 and conductive coatings165) was not related to the biomedical field, even though they were prepared by combining ECPs with biopolymers derived from natural sources. For example, cellulose–PAni membranes of micrometric thickness, which were prepared by in situ polymerization of aniline in the presence of bacterial cellulose nanofibrils, were used as electromagnetic interference shielding materials despite the biological origin of the biopolymer.166Table 3 summarizes the most relevant examples of ECP-containing FsNMs, discussed in this section.| Material | Preparation | Thickness (nm) | Propertiesa | Biomedical applications | Ref. |
|---|---|---|---|---|---|
| a σ: electrical conductivity. R: electrical resistance. E: elastic modulus. σmax: ultimate tensile strength. Fadh: adhesion force between the AFM tip and the sample surface. εg: optical band gap energy. | |||||
| PPy | Interface polymerization | From 60 to hundreds | σ = 1.14 S cm−1 for a thickness of 230 nm. Flexible, transparent, shiny and light-blue color. | Those in which films with different roughness and water wettability on each side of film fit | 149 |
| PPy | Organic crystal surface-induced polymerization | ∼21 | σ = 30.6 S cm−1. Smooth surface | Chemical sensors, successful results being obtained with HCl and NH3 vapors | 150 |
| PPy | In situ FIP | ∼100 | Extremely high electrical conductivity with σ = 2000 S cm−1. Semi-transparency and smooth surface | Fabrication of sensors | 151 |
| PPy | Interface polymerization using porphyrin as an in situ template | From 50 to 250 | Low conductivity (σ ≈ 10−5 S cm−1) that can be adjusted by doping. Mechanically strong and dense morphology | Biosensors and artificial muscles | 152 |
| PEDOT–PSS | Supporting layer | From ∼40 to ∼100 | Conductivity, (σ = 0.88–1.44 S cm−1) comparable to that of micrometric films (σ = 1.38 S cm−1). E = 0.81–1.02 GPa. Flexibility. | Bioactive platforms. Electrochemical soft microactuators (microfingers) for cell manipulation with microrobots | 155 and 158 |
| PEDOT–PSS | Spin-coating + heat-shrinking process | From 53.6 ± 1.2 to 120.9 ± 1.5 | R = 199 ± 11–586 ± 16 Ω per square presence of anisotropic topographical cues at the micro- and nanoscale on the surface | Smart scaffolds for functional cell alignment and electrical stimulation. | 160 |
| PEDOT–carbon nanotube sheets | Vapor phase polymerization | ∼66 | R = 200 Ω per square. E = 12.6 GPa, σmax = 135 MPa. Volumetric capacitance: ∼40 F cm−3 at 100 V s−1. Flexibility and transparency. | Sensors and actuators | 162 |
| PE44–P3TMA | Spin-coating | From 20 to 80 | σ = 10−4 to 10−5 S cm−1 depending on the composition of the blend. Robustness, flexibility and transparency. Yellowish color. Biodegradable and electro-compatible with cells. | Bioactive platforms with semiconducting response for tissue regeneration. | 167 and 168 |
| TPU–P3TMA | Spin-coating | From 11 to 93 | σ = from 2.23 × 10−5 to 5.19 × 10−6 S cm−1, current = from 0.43 to 1.85 pA. E = 0.9–1.7 GPa, Fadh ≈ 6 nN. εg = 2.35 eV. Robustness, flexibility and transparency. Yellowish color. Biodegradable and electro-compatible with cells. | Bioactive substrates for advanced biomedical applications | 169–171 |
| (PEDOT–PSS)–PLA | Spin-coating | ∼45 (PEDOT–PSS layer) + ∼200 (PLA layer) | σ = 180 S cm−1. Mechanical robustness and conformability. Colors from dark blue to light blue or even transparent depending on the oxidation state. Electrochemical regulation of the surface wettability. | Smart conductive biointerfaces for directing cell adhesion and differentiation. Bioelectrodes. | 172 |
| (TPU–P3TMA)–collagen | Spin-coating + adsorption | From 11 to 93 (TPU–P3TMA layer) + 73 (collagen layer) | Formation of a pseudoregular honeycomb 2D network, in which the top layer resembled a fibril structure | Three-dimensional scaffolds for tissue engineering | 171 |
Armelin et al.167 reported the preparation and characterization of FsNMs that were obtained by spin-coating mixtures of a biodegradable polyester, poly(tetramethylene succinate) (PE44), and P3TMA. The thickness of these ultra-thin membranes, which ranged from 20 to 80 nm, was precisely controlled by adjusting the spin-coating process parameters and the solution concentration. It was proved that PE44–P3TMA FsNMs retained both the biodegradability of PE44 and the semiconducting (∼10−4 S cm−1) and electrochemical properties of P3TMA. Calorimetric assays revealed that P3TMA and PE44 were only partially miscible, evidencing a phase separation. Thus, two glass transitions were identified in the mixture, even though they depended on the composition of PE44–P3TMA FsNMs. Furthermore, the presence of P3TMA hindered the crystallization of PE44 and also affected the fusion of the PE44 crystalline fraction. Nanosheets were found to be stable in air and ethanol for more than one year, which facilitates their manipulation. Preliminary cell culture results using epithelial cells (HEp-2) suggested that PE44–P3TMA FsNMs are potential candidates for the fabrication of bioactive platforms with semiconducting response.
The good results obtained for PE44–P3TMA FsNMs prepared using a 50:50 PE44:P3TMA molar ratio motivated further study on their potential application in the biomedical field. More specifically, Pérez-Madrigal et al.168 conducted an investigation devoted to quantifying the following aspects related to these nanosheets: (1) their hydrolytic and enzymatic degradability; (2) their response towards different fibroblast and epithelial cellular lines; and (3) their electrochemical response when coated with cell monolayers. Hydrolytic and enzymatic degradation assays revealed the appearance of abundant crevasses and thin grooves after 4 weeks of immersion in phosphate buffered saline solution (PBS), and these effects become more pronounced after 8 weeks (Fig. 18a). Moreover, the weight loss of samples immersed in hydrolytic and enzymatic media indicates that the degradation rate is higher for the PE44–P3TMA blend than that for the individual polyester (Fig. 18b). This was attributed to the fact that in the blend the degradation of the polyester domains produced the detachment of P3TMA domains (labelled with white circles in Fig. 18a). On the other hand, in vitro cellular adhesion and proliferation assays using HEp-2, MDCK, Cos-7 and Du-145 cell lines evidenced that PE44–P3TMA nanosheets behaved as potent cellular matrices (Fig. 18c). Thus, the viability of cultured cells was higher, in terms of adhesion and proliferation, in blended FsNMs than in individual PE44 and P3TMA ultra-thin films. Moreover, cyclic voltammetry studies reflected that PE44–P3TMA FsNMs were electro-compatible with cellular monolayers, even though there was a slight reduction in the cathodic and anodic intensities. These features combined with the outstanding flexibility and robustness of the nanomembranes, which was demonstrated through aspiration in pipette/release/shape recovery cycles that were repeated without breaking the film (Fig. 19), allowed the authors to propose the use of biodegradable PE44–P3TMA FsNMs as bioactive platforms for tissue engineering.
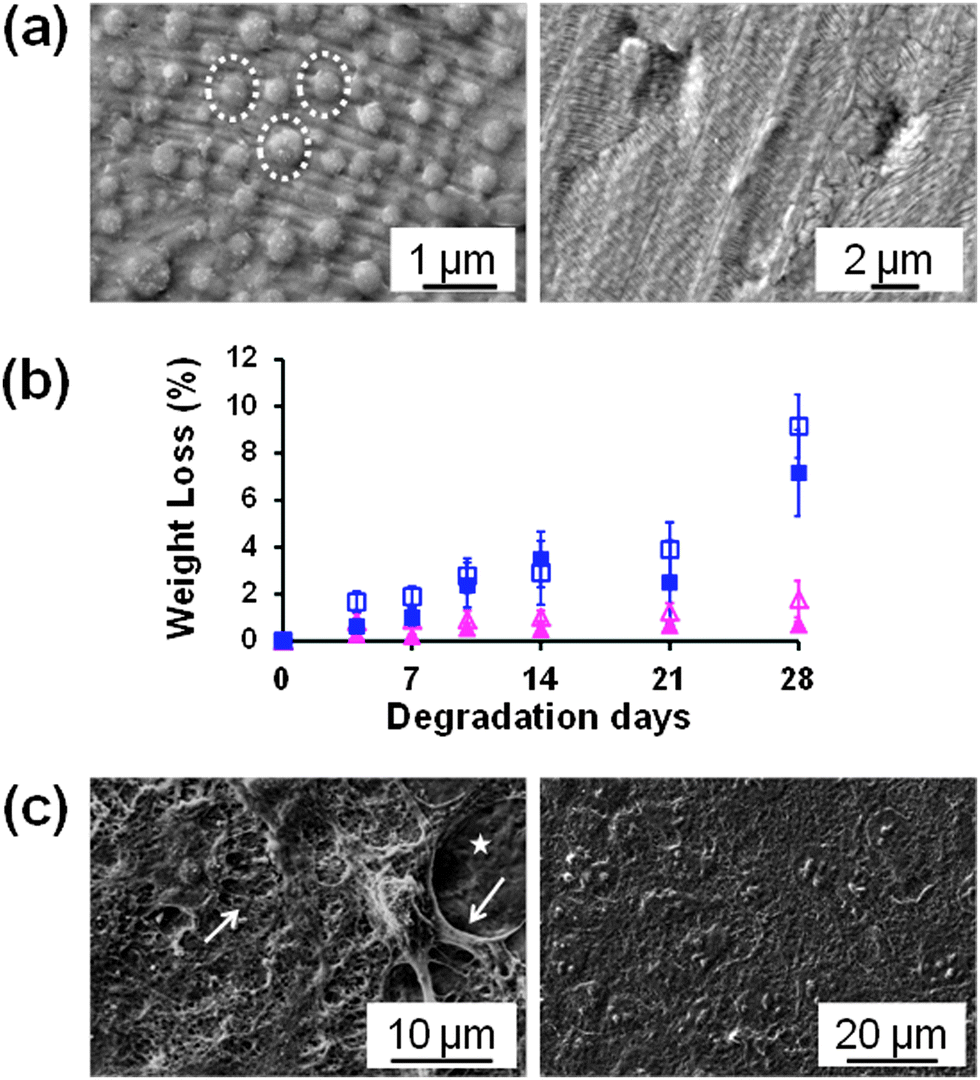 | ||
| Fig. 18 (a) SEM micrographs of hydrolytically degraded PE44–P3TMA FsNMs (blend prepared using a 50:50 molar ratio) after 4 weeks (left) and 8 weeks (right) of immersion in PBS. Circles show the conductive phase (spherical aggregates of P3TMA). (b) Plot of the weight loss (%) versus the degradation time (days) in hydrolytic (filled symbols) and enzymatic media (empty symbols) for PE44 (triangles) and PE44–P3TMA (squares) films. (c) SEM micrographs of Cos-7 (left) and Du-145 (right) cells adhered on the surface of PE44–P3TMA nanomembranes. The film surface (domains without cells) is shown by asterisks (*), while the connections or interactions between the cell and the surface or between two cells are indicated by arrows. | ||
 | ||
| Fig. 19 (a) Digital camera image of a PE44–P3TMA FsNM immersed in ethanol; (b–c) aspiration of the nanomembrane floating in ethanol into a pipette; (d–e) release of the folded nanomembrane into the ethanol solution; and (f) aspect of the nanomembrane after recovering the shape. | ||
Inspired by those promising results, Pérez-Madrigal et al.169 replaced the polyester component in the preparation of PE44–P3TMA FsNMs by an aromatic grade thermoplastic polyurethane matrix (TPU). This was expected to improve the miscibility between the two components of the blend because of the presence of aromatic moieties in both polymers. Hence, TPU–P3TMA FsNMs were prepared by spin-coating a TPU:P3TMA mixture with a 40:60 weight ratio. The surface of the resulting nanosheets was described to be the combination of the topographies of both individual components. This consisted of a homogeneous distribution of granules throughout the surface, which were associated with the P3TMA rich phase by conductive AFM (C-AFM) measurements (Fig. 20). Moreover, TPU–P3TMA nanosheets showed homogeneously distributed well-localized folds, similar to those also observed in individual TPU FsNMs. As some films did not present folds, their formation was attributed to artefacts produced during the spin-coating process. The thickness of FsNM TPU–P3TMA ranged from 11 to 93 nm, while the average roughness was 16.3 ± 0.8 nm. Analysis of the mechanical properties indicated that the Young's modulus, which was determined by applying the Derjanguin–Müller–Toporov (DMT) contact mechanics, depended on the thickness of the nanomembranes. Thus, values determined for the thicker (80–140 nm)/thinner (10–40 nm) regions of TPU, P3TMA and blended ultra-thin films were 25/35 MPa, 3.5/12 GPa and 0.9/1.7 GPa, respectively. In contrast, the adhesion force was found to be homogeneous throughout the whole surface of TPU and P3TMA films (average values: 7.2 and 5.0 nN, respectively), while it depended on the phase distribution in the case of TPU–P3TMA FsNMs. The potential utility of these FsNMs for tissue engineering applications was proved by cellular proliferation assays using Cos-7 cells. The results showed that the TPU–P3TMA FsNM was more active as a cellular matrix than each of the two individual polymers.
 | ||
| Fig. 20 C-AFM characterization for TPU–P3TMA (40:60) nanomembranes deposited onto ITO: (a) simultaneous 1 μm × 1 μm topographical image (left) and C-AFM current image (right) for the same sample. (b) Dual cross-section profile of the above images indicating variations in height (solid line) and current (dashed line). (c) Typical current–voltage curves (100 nA/div amplifier, max top current 1 μA). | ||
In a subsequent study, the same authors170 examined the electronic, electric and electrochemical response of the above mentioned TPU–P3TMA FsNM. Interestingly, the optical band gap energy of these blended nanofilms was very similar to that obtained for individual P3TMA (i.e. Eg = 2.35 and 2.32 eV, respectively). This similarity, which did not occur when comparing the Eg value of the TPU:P3TMA mixture with that of P3TMA dissolved in THF, was attributed to the influence of the spin-coating process on the π-conjugation length and packing interactions of P3TMA chains (i.e. ECP chains are not able to adopt their equilibrium conformation because the solvent is completely evaporated). On the other hand, the electrical conductivity of TPU–P3TMA FsNMs, determined by C-AFM measurements, was found to range from 2.23 × 10−5 to 5.19 × 10−6 S cm−1. These inhomogeneous values were consistent with the presence of insulating TPU chains in P3TMA-rich domains. The voltammetric response of TPU–P3TMA FsNMs and P3TMA was similar in terms of the ability to exchange charge reversibly and their electrochemical stability. On the basis of these results, the authors proposed that TPU–P3TMA FsNMs were excellent candidates to be used in tissue regeneration applications as biointerfaces to conduct electrical or electrochemical stimulation.170
Following this research line, in a very recent study,171 special emphasis was given to determine the influence of the TPU–P3TMA FsNM composition on those properties typically related to biomedical applications, such as swelling, resistance to hydrolytic and enzymatic degradation, and biocompatibility. Therefore, TPU–P3TMA FsNM samples with different TPU:P3TMA compositions (20:80, 40:60 and 60:40 weight ratios) were studied. Structural investigations based on morphological and topographical analyses evidenced that the 20:80 sample exhibited a surface similar to that obtained for individual P3TMA, while 40:60 and 60:40 FsNMs presented an irregular distribution of prominent and well-localized folds like individual TPU. The water uptake of nanosheets decreased with the concentration of TPU, even though it was relatively high in all cases. Consistently, hydrolytic and enzymatic degradation increased with the P3TMA content (Fig. 21a and b). Moreover, TPU–P3TMA blends behaved as biodegradable materials. Viability assays evidenced that, although all TPU–P3TMA compositions provided biocompatible blends, the viability of cells increased with the concentration of TPU in the composition (Fig. 21c and d). The overall results allowed the authors to conclude that the 40:60 TPU–P3TMA FsNM was the most appropriate system for tissue engineering applications.
 | ||
| Fig. 21 Weight loss of pure TPU and TPU–P3TMA (20:80, 40:60, and 60:40 ratios) films immersed in (a) PBS solution and (b) lipase-containing PBS solution. Dashed lines were added manually to help following data progression. Relative cellular adhesion (c) and viability (d) on TPU, P3TMA, and TPU–P3TMA (20:80, 40:60, and 60:40 weight ratios) nanomembranes. Assays were carried out using the following cell lines: Vero (shaded bars) and Cos-7 (empty bars). The relative viability was established in relation to the TCP control (tissue culture polystyrene). Steel was also considered as a control substrate because the individual polymers and the blends were deposited on this material. Greek letters on the columns refer to significant differences (p < 0.05) using the ANOVA and Tukey test: α vs. TCP; β vs. steel. | ||
A completely different strategy to obtain FsNMs made of an ECP and an insulating polymer is the one in which both components are arranged in a bilayered configuration. Greco et al.172 fabricated self-supported nanosheets with patterned conductivity using PEDOT–PSS and PLA, which acted as the mechanical support layer, thus maintaining continuity and robustness. In a first step, the PEDOT–PSS layer (a thickness of ∼45 nm) was spin-coated onto a substrate. Then, in a second step, and after thermal treatment, the PLA layer (a thickness of ∼200 nm) was spin-coated onto the previous one. Nevertheless, n order to obtain a patterned bilayer FsNM, an intermediate step (inject patterning) was introduced just before the deposition of the PLA layer (i.e. localized over-oxidation of PEDOT–PSS nanofilm to provoke an irreversible loss of electrical conductivity at specific spots). Moreover, to enhance its electrical conductivity, which reached a value of 180 S cm−1, DMSO was added as a secondary doping agent. The resulting bilayered FsNM is of great interest as a (bio)electrical interface and as a thin floating or ultraconformable circuit. In addition to that, the surface wettability of the bilayered FsNM was electrochemically switched through simple oxidation and reduction processes. This change was even more evident for nanosheets supported on a PS substrate. On the basis of this interesting ability, the authors proposed the application of these FsNMs as smart conductive biointerfaces for directing cell adhesion and differentiation.172
A similar approach was followed by Pérez-Madrigal et al.171 to prepare bilayered FsNMs made of TPU–P3TMA and collagen. However, in this case, the role of the collagen layer was not to provide robustness, which was an intrinsic property of TPU–P3TMA FsNMs, but to enhance the cellular response towards the TPU–P3TMA biointerface. Therefore, the TPU–P3TMA/collagen bilayer was formed by incubating a spin-coated TPU–P3TMA (40:60 weight ratio) layer in a collagen solution. Amazingly, the adsorbed collaged layer was found to form two layers (Fig. 22). The top layer exhibited a pseudoregular honeycomb 2D network with cavities of different diameters (i.e. ranging from 194 ± 55 nm to 1.2 ± 0.7 μm) and depths ca. 73 nm, whereas, in contrast, collagen adopted a much more compact structure in the bottom layer. According to previous studies,173,174 TPU–P3TMA/collagen biointerfaces were proposed as suitable scaffolds for biological and biomedical purposes. Similarly, a collagen layer was recently used to inhibit the cytotoxic effects of the remaining monomer leaking from a supported PTh3 film, the resulting PTh3/collagen biointerface behaving as bioactive platforms.132
 | ||
| Fig. 22 SEM-AFM images of TPU–P3TMA/collagen nanomembranes: (a) height image 5 × 5 μm2; (b) cross-sectional data from (a); (c) height image 1 × 1 μm2; (d) phase image 1 × 1 μm2 of (c); (e and f) SEM images (14k× and 150k×, respectively). Reproduced with permission.171 Copyright 2014, American Chemical Society. | ||
5.3. A challenge: electroactive conducting polymer free-standing nanomembranes for energy-based biomedical applications
In the 21st century, fuel cells have emerged as smart storage systems or alternative energy conversion devices due to both their efficiency and lack of pollutant emission in comparison with coal combustion engines.175 Currently, the development of new materials and fabrication processes to improve the effectivity of fuel cells and reduce their cost is an important area of research within this field. The application of ECPs in fuel cell electrodes was proposed as a promising remedy to solve some of the problems identified in such energy storage and generation systems, for example chemical long-term stability of the catalytic support and stable mechanical-structural stability.176 Four different types of applications can be identified depending on the use of ECPs in fuel cell electrodes:– Support: ECPs as obtained or structured (e.g. nanofibers and microspheres) act as a support for the catalyst. For example, Pd–PAni nanofibers showed excellent electrocatalytic activity for the oxidation of methanol, ethanol and formic acid,177 and Pt incorporated into poly(3-methylthiophene) showed effective dispersion of the catalyst and high catalytic activity in methanol oxidation.178
– Coating: the ECP acts as the structured support of another material. For example, graphene nanosheets coated with PAni and decorated with Pt nanoparticles show high activity in the reduction of O2 and oxidation of methanol, glucose and H2O2.179
– Part or a composite: for example simultaneous deposition of Pt and Ru on PEDOT doped with polystyrene-4-sulfonate yielded an active electrocatalyst for methanol oxidation.180,181
Some of these fuel cell applications of ECPs are based on membranes separating the anodic (fuel) compartment from the cathodic (oxidant) compartment. However, the thickness of polymeric membranes in current fuel cells is on the micrometre scale176,182 and, therefore, out of the scope of this review. In spite of this, recent advancements in the FsNM field should also be considered as a strong driving force to look for alternative strategies for energy production. The biocompatibility, chemical and dimensional stability, and mechanical properties of FsNMs discussed in previous sub-sections should motivate the scientific community to develop inexpensive and high-performing membranes for the fabrication of biological fuel cells. These devices could be considered from two points of view: (1) macroscopic devices with biological microorganisms participating in the energy production; or (2) biological microorganisms with nanosheets, incorporated as electrode membranes, acting as micrometric biofuel cells. The latter may result in a new field originated by the combination of energy and biomedicine.
Another promising field is the development of artificial muscles using ECP films. In this application, which is based on the transduction of electrical or electrochemical energy into mechanical work, electrochemical reactions in ECP films provoke dimensional variations that result in bending or linear macroscopic movements. The simple way to transduce reversible length variations in films of ECPs into macroscopic movement is through a bilayer ECP/passive layer. The passive layer can be a tape,183,184 a sputtered metal,185,186 a piece of paper,187 ECP/plastic,188 or a solid state electrolyte.189 Electrochemically driven length variations in the film of ECP produce stress gradients across the two film interface and subsequently result in macroscopic bending, as schematically illustrated in Fig. 23. More specifically, for ECPs exchanging anions with the electrolyte the film gives anticlockwise movement by oxidation (swelling) and clockwise during the reduction, while ECPs exchanging cations produce clockwise movement by oxidation (shrinking) and anticlockwise by reduction of the ECP.190 Artificial muscles based on ECPs were recently reviewed by Otero and co-workers.191 Unfortunately, at present time all biomimetic artificial muscles based on ECPs have been prepared using free-standing membranes of micrometric thickness, as occurred for fuel cell electrodes. However, the increasing evolution in biomimetic reactive devices has resulted in the opening of new technological forefronts and challenges, including the application of FsNMs.191
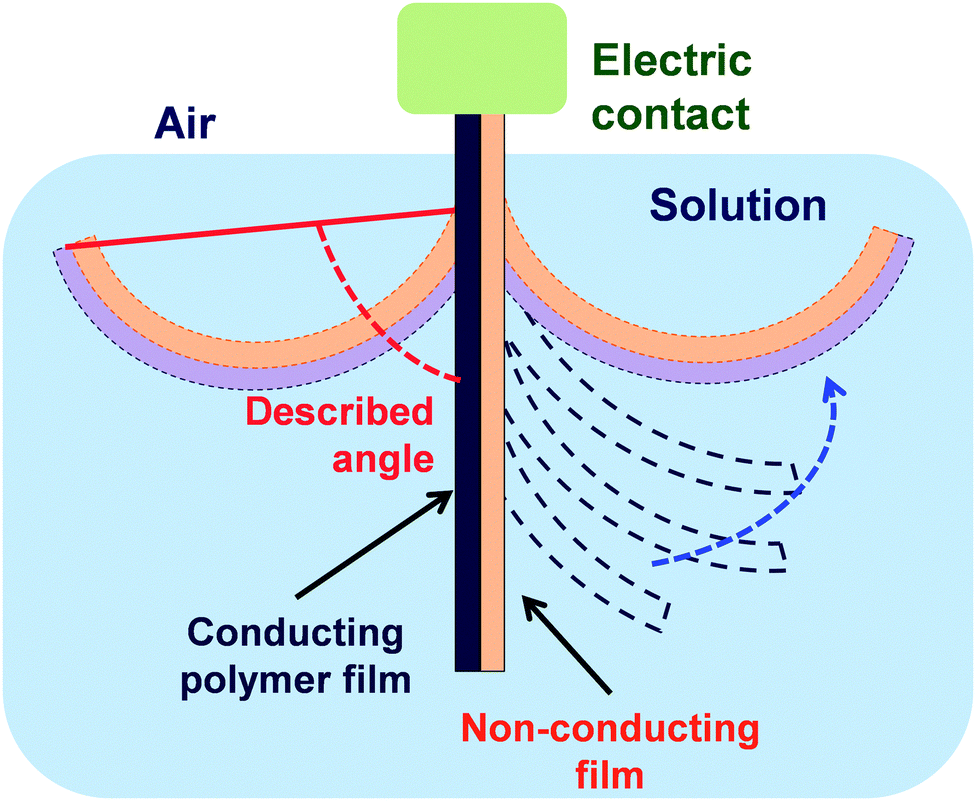 | ||
| Fig. 23 Scheme of a bilayered artificial muscle device made of ECP adhered on a non-conductive tape. Clockwise and anticlockwise movements are schematically represented by bended dashed lines at left and right, respectively. | ||
6. Conclusions and perspectives
This review of FsNMs for biomedical applications demonstrates the versatility of these simple nanostructures, in which molecules organize in a 2D configuration. Moreover, it highlights several ideas: (i) the variety of techniques available for their preparation, (ii) the concept that the rational design of nanostructured materials can be utilized to obtain tailored properties, and (iii) the relationship between the features of the nanosheets and their applications. Although the list of biomedical applications for self-supported nanosheets is very extensive (e.g. wound dressing, patches for bone or tendon repair, scaffolds for regenerative medicine and tissue engineering, magnetically controllable bioactive platforms, drug delivery systems, platelet substitutes, devices for overlapping therapy, artificial soft tissues, biosensors, supports for biological samples, biointerfaces, coatings with microbicide properties, artificial muscles, microactuators for cell manipulation, and bioelectrodes), investigation on such nanostructured organizations is still in its early stages, and many treasures still await scientific discovery.Research on FsNMs for biomedical applications, which began about one decade ago, has been essentially focused in using insulating polymers. From a practical point of view, studies have been conducted considering only two families of biodegradable materials: (1) polyesters, mainly PLA (i.e. PLGA and PE44 nanosheets were also sporadically chosen), and (2) polysaccharides. The inflammatory response provoked by FsNMs made of these materials tends to be very low (i.e. lower than that of sutures), which is of particular importance for therapeutic applications. Even though a few studies with other materials have been reported (e.g. PEG, PAA, PDDA, P4VPPS and PMMA), these involve polymeric families with very different and disperse properties and, therefore, a general rationalization of the derived conclusions is not an easy task. Within this context, comparative studies to extract general conclusions are imperative. Biomedical applications of FsNMs made of ECPs have emerged with great interest in the last few years (i.e. the first work appeared in 2010, even though most studies are from 2013 onwards). Consequently, the number of ECPs studied so far, individually or combined with insulating polymers, is still limited and much more effort is necessary in this direction.
In spite of the studies based on polysaccharides, the preparation of FsNMs by combining synthetic polymers and biomolecules, such as proteins and DNA, is an unexplored field. Depending on the desired application, nanosheets with a heterogeneous or homogeneous distribution of the two components (i.e. polymer/biomolecule layers or polymer–biomolecule composites, respectively) may be designed. Not only the role played by the polymer in these FsNMs could provide mechanical robustness but also could protect biomolecules against too fast biodegradation. Besides, if it was an ECP, it could also induce electrochemical, electrical or optical activity. However, the biological component would be the main protagonist and, therefore, responsible for the functionality and biomedical applications of such nanosheets. For this purpose, both the number of possibilities and the palette of biocompounds offered by nature are immense. In addition, novel and effective advanced biomedical applications that reach the society cannot be obtained using synthetic polymers alone, thus significant studies related to the combination of synthetic and natural compounds are desirable, and should be a main research focus.
Regarding ECP-containing nanostructured materials, biomedical applications centred on electrophysiology is a field that needs to be further explored. One of the central goals of electrophysiology is to offer tools that can monitor and manipulate bioelectrical activities in the human brain. In particular, implantable neural prostheses aim to replace or restore lost motor functions after disease or disability. Since the brain has soft curvilinear surfaces, properties of conductive FsNMs discussed above are appropriate for the development of flexible bioelectronic interfaces. Hence, by a rational design, flexible ECP-containing FsNMs may be used to establish conformal contact in the cerebral cortex. Another electrophysiological application of these flexible nanosheets is the integration of nanoelectronics in 3D polymer scaffolds for cell cultures. The merging of flexible nanoelectronics and cells would allow the monitoring of biological signals for clinical use.
FsNMs made with unique optical, electrical and magnetic properties can be fabricated using organic–inorganic composites, in which nanoparticles, nanowires, carbon nanotubes, clay platelets or other nanocolloids are combined with insulating or conducting polymers. Although a few studies have been reported in this field, developments are still at a very early stage. For example, organic–inorganic nanosheets could be interfaced with proteins and cells to produce complexes of nanostructured and biological entities with specific and finely tuned functions for a broad range of applications in diagnosis and treatment.
Finally, concerning the architecture of FsNMs, it is necessary to explore the advantages provided by new techniques. For example, a procedure to obtain perforated nanosheets at the nanometric scale has been recently reported, as discussed above. However, the enormous potentiality of these FsNMs remains completely unknown from a technological point of view. Moreover, much effort needs to be devoted to integrating new techniques for modifying or patterning the surface of nanosheets not only in already developed biomedical applications, but also in the development of new ones. For example, surface modification to functionalize and/or re-orient polymer molecules in ECP-containing nanosheets is expected to offer numerous opportunities as active biomedical interfaces.
In summary, numerous scientific and technical challenges for the design of FsNM abound, and it can be foreseen that their development for biomedical applications will continue to be a research area of growing interest.
Acknowledgements
The authors are in debt to support from MINECO and FEDER (MAT2012-34498 and MAT2012-36205) and Generalitat de Catalunya (XRQTC). M.M.P.-M. thanks financial support through a FPI-UPC grant.References
- R. Langer, J. Biomed. Mater. Res., Part A, 2013, 101, 2449 CrossRef PubMed.
- S. Fleischer and T. Dvir, Curr. Opin. Biotechnol., 2013, 24, 664 CrossRef CAS PubMed.
- T. Cohen-Karni, R. Langer and D. S. Kohane, ACS Nano, 2012, 6, 6541 CrossRef CAS PubMed.
- C. Jiang, S. Markutsya, Y. Pikus and V. V. Tsukruk, Nat. Mater., 2004, 3, 721 CrossRef CAS PubMed.
- J. D. Kittle, C. Wang, C. Qian, Y. Zhang, M. Zhang, M. Roman, J. R. Morris, R. B. Moore and A. R. Esker, Biomacromolecules, 2012, 13, 714 CrossRef CAS PubMed.
- M. G. Bellino, I. Tropper, H. Duran, A. E. Regazzoni and G. J. A. A. Soler-Illia, Small, 2010, 6, 1221 CrossRef CAS PubMed.
- J. A. Jaber, P. B. Chase and J. B. Schlenoff, Nano Lett., 2003, 3, 1505 CrossRef CAS.
- C. S. Hajicharalambous, J. Lichter, W. T. Hix, M. Swierczewska, M. F. Rubner and P. Rajagopalan, Biomaterials, 2009, 30, 4029 CrossRef CAS PubMed.
- Z. Tai, H. Ma, B. Liu, X. Yan and Q. Xue, Colloids Surf., B, 2012, 89, 147 CrossRef CAS PubMed.
- S. Y. Wong, Q. Li, J. Veselinovic, B.-S. Kim, A. M. Klibanov and P. T. Hammond, Biomaterials, 2010, 31, 4079 CrossRef CAS PubMed.
- M. C. Berg, L. Zhai, R. E. Cohen and M. F. Rubner, Biomacromolecules, 2006, 7, 357 CrossRef CAS PubMed.
- K. C. Wood, J. Q. Boedicker, D. M. Lynn and P. T. Hammond, Langmuir, 2005, 21, 1603 CrossRef CAS PubMed.
- H. Watanabe, E. Muto, T. Ohzono, A. Nakao and T. Kunitake, J. Mater. Chem., 2009, 19, 2425 RSC.
- Handbook of Nanophysics, Functional Nanomaterials, ed. K. D. Sattler, CRC Press, Taylor and Francis Group, Boca Raton, FL, 2011, vol. 5 Search PubMed.
- G. Decher, J. D. Hong and J. Schmitt, Thin Solid Films, 1992, 210–211, 831 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232 CrossRef CAS.
- G. Decher, Y. Lvov and J. Schmitt, Thin Solid Films, 1994, 244, 772 CrossRef CAS.
- R. W. Corkery, Langmuir, 1997, 13, 3591 CrossRef CAS.
- D. B. Hall, P. Underhill and J. M. Torkelson, Polym. Eng. Sci., 1998, 38, 2039 CAS.
- I. Gurrappa and L. Binder, Sci. Technol. Adv. Mater., 2008, 9, 043001 CrossRef.
- A. D. Stroock, R. S. Kane, M. Weck, S. J. Metallo and G. M. Whitesides, Langmuir, 2002, 19, 2466 CrossRef.
- W. Cheng, M. J. Campolongo, S. J. Tan and D. Luo, Nano Today, 2009, 4, 482 CrossRef CAS PubMed.
- C. Jiang and V. V. Tsukruk, Adv. Mater., 2006, 18, 829 CrossRef CAS PubMed.
- T. A. Skotheim, R. L. Elsenbaumer and J. R. Reynolds, Handb. Conduct. Polym., Marcel Dekker, New York, 1998 Search PubMed.
- C. Li, C. Bai and G. Shi, Chem. Soc. Rev., 2009, 38, 2397 RSC.
- D. W. Hatchett and M. Josowicz, Chem. Rev., 2008, 108, 746 CrossRef CAS PubMed.
- J. L. Bredas and G. B. Street, Acc. Chem. Res., 1985, 18, 309 CrossRef CAS.
- B. A. Bolto, R. McNeill and D. E. Weiss, Aust. J. Chem., 1963, 16, 1090 CrossRef CAS.
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578 RSC.
- Y. Xu, F. Zhang and X. Feng, Small, 2011, 7, 1338 CrossRef CAS PubMed.
- B. Kippelen and J.-L. Brédas, Energy Environ. Sci., 2009, 2, 251 CAS.
- Y. Xuan, M. Sandberg, M. Berggren and X. Crispin, Org. Electron., 2012, 13, 632 CrossRef CAS PubMed.
- D. Aradilla, F. Estrany, F. Casellas, J. I. Iribarren and C. Alemán, Org. Electron., 2014, 15, 40 CrossRef CAS PubMed.
- D. Aradilla, J. Casanovas, F. Estrany, J. I. Iribarren and C. Alemán, Polym. Chem., 2012, 3, 436 RSC.
- H.-E. Yin, C.-H. Wu, K.-S. Kuo, W.-Y. Chiu, C.-F. Lee, N.-T. Li and P.-J. Chen, Synth. Met., 2011, 161, 1878 CrossRef CAS PubMed.
- N. K. Guimard, N. Gomez and C. E. Schmidt, Prog. Polym. Sci., 2007, 32, 876 CrossRef CAS PubMed.
- A.-D. Bendrea, L. Cianga and I. Cianga, J. Biomater. Appl., 2011, 26, 3 CrossRef CAS PubMed.
- R. Ravichandran, S. Sundarrajan, J. R. Venugopal, S. Mukherjee and S. Ramakrishna, J. R. Soc., Interface, 2010, 7, S559 CrossRef CAS PubMed.
- J. Jaguar-Grodzinski, e-Polym., 2012, 12, 722 Search PubMed.
- C. Rincón and J. C. Meredith, Macromol. Biosci., 2010, 10, 258 CrossRef PubMed.
- L. J. del Valle, F. Estrany, E. Armelin, R. Oliver and C. Alemán, Macromol. Biosci., 2008, 8, 1144 CrossRef CAS PubMed.
- L. J. del Valle, D. Aradilla, R. Oliver, F. Sepulcre, A. Gamez, E. Armelin, C. Alemán and F. Estrany, Eur. Polym. J., 2007, 43, 2342 CrossRef CAS PubMed.
- B. Guo, L. Glavas and A.-C. Albertsson, Prog. Polym. Sci., 2013, 38, 1263 CrossRef CAS PubMed.
- G. Shi, M. Rouabhia, Z. Wang, L. H. Dao and Z. Zhang, Biomaterials, 2004, 25, 2477 CrossRef CAS PubMed.
- L. Huang, X. Zhuang, J. Hu, L. Lang, P. Zhang, Y. Wang, X. Chen, Y. Wei and X. Jing, Biomacromolecules, 2008, 9, 850 CrossRef CAS PubMed.
- S. I. Jeong, I. D. Jun, M. J. Choi, Y. C. Nho, Y. M. Lee and H. Shin, Macromol. Biosci., 2008, 8, 627 CrossRef CAS PubMed.
- E. Llorens, M. M. Pérez-Madrigal, E. Armelin, L. J. del Valle, J. Puiggalí and C. Alemán, RSC Adv., 2014, 4, 15245 RSC.
- M. Planellas, M. M. Pérez-Madrigal, L. J. del Valle, S. Kobauri, R. Katsarava, C. Alemán and J. Puiggalí, Polym. Chem., 2015, 6, 925 RSC.
- J. H. Hardy, D. J. Mouser, N. Arroyo-Currás, S. Geissler, J. K. Chow, L. Nguy, J. M. Kim and C. E. Schmidt, J. Mater. Chem. B, 2014, 2, 6809 RSC.
- K. M. Nampoothiri, N. R. Nair and R. P. John, Bioresour. Technol., 2010, 101, 8493 CrossRef PubMed.
- K. Odelius, P. Plikk and A.-C. Albertsson, Biomacromolecules, 2005, 6, 2718 CrossRef CAS PubMed.
- F. Khan and S. R. Ahmad, Macromol. Biosci., 2013, 13, 395 CrossRef CAS PubMed.
- R. Parenteau-Bareil, R. Gauvin and F. Berthod, Materials, 2010, 3, 1863 CrossRef CAS PubMed.
- S. Martino, F. D'Angelo, I. Armentano, J. M. Kenny and A. Orlacchio, Biotechnol. Adv., 2012, 30, 338 CrossRef CAS PubMed.
- C. Zhao, A. Tan, G. Pastorin and H. K. Ho, Biotechnol. Adv., 2013, 31, 654 CrossRef CAS PubMed.
- Y. Okamura, K. Kabata, M. Kinoshita, D. Saitoh and S. Takeoka, Adv. Mater., 2009, 21, 4388 CrossRef CAS PubMed.
- K. Fujino, M. Kinoshita, A. Saitoh, H. Yano, K. Nishikawa, T. Fujie, K. Iwaya, M. Kakihara, S. Takeoka, D. Saitoh and Y. Tanaka, Surg. Endosc., 2011, 25, 3428 CrossRef PubMed.
- H. Miyazaki, M. Kinoshita, A. Saito, T. Fujie, K. Kabata, E. Hara, S. Ono, S. Takeoka and D. Saitoh, Wound Repair Regen., 2012, 20, 573 Search PubMed.
- Y. Okamura, K. Kabata, M. Kinoshita, H. Miyazaki, A. Saito, T. Fujie, S. Ohtsubo, D. Saitoh and S. Takeoka, Adv. Mater., 2013, 25, 545 CrossRef CAS PubMed.
- V. Pensabene, S. Taccola, L. Ricotti, G. Ciofani, A. Menciassi, F. Perut, M. Salerno, P. Dario and N. Baldini, Acta Biomater., 2011, 7, 2883 CrossRef CAS PubMed.
- L. Ricotti, S. Taccola, V. Pensabene, V. Mattoli, T. Fujie, S. Takeoka, A. Menciassi and P. Dario, Biomed. Microdevices, 2010, 12, 809 CrossRef CAS PubMed.
- T. Fujie, L. Ricotti, A. Desii, A. Menciassi, P. Dario and V. Mattoli, Langmuir, 2011, 27, 13173 CrossRef CAS PubMed.
- D. Niwa, T. Fujie, T. Lang, N. Goda and S. Takeoka, J. Biomater. Appl., 2012, 27, 131 CrossRef PubMed.
- S. Taccola, A. Desii, V. Pensabene, T. Fujie, A. Saito, S. Takeoka, P. Dario, A. Menciassi and V. Mattoli, Langmuir, 2011, 27, 5589 CrossRef CAS PubMed.
- D. Chen, J. Chen, M. Wu, H. Tian, X. Chen and J. Sun, Langmuir, 2013, 29, 8328 CrossRef CAS PubMed.
- Y. Okamura, Y. Fukui, K. Kabata, H. Suzuki, M. Handa, Y. Ikeda and S. Takeoka, Bioconjugate Chem., 2009, 20, 1958 CrossRef CAS PubMed.
- S. Boddohi and M. J. Kipper, Adv. Mater., 2010, 22, 2998 CrossRef CAS PubMed.
- T. Fujie, Y. Okamura and S. Takeoka, Adv. Mater., 2007, 19, 3549 CrossRef CAS PubMed.
- S. Takeoka, Y. Okamura, T. Fujie and Y. Fukui, Pure Appl. Chem., 2008, 80, 2259 CrossRef CAS.
- T. Fujie, N. Matsutani, M. Kinoshita, Y. Okamura, A. Saito and S. Takeoka, Adv. Funct. Mater., 2009, 19, 2560 CrossRef CAS PubMed.
- T. Fujie, M. Kinoshita, S. Shono, A. Saito, Y. Okamura, D. Saitoh and S. Takeoka, Surgery, 2010, 148, 48 CrossRef PubMed.
- T. Fujie, A. Saito, M. Kinoshita, H. Miyazaki, S. Ohtsubo, D. Saitoh and S. Takeoka, Biomaterials, 2010, 31, 6269 CrossRef CAS PubMed.
- A. Saito, H. Miyazaki, T. Fujie, S. Ohtsubo, M. Kinoshita, D. Saitoh and S. Takeoka, Acta Biomater., 2012, 8, 2932 CrossRef CAS PubMed.
- K. Hagisawa, A. Saito, M. Kinoshita, T. Fujie, N. Otani, S. Shono, Y.-K. Park and S. Takeoka, J. Vasc. Surg. Venous Lymphat. Disord., 2013, 1, 289 CrossRef PubMed.
- N. Otani, M. Kinoshita, T. Fujie, A. Saito, S. Takeoka, D. Saitoh, K. Hagisawa, H. Nawashiro and K. Shima, J. Clin. Neurosci., 2013, 20, 301 CrossRef CAS PubMed.
- T. Fujie, S. Furutate, D. Niwa and S. Takeoka, Soft Matter, 2010, 6, 4672 RSC.
- C. Picart, J. Mutterer, L. Richert, Y. Luo, G. D. Prestwich, P. Schaaf, J.-C. Voegel and P. Lavalle, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12531 CrossRef CAS PubMed.
- J. Chen, X. Qiu, L. Wang, W. Zhong, J. Kong and M. M. Q. Xing, Adv. Funct. Mater., 2014, 24, 2216 CrossRef CAS PubMed.
- P. T. Hammond, Adv. Mater., 2004, 16, 1271 CrossRef CAS PubMed.
- F. Caruso, R. A. Caruso and H. Möhwald, Science, 1998, 282, 1111 CrossRef CAS.
- M. Matsusaki, K. Kadowaki, T. Nakahara and M. Akashi, Angew. Chem., Int. Ed., 2007, 46, 4689 CrossRef CAS PubMed.
- L. Ma, D. Li and J. Chen, Drug Dev. Ind. Pharm., 2014, 40, 845 CrossRef CAS PubMed.
- B. L. Seal, T. C. Otero and A. Panitch, Mater. Sci. Eng., R, 2001, 34, 147 CrossRef.
- L. Moroni, J. R. de Wijn and C. A. van Blitterswijk, J. Biomater. Sci., Polym. Ed., 2008, 19, 543 CrossRef CAS PubMed.
- J. L. Lutkenhaus, K. D. Hrabak, K. McEnnis and P. T. Hammond, J. Am. Chem. Soc., 2005, 127, 17228 CrossRef CAS PubMed.
- S. S. Ono and G. Decher, Nano Lett., 2006, 6, 592 CrossRef CAS PubMed.
- N. Meyerbröker, Z.-A. Li, W. Eck and M. Zharnikov, Chem. Mater., 2012, 24, 2965 CrossRef.
- N. Meyerbröker and M. Zharnikov, Adv. Mater., 2014, 26, 3328 CrossRef PubMed.
- N. Meyerbröker, T. Kriesche and M. Zharnikov, ACS Appl. Mater. Interfaces, 2013, 5, 2641 Search PubMed.
- F. Mallwitz and W. A. Goedel, Angew. Chem., Int. Ed., 2001, 40, 2645 CrossRef CAS.
- Y. Ma, J. Sun and J. Shen, Chem. Mater., 2007, 19, 5058 CrossRef CAS.
- Z. Gui, J. Qian, B. Du, M. Yin and Q. An, J. Colloid Interface Sci., 2009, 340, 35 CrossRef CAS PubMed.
- S. T. Dubas, T. R. Farhat and J. B. Schlenoff, J. Am. Chem. Soc., 2001, 123, 5368 CrossRef CAS.
- M. Kohri, Y. Shinoda, H. Kohma, Y. Nannichi, M. Yamauchi, S. Yagai, T. Kojima, T. Taniguchi and K. Kishikawa, Macromol. Rapid Commun., 2013, 34, 1220 CrossRef CAS PubMed.
- T. Fujie, H. Haniuda and S. Takeoka, J. Mater. Chem., 2011, 21, 9112 RSC.
- T. Fujie, A. Desii, L. Ventrelli, B. Mazzolai and V. Mattoli, Biomed. Microdevices, 2012, 14, 1069 CrossRef CAS PubMed.
- H. Zhang and S. Takeoka, Macromolecules, 2012, 45, 4315 CrossRef CAS.
- T. Fujie, S. Ahadian, H. Liu, H. Chang, S. Ostrovidov, H. Wu, H. Bae, K. Nakajima, H. Kaji and A. Khademhosseini, Nano Lett., 2013, 13, 3185 CrossRef CAS PubMed.
- C. Jiang, S. Markutsya and V. V. Tsukruk, Adv. Mater., 2004, 16, 157 CrossRef CAS PubMed.
- L. Shen, B. Wang, J. Wang, J. Fu, C. Picart and J. Ji, ACS Appl. Mater. Interfaces, 2012, 4, 4476 CAS.
- S. Y. Wong, Q. Li, J. Veselinovic, B.-S. Kim, A. M. Klibanov and P. T. Hammond, Biomaterials, 2010, 31, 4079 CrossRef CAS PubMed.
- S. H. Baxamusa, M. Stadermann, C. Aracne-Ruddle, A. J. Nelson, M. Chea, S. Li, K. Youngblood and T. I. Suratwala, Langmuir, 2014, 30, 5126 CrossRef CAS PubMed.
- L. B. Freund and S. Suresh, Thin Film Materials: Stress, Defect Formation and Surface Evolution, Cambridge University Press, Cambridge, UK, 2003 Search PubMed.
- H. Qin, D. Wang, X. Xiong and J. Jin, Macromol. Rapid Commun., 2014, 35, 1055 CrossRef CAS PubMed.
- P. R. Bidez, S. Li, A. G. MacDiarmid, E. C. Venancio, Y. Wei and P. I. Lelkes, J. Biomater. Sci., Polym. Ed., 2006, 17, 199 CrossRef CAS PubMed.
- I. Jun, S. Jeong and H. Shin, Biomaterials, 2009, 30, 2038 CrossRef CAS PubMed.
- J. Huang, X. Hu, L. Lu, Z. Ye, Q. Zhang and Z. Luo, J. Biomed. Mater. Res., Part A, 2010, 93, 164 Search PubMed.
- J. Y. Lee, C. A. Bashur, C. A. Milroy, L. Forciniti, A. S. Goldstein and C. E. Schmidt, IEEE Trans. Nanobiosci., 2012, 11, 15 CrossRef PubMed.
- C. R. Broda, J. Y. Lee, S. Sirivisoot, C. E. Schmidt and B. S. Harrison, J. Biomed. Mater. Res., Part A, 2011, 98, 509 CrossRef PubMed.
- A.-D. Bendrea, G. Fabregat, J. Torras, S. Maione, L. Cianga, L. J. del Valle, I. Cianga and C. Alemán, J. Mater. Chem. B, 2013, 1, 4135 RSC.
- E. Llorens, M. M. Pérez-Madrigal, E. Armelin, L. J. del Valle, J. Puiggalí and C. Alemán, RSC Adv., 2014, 4, 15245 RSC.
- D. E. López-Pérez, D. Aradilla, L. J. del Valle and C. Alemán, J. Phys. Chem. C, 2013, 117, 6607 Search PubMed.
- B. Teixeira-Dias, L. J. del Valle, D. Aradilla, F. Estrany and C. Alemán, Macromol. Mater. Eng., 2012, 297, 427 CrossRef CAS PubMed.
- G. Fabregat, B. Teixeira-Dias, L. J. del Valle, E. Armelin, F. Estrany and C. Alemán, ACS Appl. Mater. Interfaces, 2014, 6, 11940 CAS.
- G. Fabregat, E. Armelin and C. Alemán, J. Phys. Chem. B, 2014, 118, 4669 CrossRef CAS PubMed.
- M. Martí, G. Fabregat, F. Estrany, E. Armelin and C. Alemán, J. Mater. Chem., 2010, 20, 10652 RSC.
- G. Fabregat, E. Córdova-Mateo, E. Armelin, O. Bertran and C. Alemán, J. Phys. Chem. C, 2011, 115, 14933 CAS.
- S. Radhakrishnan, C. Sumathi, A. Umar, S. J. Kim, J. Wilson and V. Dharuman, Biosens. Bioelectron., 2013, 47, 133 CrossRef CAS PubMed.
- B. Teixeiras-Dias, D. Zanuy, J. Poater, M. Solà, F. Estrany, L. J. del Valle and C. Alemán, Soft Matter, 2011, 7, 9922 RSC.
- E. Llorens, E. Armelin, M. M. Pérez-Madrigal, L. J. del Valle and C. Alemán, Polymers, 2013, 5, 1115 CrossRef PubMed.
- X. F. Lu, W. J. Zhang, C. Wang, T.-C. Wen and Y. Wei, Prog. Polym. Sci., 2011, 36, 671 CrossRef CAS PubMed.
- G. Fabregat, E. Córdova-Mateo, E. Armelin, O. Bertran and C. Alemán, J. Phys. Chem. C, 2011, 115, 14933 CAS.
- C. Z. Liao and F. Yan, Polym. Rev., 2013, 3, 352–406 CrossRef PubMed.
- M. Marti, G. Fabregat, F. Estrany, C. Alemán and E. Armelin, J. Mater. Chem., 2010, 20, 10652 RSC.
- L. Li, Y. Shi, L. J. Pan, Y. Shi and G. H. Yu, J. Mater. Chem. B, 2015, 3, 2920 RSC.
- L. Kergoat, B. Piro, D. T. Simon, M. C. Pham, V. Noel and M. Berggren, Adv. Mater., 2014, 26, 5658 CrossRef CAS PubMed.
- P. Humpolicek, V. Kasparkova, P. Saha and J. Stejskal, Synth. Met., 2012, 162, 722 CrossRef CAS PubMed.
- C. H. Wang, Y. Q. Dong, K. Sengothi, K. L. Tan and E. T. Kang, Synth. Met., 1999, 102, 1313 CrossRef CAS.
- M. Mattioli-Belmonte, G. Giavaresi, G. Biagini, L. Virgili, M. Giacomini, M. Fini, F. Giantomassi, D. Natali, P. Torricelli and R. Giardino, Int. J. Artif. Organs, 2003, 23, 1077 Search PubMed.
- X. Wang, X. Gu, C. Yuan, S. Chen, P. Zhang, T. zhang, J. Yao, F. Chen and G. Chen, J. Biomed. Mater. Res., Part A, 2004, 68, 411 CrossRef PubMed.
- P. M. George, A. w. Lyckman, D. A. LaVan, A. Hedge, Y. Leung, R. Avasare, C. Testa, P. M. Alexander, R. Langer and M. Sur, Biomaterials, 2005, 26, 3511 CrossRef CAS PubMed.
- S. Maione, G. Fabregat, L. J. del Valle, A.-D. Bendrea, L. Cianga, I. Cianga, I. F. Estrany and C. Alemán, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 239 CrossRef CAS PubMed.
- F. Wang, M. Li, B. Wang, J. Zhang, Y. Cheng, L. Liu, F. Lv and S. Wang, Sci. Rep., 2015, 5, 7617 CrossRef CAS PubMed.
- S. Yin, Y. Goldovsky, M. Herzberg, L. Liu, H. Sun, Y. Zhang, F. Meng, X. Cao, D. D. Sun, H. Chen, A. Kushmaro and X. Chen, Adv. Funct. Mater., 2013, 23, 2971 CrossRef PubMed.
- T. Nishikawa, R. Ookura, J. Nishida, K. Arai, J. Hayashi, N. Kurono, T. Sawadaishi, M. Hara and M. Shimomura, Langmuir, 2002, 18, 5734 CrossRef CAS.
- Y. Zhang, T. R. Nayak, H. Hong and W. Cai, Nanoscale, 2012, 4, 3833 RSC.
- Y. Yang, A. M. Asiri, Z. Tang, D. Du and Y. Lin, Mater. Today, 2013, 16, 365 CrossRef CAS PubMed.
- J. Lei, Z. Li, X. Lu, W. Wang, X. Bian, T. Zheng, Y. Xue and C. Wang, J. Colloid Interface Sci., 2011, 364, 555 CrossRef CAS PubMed.
- T. F. Otero and M. J. Ariza, J. Phys. Chem. B, 2003, 107, 13954 CrossRef CAS.
- D. Chowdhury, A. Paul and A. Chattopadhyay, Langmuir, 2005, 21, 4123 CrossRef CAS PubMed.
- Y. Lu, G. Shi, C. Li and Y. Liang, J. Appl. Polym. Sci., 1998, 70, 2169 CrossRef CAS.
- J. Song, H. Liu, M. Wan, Y. Zhu and L. Jiang, J. Mater. Chem. A, 2013, 1, 1740 CAS.
- B. Zhao, K. G. Neoh, F. T. Liu and E. T. Kang, Langmuir, 1999, 15, 8259 CrossRef CAS.
- I. Mihai, F. Addiego, D. Ruch and V. Ball, Sens. Actuators, B, 2014, 192, 769 CrossRef CAS PubMed.
- G.-W. Huang, H.-M. Xiao, H.-Q. Shi and S.-Y. Fu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2794 CrossRef CAS PubMed.
- R. Yue, S. Chen, B. Lu, C. Liu and J. Xu, J. Solid State Electrochem., 2011, 15, 539 CrossRef CAS PubMed.
- H. Zhu, L. Gao, M. Li, H. Yin and D. Wang, Electrochem. Commun., 2011, 13, 1479 CrossRef CAS PubMed.
- G. Shi, S. Jin, G. Xue and C. Li, Science, 1995, 267, 994 CrossRef CAS PubMed.
- D. Wang, Y.-X. Li, Z. Shi, H.-L. Qin, L. Wang, X.-F. Pei and J. Jin, Langmuir, 2010, 26, 14405 CrossRef CAS PubMed.
- S. S. Jeon, H. H. An, C. S. Yoon and S. S. Im, Polymer, 2011, 52, 652 CrossRef CAS PubMed.
- G. Qi, L. Huang and H. Wang, Chem. Commun., 2012, 48, 8246 RSC.
- P. Jha, S. P. Koiry, V. Saxena, P. Veerender, A. K. Chauhan, D. K. Aswal and S. K. Gupta, Macromolecules, 2011, 44, 4583 CrossRef CAS.
- Z. Tong, Y. Yang, J. Wang, J. Zhao, B.-L. Su and Y. Li, J. Mater. Chem. A, 2014, 2, 4642 CAS.
- Z. Niu, P. Luan, Q. Shao, H. Dong, J. Li, J. Chen, D. Zhao, L. Cai, W. Zhou, X. Chen and S. Xie, Energy Environ. Sci., 2012, 5, 8726 CAS.
- F. Greco, A. Zucca, S. Taccola, A. Menciassi, T. Fujie, H. Haniuda, S. Takeoka, P. Dario and V. Mattoli, Soft Matter, 2011, 7, 10642 RSC.
- M. H. Bolin, K. Svennersten, X. Wang, I. S. Chronakis, A. Richter-Dahlfors, E. W. H. Jager and M. Berggren, Sens. Actuators, B, 2009, 142, 451 CrossRef CAS PubMed.
- K. Svennersten, M. H. Bolin, E. W. H. Jager, M. Berggren and A. Richter-Dahlfors, Biomaterials, 2009, 30, 6257 CrossRef CAS PubMed.
- S. Taccola, F. Greco, B. Mazzolai, V. Mattoli and E. W. H. Jager, J. Micromech. Microeng., 2013, 23, 117004 CrossRef.
- S. Taccola, F. Greco, A. Zucca, C. Innocenti, C. de Julián Fernández, G. Campo, C. Sangregorio, B. Mazzolai and V. Mattoli, ACS Appl. Mater. Interfaces, 2013, 5, 6324 CAS.
- F. Greco, T. Fujie, L. Ricotti, S. Taccola, B. Mazzolai and V. Mattoli, ACS Appl. Mater. Interfaces, 2013, 5, 573 CAS.
- C.-C. Fu, A. Grimes, M. Long, C. G. L. Ferri, B. D. Rich, S. Ghosh, S. Ghosh, L. P. Lee, A. Gopinathan and M. Khine, Adv. Mater., 2009, 21, 4472 CrossRef CAS PubMed.
- J. A. Lee, M. K. Shin, S. H. Kim, S. J. Kim, G. M. Spinks, G. G. Wallace, R. Ovalle-Robles, M. D. Lima, M. E. Kozlov and R. H. Baughman, ACS Nano, 2012, 6, 327 CrossRef CAS PubMed.
- O. Ahumada, M. M. Pérez-Madrigal, J. Ramirez, D. Curcó, C. Esteves, A. Salvador-Matar, G. Luongo, E. Armelin, J. Puiggalí and C. Alemán, Rev. Sci. Instrum., 2013, 84, 053904 CrossRef CAS PubMed.
- A. L. Gomes, J. Casanovas, O. Bertran, J. S. de C. Campos, E. Armelin and C. Alemán, J. Polym. Res., 2011, 18, 1509 CrossRef CAS.
- R. A. Khalkhali and M. B. Keivani, Asian J. Chem., 2005, 17, 835 CAS.
- J. A. Marins, B. G. Soares, M. Fraga, D. Müller and G. M. O. Barra, Cellulose, 2014, 21, 1409 CrossRef CAS PubMed.
- E. Armelin, A. L. Gomes, M. M. Pérez-Madrigal, J. Puiggalí, L. Franco, L. J. del Valle, A. Rodríguez-Galán, J. S. d. C. Campos, N. Ferrer-Anglada and C. Alemán, J. Mater. Chem., 2012, 22, 585 RSC.
- M. M. Pérez-Madrigal, E. Armelin, L. J. del Valle, F. Estrany and C. Alemán, Polym. Chem., 2012, 3, 979 RSC.
- M. M. Pérez-Madrigal, M. I. Giannotti, G. Oncins, L. Franco, E. Armelin, J. Puiggalí, F. Sanz, L. J. del Valle and C. Alemán, Polym. Chem., 2013, 4, 568 RSC.
- M. M. Pérez-Madrigal, M. I. Giannotti, E. Armelin, F. Sanz and C. Alemán, Polym. Chem., 2014, 5, 1248 RSC.
- M. M. Pérez-Madrigal, M. I. Giannotti, L. J. del Valle, L. Franco, E. Armelin, J. Puiggalí, F. Sanz and C. Alemán, ACS Appl. Mater. Interfaces, 2014, 6, 9719 Search PubMed.
- F. Greco, A. Zucca, S. Taccola, B. Mazzolai and V. Mattoli, ACS Appl. Mater. Interfaces, 2013, 5, 9461 CAS.
- J. George, J. Onodera and T. Miyata, J. Biomed. Mater. Res., Part A, 2008, 87, 1103 CrossRef PubMed.
- J. George, Y. Kuboki and T. Miyata, Biotechnol. Bioeng., 2006, 95, 404 CrossRef CAS PubMed.
- D. J. Kim, M. J. Jo and S. Y. Nam, J. Ind. Eng. Chem., 2015, 21, 36 CrossRef CAS PubMed.
- R. Holze and Y. P. Wu, Electrochim. Acta, 2014, 122, 93 CrossRef CAS PubMed.
- R. K. Pandey and V. Lakshminarayanan, J. Phys. Chem. C, 2009, 113, 21596 CAS.
- B. Rajesh, K. R. Thampi, J. M. Bonard, A. J. McEvoy, N. Xanthopoulus, H. J. Mathieu and B. Viswanathan, J. Power Sources, 2004, 133, 155 CrossRef CAS PubMed.
- J.-D. Qiu, L. Shi, R. P. Liang, G.-C. Wang and X. H. Xia, Chem. – Eur. J., 2012, 18, 7950 CrossRef CAS PubMed.
- C. Arbizzani, M. Biso, E. Manferrari and M. Mastragostino, J. Power Sources, 2008, 180, 41 CrossRef CAS PubMed.
- C. Arbizzani, M. Biso, E. Manferrari and M. Mastragostino, J. Power Sources, 2008, 178, 584 CrossRef CAS PubMed.
- A. Kraytsbergs and Y. Ein-Eli, Energy Fuels, 2014, 28, 7303 CrossRef.
- T. F. Otero, E. Angulo, J. Rodriguez and C. Santamaria, J. Electroanal. Chem., 1992, 341, 369 CrossRef CAS.
- Q. B. Pei and O. Inganäs, Adv. Mater., 1992, 4, 277 CrossRef CAS PubMed.
- E. W. H. Jager, E. Smela and O. Inganäs, Science, 2000, 290, 1540 CrossRef CAS.
- E. W. H. Jager, O. Inganäs and I. Lundstrom, Science, 2000, 288, 2335 CrossRef CAS.
- S. D. Deshpande, J. Kim and S. R. Yun, Smart Mater. Struct., 2005, 14, 876 CrossRef CAS.
- S. J. Higgins, K. V. Lovell, R. M. G. Rajapakse and N. M. Walsby, J. Mater. Chem., 2003, 13, 2485 RSC.
- G. Alici, A. Punning and H. R. Shea, Sens. Actuators, B, 2011, 157, 72 CrossRef CAS PubMed.
- L. Valero Conzuelo, J. Arias-Pardilla, J. V. Cauigh-Rodríguez, M. A. Smit and T. F. Otero, Sensors, 2010, 10, 2638 CrossRef PubMed.
- T. F. Otero, J. G. Martínez and J. Arias-Pardilla, Electrochim. Acta, 2012, 84, 112–128 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |