
A biocompatible and functional adhesive amine-rich coating based on dopamine polymerization†
Ying
Yang
ab,
Pengkai
Qi
ab,
Yonghui
Ding
e,
Manfred F.
Maitz
ad,
Zhilu
Yang
*ab,
Qiufen
Tu
ac,
Kaiqin
Xiong
ab,
Yang
Leng
e and
Nan
Huang
*ab
aKey Lab. of Advanced Technology for Materials of Education Ministry, Southwest Jiaotong University, Chengdu, 610031, China. E-mail: zhiluyang1029@126.com; nhuang@263.net; Fax: +86 28 87600625; Tel: +86 28 87600625
bThe Institute of Biomaterials and Surface Engineering, School of Materials Science and Engineering, Southwest Jiaotong University, Chengdu, 610031, China
cLaboratory of Biosensing and MicroMechatronics, Southwest Jiaotong University, Chengdu, 610031, China
dMax Bergmann Center of Biomaterials, Leibniz Institute of Polymer Research Dresden, Hohe Strasse 6, 01069 Dresden, Germany
eDepartment of Mechanical and Aerospace Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China
First published on 10th September 2014
Abstract
Amine groups physiologically play an important role in regulating the growth behavior of cells and they have technological advantages for the conjugation of biomolecules. In this work, we present a method to deposit a copolymerized coating of dopamine and hexamethylendiamine (HD) (PDAM/HD) rich in amine groups onto a target substrate. This method only consists of a simple dip-coating step of the substrate in an aqueous solution consisting of dopamine and HD. Using the technique of PDAM/HD coating, a high density of amine groups of about 30 nmol cm−2 was obtained on the target substrate surface. The PDAM/HD coating showed a high cross-linking degree that is robust enough to resist hydrolysis and swelling. As a vascular stent coating, the PDAM/HD presented good adhesion strength to the substrate and resistance to the deformation behavior of compression and expansion of a stent. Meanwhile, the PDAM/HD coating exhibited good biocompatibility and attenuated the tissue response compared with 316L stainless steel (SS). The primary amine groups of the PDAM/HD coating could be used to effectively immobilize biomolecules containing carboxylic groups such as heparin. These data suggested the promising potential of this PDAM/HD coating for application in the surface modification of biomedical devices.
1. Introduction
The unique advantage of surface modification is that the surface properties can be selectively improved while the bulk properties of the materials are maintained. Biomolecule conjugation is the most important method of surface modification and has been widely applied in molecular biology, analytical chemistry, bioprocess engineering, medical diagnostics, and tissue engineering and on biomedical devices.1–6 However, conjugation requires the presence of reactive groups on the surfaces of the target material, which is not the case for most metal, inorganic and many polymeric materials.In recent years, there has been rapidly increasing interest in the deposition of organic–polymeric thin films with reactive groups (e.g., plasma polymer films and polyelectrolyte films) onto the surface of bulk materials for the covalent conjugation of bioactive molecules.7–10 In particular, the attachment strategy based on the mussel-inspired surface chemistry for polydopamine (PDAM) coating has aroused extensive interest.11–15 PDAM has attracted much attention in the modification of biomaterials due to its outstanding ability to bind strongly to virtually all substrates and provide secondary reactivity for conjugating biomolecules.12,16–18 It opens a new approach to modify various substrates and synthesize functional composite materials through simple chemistry.19–21 However, the secondary reactivity of PDAM is limited to bind biomolecules containing sulfhydryl or amine groups, because its specific reactivity is based on Michael addition or Schiff base reaction between the phenolic hydroxyl/quinone groups of PDAM and sulfhydryl/amine groups of biomolecules.22 For the conjugation of biomolecules containing carboxyl groups such as heparin, PDAM lacks suitable reactive functional groups e.g. primary amine groups.
To expand the ability to immobilize this type of biomolecule containing carboxyl groups, here we report a method to prepare a functional polymer coating rich in amine groups inspired by the adhesive proteins secreted by marine mussels for attachment to wet surfaces.23Mytilus edulis foot protein-3 (Mefp)-3 and Mefp-5 of mussel have been believed to be the key adhesives that tether the organism to the surface of the substrate. These two proteins contain the highest 3,4-dihydroxy-L-phenylalanine (DOPA) content and are rich in lysine.24 It has been demonstrated that the strong adhesion of mussels to a surface is based on the strong covalent and noncovalent interactions with substrates produced by phenolic hydroxyl/quinone groups of DOPA.25,26 In addition to this, lysine plays an important role in cross-linking, resulting in solidification of the secreted liquid protein adhesive.24,27 Inspired by this, dopamine and hexamethylendiamine (HD) are used to synthesize an amine-rich, highly cross-linked copolymer.
As a coating that is applied on a long-term implanted device, robust adhesion and good stability are pre-conditions for the safety property in vivo. In this work, stent expansion and dynamic dissolution assay were performed to test the adhesion strength and stability of the PDAM/HD coatings. The growth behavior of human umbilical vein endothelial cells (HUVECs) was investigated to evaluate the cytocompatibility of the PDAM/HD coating. Good tissue-compatibility is also an important aspect for biomedically modified coating. Thus, the tissue response of PDAM/HD was evaluated in vivo. Additionally, heparin, as a typical molecule with carboxylic groups, was chosen to demonstrate the conjugation to the amine groups of PDAM/HD. Covalent immobilization of heparin via carbodiimide activated carboxylic groups to the amine groups of PDAM/HD through the formation of amide bonds was demonstrated.
2. Materials and experiments
2.1 Preparation of the PDAM/HD copolymer coating
PDAM/HD coatings were deposited on mirror polished 316L SS (Φ 10 mm) based on a simple one-step dip-coating in the reaction solution at 20 °C.12 The reaction solution was obtained by dissolving dopamine hydrochloride (1 mg mL−1) and hexamethylendiamine (HD) (2.44 mg mL−1) into Tris buffer (pH 8.5). After being deposited for 4, 8, 16, 24 and 48 h respectively, the samples were washed with distilled water. The control PDAM coating was also fabricated using the same way. The as-deposited coatings were subsequently tempered at 120 °C for 1 h under 5 × 10−4 Pa.2.2 Characterization of the PDAM/HD copolymer coating
A grazing incidence attenuated total reflection Fourier transform infrared spectroscopy (GATR-FTIR, NICOLET 5700) spectrometer was used to analyze the surface chemical structures of the coatings.The density of amine groups was determined using the Acid Orange II (AO II) colorimetric method. The details has been described elsewhere.28
The thickness of the coating was detected using a spectroscopic ellipsometer (M-2000V, J.A. Woollam, USA) operating in the Cauchy model, with Δ and Ψ values measured at a wavelength of 370–1000 nm.
The zeta potential of samples was measured using a commercial electrokinetic analyzer (EKA, Anton Paar GmbH, Graz, Austria) with a solution of 0.001 M KCl (pH = 7.4) at room temperature.
The matrix assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS) measurement was performed using a MALDI micro MX time-of-flight mass spectrometer (Waters, Milford, MA) operating in reflection mode. The MALDI source was equipped with a 337 nm N2 laser operating with a 4 ns-duration pulse. The laser pulse energy was set as 200 arbitrary units (AUs) when using 2,5-dihydroxybenzoic acid (DHB) as the matrix. All mass spectra were recorded in the m/z range of 50–4000. Each mass spectrum was obtained by averaging the ions from ten laser shots. A total of 10 mass spectra were combined for data analysis of each sample. The matrix solution was prepared by dissolving DHB in 0.1% trifluoroacetic acid (TFA)/acetonitrile (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (v/v)) at a concentration of 20 mg mL−1. For the MALDI analysis of PDAM/HD and control PDAM coatings, the MALDI target plates (stainless steel) were directly modified with the target coatings. Before analysis, 1 μL of the matrix solution was added to the plates modified by target coatings and allowed to air-dry.
:4 (v/v)) at a concentration of 20 mg mL−1. For the MALDI analysis of PDAM/HD and control PDAM coatings, the MALDI target plates (stainless steel) were directly modified with the target coatings. Before analysis, 1 μL of the matrix solution was added to the plates modified by target coatings and allowed to air-dry.
The surface morphology and roughness of coatings were analyzed by atomic force microscopy (AFM, SPI 3800, NSK Inc.).
The water contact angle (WCA) was measured using a Krüss GmbH DSA 100 Mk 2 goniometer (Hamburg, Germany) at room temperature. The image sessile drop processing of 5 μL test ultrapure water was recorded using DSA 1.8 software. The WCA calculation was carried out using a circle segment function.
The surface morphology of PDAM/HD coated 316L SS vascular stents (1.65 mm × 18 mm) was examined by scanning electron microscopy (SEM, Quanta 200, FEI, Holland). The stent was dilated from 1.65 mm to 3.0 mm (diameter) at a pressure of 8 atm using an angioplasty balloon.
For the stability evaluation of the PDAM and PDAM/HD coatings, the surface microstructures of the coatings before and after immersing into Phosphate Buffer Solution (PBS) at 37 °C in the dynamic state were observed by SEM.
2.3 HUVEC culture, staining and proliferation
Primary HUVECs were derived from the human umbilical vein.7 HUVECs were seeded on the samples at a density of 5 × 104 cells per cm2. After being cultured for 2 h, 1 and 3 days respectively, the samples were sequentially washed, fixed and stained by Rhodamine 123. Then the cells were examined and recorded using a Leica DMRX fluorescence microscope (DMRX, Leica, Germany). The coverage area of adherent HUVECs was calculated from at least 12 images. The minor/major axis ratio and the projected area per cell were calculated from more than 120 cells.The HUVEC proliferation was examined using Cell Counting Kit-8 (CCK-8) after incubation for 1 and 3 days respectively. The details have been described elsewhere.7 The proliferation experiments were performed in triplicate.
2.4 In vivo tissue response test
All procedures were in compliance with the China Council on Animal Care and Southwest Jiaotong University animal use protocol, following all the ethical guidelines for experimental animals. In this study, four adult New Zealand white rabbits weighing about 4 kg were used. The double mirror-polished bare 316L SS disks (3 × 4 mm2, n = 3) and PDAM/HD coated 316L SS disks (n = 3) were subcutaneously implanted on the back of both sides. After 2 and 9 weeks, the tissue around the implanted samples was harvested to evaluate the in situ body response reaction. The harvested tissue was fixed in 10% formaldehyde for 3–5 days. After washing with PBS, the harvested tissue was then dehydrated in graded ethanol, saturated with xylene and embedded in paraffin. Hematoxylin and eosin (HE) and Masson's trichrome staining of paraffin sections were performed for further histological analysis.2.5 Heparin conjugation and quantitative determination
1 mg of heparin sodium salt (potency ≥ 150 IU mg−1, Sigma-Aldrich) was first activated in 1 mL of water-soluble carbodiimide (WSC, pH 5.4) solution for 30 min.28 Then the PDAM/HD coated samples were immersed into the above mixed solution at room temperature. After 12 h reaction, the samples were rinsed with PBS and distilled water respectively.X-ray photoelectron spectroscopy (XPS, XSAM800, Kratos Ltd, UK) was used to determine the chemical compositions of the samples. The XPS instrument was equipped with a monochromatic Al Kα (1486.6 eV) X-ray source operating at 12 kV × 15 mA at a pressure of 2 × 10−7 Pa. The C1s peak (binding energy 284.8 eV) was used as a reference for charge correction.
The amount of heparin bound to the surface of coating was determined by a toluidine blue-O (TBO) method.28
2.6 APTT assay
Platelet poor plasma (PPP) was obtained by centrifuging fresh human whole blood (3000 rpm, 15 min). The samples were incubated with 350 μL PPP for 30 min at 37 °C. The clotting time of incubated PPP was detected using a coagulometer (ACL 200, USA) with the APTT Reagent Kit.292.7 Statistical analysis
All of the blood and cell tests, AO II and TBO assay were carried out at least three times with more than four parallel samples. The data were provided as mean ± standard error (SD). Statistical analysis was performed by a one-way ANOVA, *p < 0.05 indicated significant difference.3. Results and discussion
We first investigated the influence of reaction time on the surface density of the amine groups of the PDAM/HD copolymer coatings. Five periods of time, 4, 8, 16, 24 and 48 h, were used for analysis, respectively. To investigate the chemical structures and compositions of the PDAM/HD coatings, FTIR and XPS analyses were performed. Here, FTIR spectra of PDAM/HD were compared at the typical time points of 4 h and 48 h and the control PDAM coating. As shown in Fig. 1B, the basic features of the chemical structure of the PDAM coating were well maintained in the PDAM/HD coatings.30 However, some differences in the C–H and N–H peaks were apparent. Peaks related to the stretching vibrations of ν–N–H at 3340 and 3270 cm−1 and deformation vibrations of γ (NH) at 700 cm−1 were enhanced. This not only indicated the copolymerization between dopamine and HD, but also indicated a good retention of –NH2 of HD or dopamine (Fig. 1C). In addition, the remarkable reinforcement in the C–H peaks around 2850 and 2930 cm−1 was a strong evidence for the introduction of HD during the copolymerization of dopamine and HD. XPS analysis revealed significant differences in the chemical composition of PDAM and PDAM/HD. The addition of HD significantly altered the surface chemical compositions of the coatings, as measured by a remarkable increase in the content of nitrogen, and a reduction in the contents of carbon and oxygen of the PDAM/HD (Table S1†). Note that, the contents of nitrogen of PDAM/HD coatings tended to increase with a prolonged reaction time. To further understand the chemical components, XPS C1s peak fitting was thereby carried out (Fig. S1A†). It was found that the peak consisting of C–N, –OH, aromatic C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N at 286.4 eV of PDAM shifted to a lower binding energy of 286 eV of PDAM/HD. Additionally, the PDAM/HD spectrum showed a significant increase in the ratio of this component compared to pure PDAM (Fig. S1D†). This was attributed to the introduction of C–N, aromatic C–N and CN based on the Schiff base and Michael addition reaction of dopamine with HD. The new formation of aromatic C–N and CN in PDAM/HD produced a strong inductive and conjugation effect, which shifted the shoulder of quinone (CO) to lower binding energy (Fig. S1A†). There were also remarkable differences in the high-resolution N1s and O1s XPS spectra of the PDAM and PDAM/HD coatings (Fig. S1B and S1C†). Furthermore, AO II test and ellipsometry were performed to determine the density of amine groups and thickness of the PDAM/HD copolymerized coatings. We found that the density of amine groups and thickness of the PDAM/HD coatings both were a function of the reaction time of dopamine and HD and reached the values of 30 nmol cm−2 (Fig. 1C) and 140 nm (Fig. 1D) after 48 h. Besides, all the coatings were negatively charged at pH 7.4. With the extension of the reaction time, the absolute value of the zeta potential decreased, as a result of a higher surface density of amine groups of the PDAM/HD coatings.29 A larger amine group density was correlated with a less negative value of the zeta potential. Therefore, it was concluded that a copolymerized coating of PDAM/HD rich in amine groups was synthesized on the 316L SS surface.
N at 286.4 eV of PDAM shifted to a lower binding energy of 286 eV of PDAM/HD. Additionally, the PDAM/HD spectrum showed a significant increase in the ratio of this component compared to pure PDAM (Fig. S1D†). This was attributed to the introduction of C–N, aromatic C–N and CN based on the Schiff base and Michael addition reaction of dopamine with HD. The new formation of aromatic C–N and CN in PDAM/HD produced a strong inductive and conjugation effect, which shifted the shoulder of quinone (CO) to lower binding energy (Fig. S1A†). There were also remarkable differences in the high-resolution N1s and O1s XPS spectra of the PDAM and PDAM/HD coatings (Fig. S1B and S1C†). Furthermore, AO II test and ellipsometry were performed to determine the density of amine groups and thickness of the PDAM/HD copolymerized coatings. We found that the density of amine groups and thickness of the PDAM/HD coatings both were a function of the reaction time of dopamine and HD and reached the values of 30 nmol cm−2 (Fig. 1C) and 140 nm (Fig. 1D) after 48 h. Besides, all the coatings were negatively charged at pH 7.4. With the extension of the reaction time, the absolute value of the zeta potential decreased, as a result of a higher surface density of amine groups of the PDAM/HD coatings.29 A larger amine group density was correlated with a less negative value of the zeta potential. Therefore, it was concluded that a copolymerized coating of PDAM/HD rich in amine groups was synthesized on the 316L SS surface.
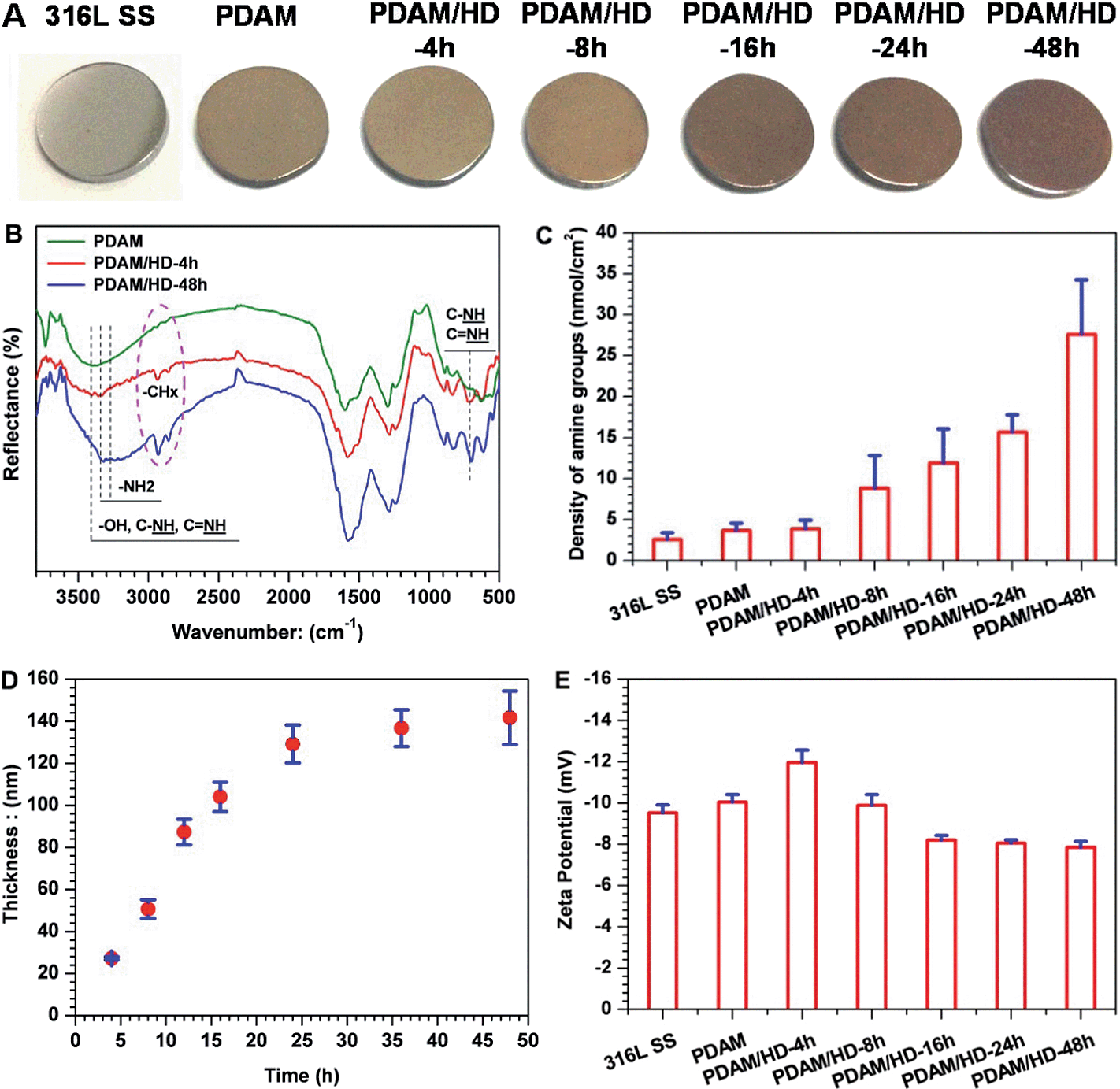 | ||
| Fig. 1 (A) Photos of 316L SS, PDAM and PDAM/HD coatings taken after defined periods of co-polymerization; (B) FTIR spectra of the PDAM and PDAM/HD coatings; (C and D) the density of amine groups and thickness of 316L SS, PDAM and PDAM/HD coatings as a function of the polymerization time; (E) the zeta potential of 316L SS, PDAM and PDAM/HD coatings obtained after defined polymerization times. | ||
Before the analysis of polymerization of the PDAM/HD coating, the analysis of the MALDI-TOF MS spectrum of PDAM was first carried out. For the polymerization mechanism of dopamine, previous studies considered the oxidation of the catechol to a quinone as the likely step, followed by polymerization in a manner reminiscent of melanin formation.32 Recent studies showed that the PDAM formation was associated with a series of complex polymer structures and mechanisms.27,33 Lee et al.25 have reported that a variety of possible inter-molecular interactions such as ionic, cation–π, π–π, quadrupole–quadrupole and hydrogen bonding (H-bonding) interactions contributed to the physical self-assembly pathway of PDAM polymerization. Vecchia et al.26 found that PDAM consisted of three main building blocks, uncyclized catecholamine/quinones, cyclized DHI units, and pyrrolecarboxylic acid moieties. There were several models built up, such as the polycatecholamine model, quinhydrone model, polyindole model and physical trimer model.27 Our result of the MALDI-TOF MS spectrum of PDAM also confirmed such a polymerization mechanism, and the incorporation of the buffer substance Tris(hydroxymethyl)-aminomethane (Tris) into PDAM was also demonstrated by the detection of the peaks at 273, 391 and 409 m/z (Fig. 2A and B).
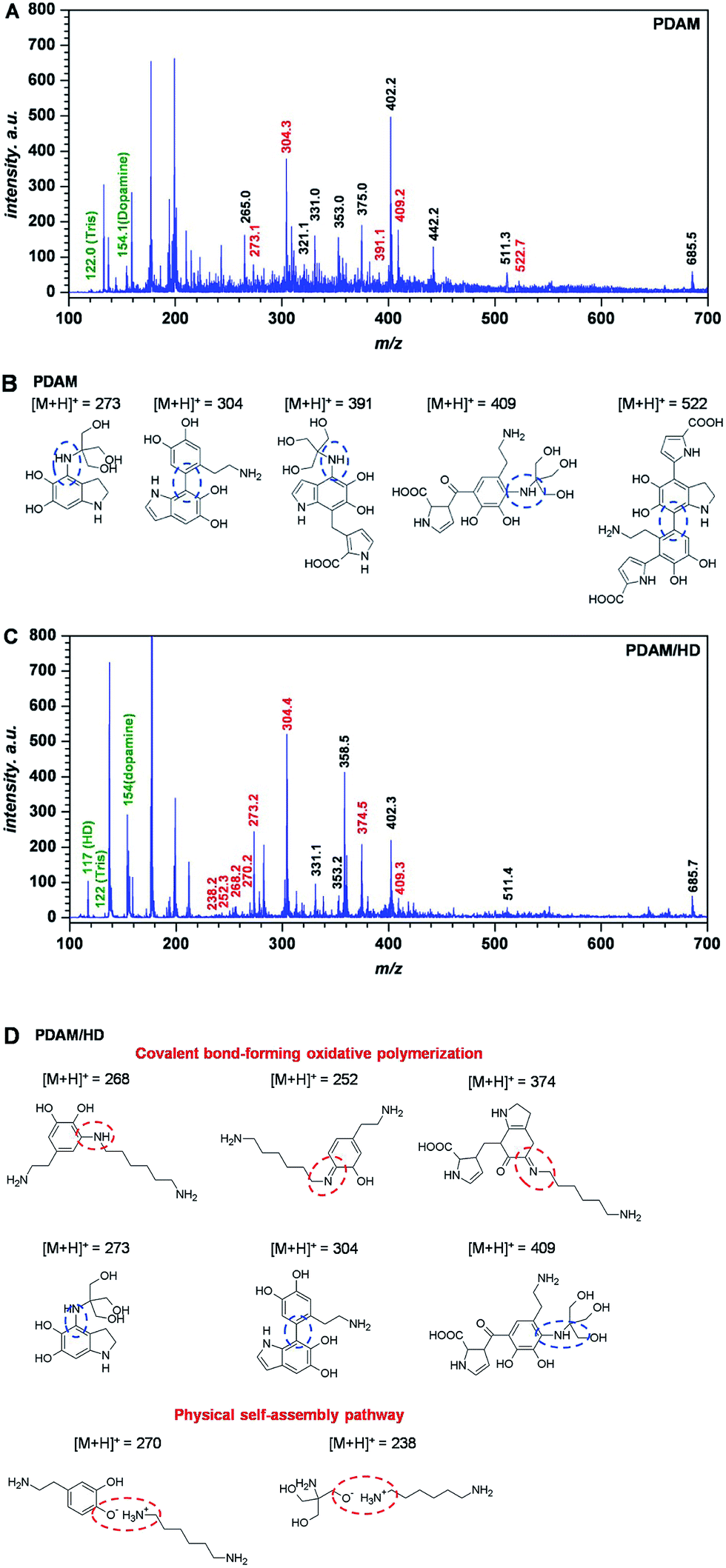 | ||
| Fig. 2 MALDI-TOF MS spectrum of the PDAM (A) and PDAM/HD (C) coating, with tentative structures assigned to main peaks (B) and (D), respectively. | ||
The introduction of HD in the PDAM/HD synthesis significantly influenced the self-polymerization manner of dopamine. The incorporation of HD into PDAM was mainly based on oxidative polymerization with the formation of covalent bondings and physical self-assembly (Fig. 2C and D). Covalent bond-forming oxidative polymerization was attributed to Michael addition and Schiff-base reactions, as evidenced by the presence of the peak at 268 252, and 374 m/z. The physical self-assembly involved the electrostatic interaction of –NH3+ (as evidenced by the presence of the peak of –NH3+ around 402 eV, as shown in Fig. S2B†) of HD and O− of dopamine or Tris, as evidenced by the presence of the peak at 270 and 238 m/z. Although the exact polymerization mechanism needs further and more studies, it is likely that mainly the oxidation of the phenolic hydroxyl group of dopamine to a quinone is involved, followed by a reaction with HD and Tris via Shiff-base and Michael addition chemistries. There was another important non-covalent electrostatic interaction between –NH3+ (derived from primary amine groups of HD) and Ph–O− (derived from dopamine) or –O− of Tris, which was formed via spontaneous proton transfer from the Ph–OH of dopamine or –OH of Tris to –NH2.
The surface uniformity of biomedical implants such as stents and artificial heart valves (AHVs) is believed to be a very important factor for biocompatibility.34 Ratner et al.34 reported that, a smooth surface with roughness less than 50 nm was required, otherwise platelets aggregation and thrombogenesis might be more prone to happen on the surface. Herein, AFM was used for the surface analysis of PDAM/HD coatings. To ensure homogeneity, roughness detection was further carried out on different areas of a scan size of 20 × 20 μm2 on the same sample. As shown in Fig. 3A, the AFM image revealed that the 316L SS surface was homogeneous, with a root mean square (RMS) roughness of 3.4 ± 0.2 nm. After the deposition of PDAM, the presence of the nanometer-scale PDAM grains resulted in a rougher surface and the RMS increased to 13.9 ± 2.5 nm.30,35 For the copolymerized coatings of PDAM/HD, the RMS was a function of reaction time: the RMS of PDAM/HD decreased during the first 16 h of reaction time and reached the lowest value of 7.5 nm, and then increased again with the increase of reaction time to 10.8 nm after 48 h. The biggest value of RMS of the PDAM/HD coating was 11.7 nm after 4 h, which was much lower than the threshold value for thrombogenicity of 50 nm. In total, it can be concluded that the RMS roughness of the coating was within the biocompatible range (below 50 nm), ranging from 7.5 to 11.7 nm.
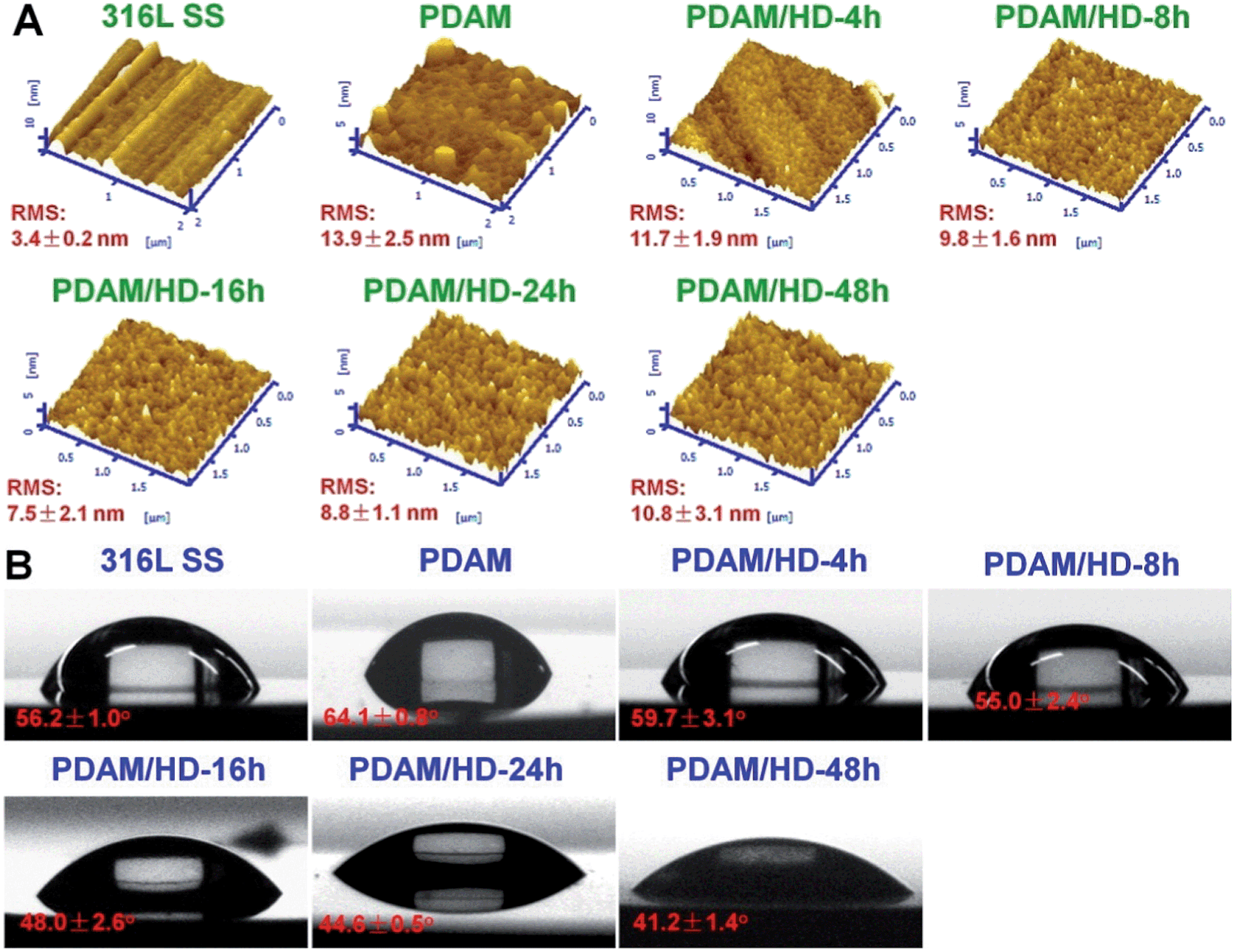 | ||
| Fig. 3 (A) AFM tapping mode images and (B) water contact angles of the control 316L SS, PDAM and PDAM/HD coatings deposited on 316L SS. | ||
The surface hydrophilicity of materials also can greatly affect biocompatibility.34 Functional groups like amine, carboxyl, and hydroxyl groups are hydrophilic. Compared with PDAM, the PDAM/HD coatings had increased hydrophilicity (Fig. 3B), which may be attributed to the higher density of hydrophilic amine groups (Fig. 1C). It was found that the density of amine groups of the PDAM/HD coating correlated with the hydrophilicity of the coating. As reported, the water contact angle (WCA) of 65° frequently is regarded as the threshold in the definition of hydrophilic or hydrophobic phases.36 Clearly, the water contact angles (WCAs) of the PDAM/HD coatings were below this value, indicating that PDAM/HD coatings were hydrophilic and favorable for improving biocompatibility.
In this work, stent dilation analysis was performed to investigate the mechanical behavior and adhesion strength of the PDAM/HD coating to a substrate.30 As shown in Fig. 4, the PDAM/HD coating on the stent was compact and homogeneous, with inevitable aggregate formation of nano-/microparticulates.35 After dilation, the PDAM/HD coating still firmly adhered on the stent without any peeling, cracking, delamination or destruction. This indicates that the PDAM/HD coating was flexible enough to bear the deformation and confirms the potential for application in surface modification of biomedical devices with a complex geometry.12
 | ||
| Fig. 4 Micrographs of the 316L SS stents coated with PDAM/HD-48h. (A) Post-dilation image; (B–F) post-dilation SEM images. | ||
Good stability of a coating is not only the guarantee for a durable retention of biomolecules bound to its surface but also the key factor to ensure the long-term safe performance in vivo. There are three typical subsequent reactions of a polymer after immersion in aqueous solution, swelling, dissolution or hydrolysis.37 However, strong swelling and water absorption of a polymer coating make it extremely difficult to quantify the stability based on e.g. the change of thickness or mass. Herein, a dynamic dissolution assay was used to indirectly evaluate the stability of the PDAM/HD coatings. The PDAM and PDAM/HD coating with the highest density of amine groups were evaluated. Fig. 5 presents the SEM images of the PDAM and PDAM/HD coatings before and after continuous immersion into PBS for 7, 15 and 30 days, respectively. Clearly, swelling occurred on both PDAM and PDAM/HD coatings after immersion for 7 days, and the swelling degree gradually enhanced along with the increase of immersion time. Meanwhile, there was still only swelling observed on the surfaces even when the immersion time prolonged to 30 days. This suggests that both PDAM and PDAM/HD coatings showed the high cross-linking degree to resist dissolution or hydrolysis.30
 | ||
| Fig. 5 Surface SEM images of the PDAM and PDAM/HD-48h coatings before and after immersion into PBS in the dynamical state for various periods of time. | ||
The cytocompatibility of the PDAM/HD coatings has to be evaluated before the application on vascular devices.38 Herein, the growth behavior of HUVECs on PDAM/HD was systematically evaluated. The cell attachment revealed good affinity of HUVECs to the PDAM/HD coatings (Fig. 6B). The PDAM/HD surfaces increased the number of adherent HUVECs by 16.4–25.4% as compared with the control 316L SS substrate. This may be ascribed to the amine and phenolic hydroxyl/quinone groups on the surface.6,30,31,39 Some previous studies demonstrated that the surface equipped with amine groups has good affinity to ECs due to their positively charged surface.6,31 Phenolic hydroxyl/quinone groups on the surface of the substrate enhance the adsorption of serum proteins from the cell culture medium and hence serve as EC adhesion sites.30,39
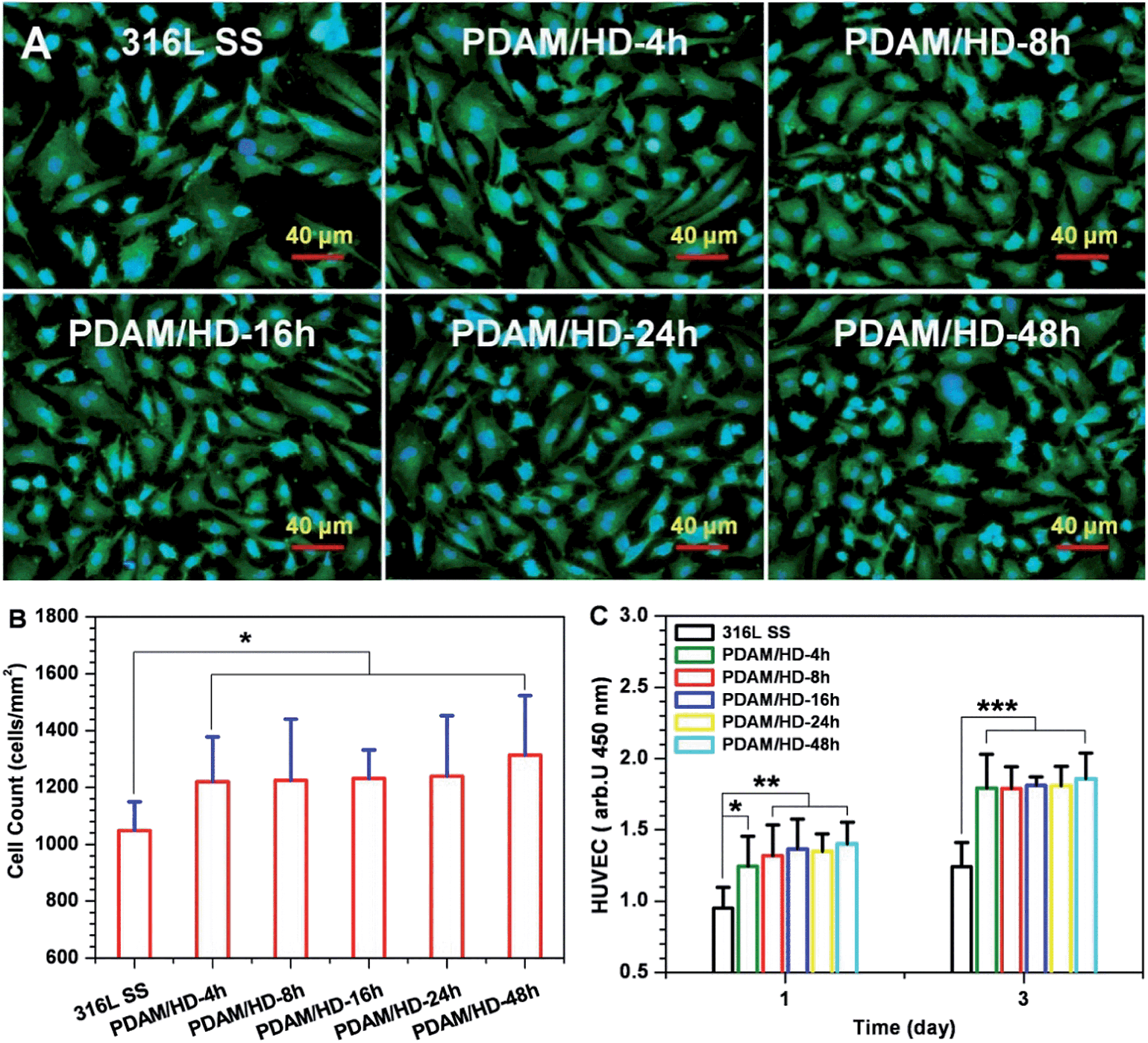 | ||
| Fig. 6 (A) Cytoskeletal actin (green) and nuclear (blue) stains of HUVECs on the on the 316L SS and PDAM/HD surfaces after 1 day of culture; (B) amounts of HUVECs attached onto the 316L SS and PDAM/HD surfaces after being cultured for 2 h are calculated from at least 12 images; (C) proliferation of HUVECs cultured on the 316L SS and PDAM/HD surfaces for 1 and 3 days detected by the CCK-8 Kit. Data presented as mean ± SD (n = 4) and analyzed using a one-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001. | ||
To further understand the interactions of the cell with the PDAM/HD coating, the analysis for HUVEC morphology was carried out. Fluorescence microscopy revealed a significant effect of the PDAM/HD coating on cellular growth behavior (Fig. 6A), which was further characterized numerically. The HUVECs grown on the PDAM/HD surface were more spread and elongated than that adhered on 316L SS. The projected area per cell of the 316L SS was 1211 μm2 in average, but the projected area per cell of PDAM/HD remarkably increased to 1639–1987 μm2 which was 1.35–1.64 fold for 316L SS (Fig. S2A†). The minor/major axis ratios of HUVECs of the PDAM/HD surfaces were much lower than that of 316L SS (Fig. S2B†), revealing a more elongated, unidirectional, and polarized shape. This indicated that HUVECs grown on PDAM/HD surfaces had a larger spread area and lower shape index (0 for a straight line and 1 for perfectly round) and suggested the accelerated cell spreading and development of a cell cytoskeleton. The significant increase in the cell area coverage (Fig. S2C†) and cell proliferation (Fig. 6C) well confirmed these results. Elongated, unidirectional, and polarized arrangement of cells generally is an indicator for increased proliferation and extracellular matrix production.40,41 The above data thus indicate that the PDAM/HD coatings significantly enhanced cell adhesion and proliferation (Fig. 6, and S2†) and good cytocompatibility. Besides, there were no significant differences in HUVEC attachment and proliferation between PDAM and PDAM/HD-48h coating (Fig. S3†).
Good tissue compatibility is also demanded for an implanted biomedical device. As the local tissue response may influences its safety and performance in vivo.42 After the implantation, the body cannot clear the implant by natural mechanisms from the tissue site.43 Thus, the implant alters the local tissue response towards inflammatory reactions and forms fibrous encapsulation. This capsule is compact and usually separates the implant from host tissue sites. Granulation tissue is the precursor to fibrous encapsulation, which is isolated from the implant by cellular components.42,44,45 Higher infiltration of inflammatory cells, stronger development of granulation tissue and thicker fibrous encapsulation indicate a more serious tissue response.42,46,47
Since macrophage is an important kind of inflammatory cell associated with tissue responses and their activities are closely related to the immune system, the in vitro interaction of the PDAM/HD surface with macrophages was evaluated for the first time.48 The results showed that the PDAM/HD modified 316L SS has an anti-inflammatory effect on macrophages (Fig. S4†). The amount of adherent macrophages has been reduced and the cytokine interleukin-6 (IL-6) release has been down-regulated. Then, to fully rate the tissue response to PDAM/HD coating, samples were implanted subcutaneously for implantation in rabbits. After 2 and 9 weeks, the tissue around the implant site was harvested for in situ analysis. Hematoxylin and eosin (HE) (Fig. 7A) and Masson's trichrome staining (Fig. 7B) of sections were performed for histological analysis. Neither bare 316L SS nor PDAM/HD coated 316L SS induced local toxic reactions. There were less inflammatory cell infiltration and granulation tissue development presented on PDAM/HD coated 316L SS. The introduction of PDAM/HD altered the tissue response, but did not completely suppress fibrous capsule formation. For the control 316L SS, the thickness of fibrous encapsulation surrounding the implanted site was 160 ± 34 μm (Fig. 7B1) at 2 weeks. For PDAM/HD coated 316L SS, the thickness reduced to 109 ± 29 μm (Fig. 7B2), suggesting a milder tissue response. As the implantation time prolonged to 9 weeks, fibrous encapsulation thickness of the control 316L SS and PDAM/HD coated 316L SS reduced to 78 ± 11 μm and 73 ± 8 μm, respectively (Fig. 7B3 and B4). It is included that the PDAM/HD coating showed an acceptable and attenuated tissue response compared to 316L SS, with less inflammatory cell infiltration, granulation tissue formation and thinner fibrous capsule development. This is a valuable characteristic for a functional coating, used for surface modification of biomedical devices.
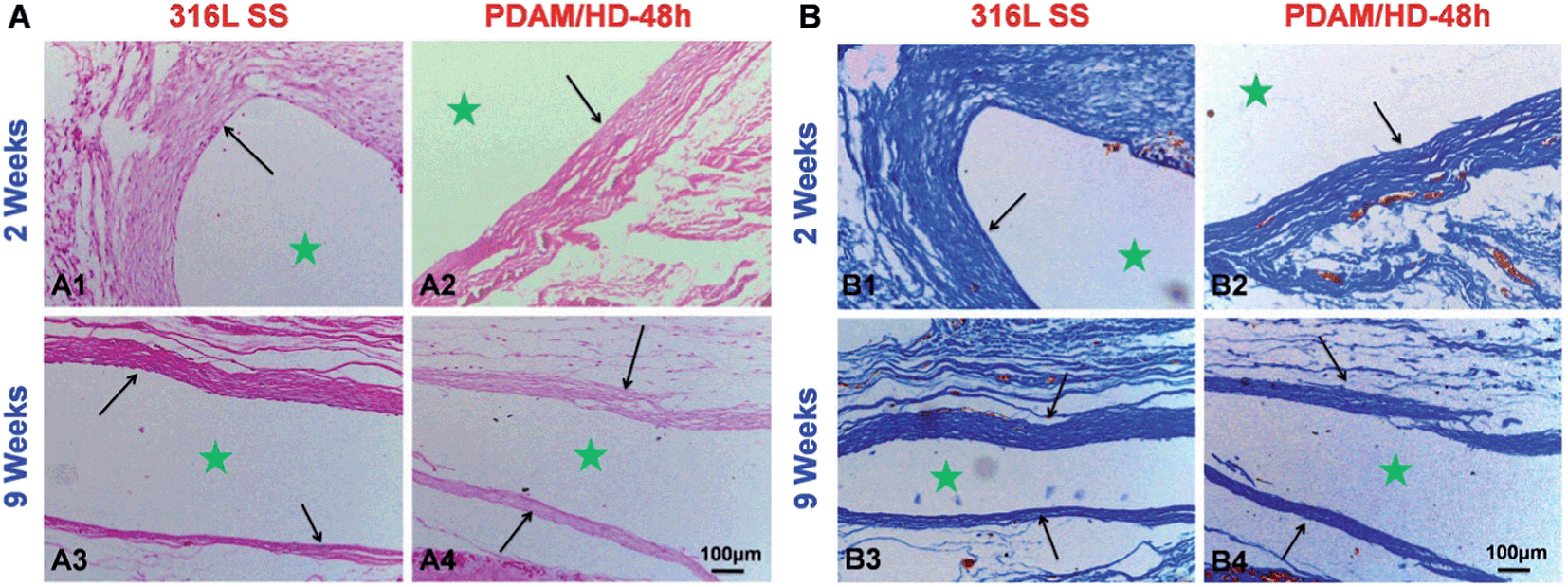 | ||
| Fig. 7 (A) HE and (B) Masson's trichrome staining for subcutaneous tissues around bare and PDAM/HD coated 316L SS implants after 2 and 9 weeks, respectively. ★ Represents the implant site. | ||
Subsequently, the chemical reactivity of the primary amine groups on the PDAM/HD coating was demonstrated through the covalent immobilization of heparin using the carbodiimide (EDC/NHS) coupling procedure.28 Here, XPS analysis, the quantitative determination of the amount of heparin bound to the PDAM/HD coatings and APTTs test were carried out to confirm the immobilization of heparin. XPS results (Fig. 8A and B) revealed the presence of a new peak for S2p (168.5 eV) on the Hep-PDAM/HD-48h that did not exist on the PDAM/HD-48h. The immobilization of heparin was also confirmed by a remarkable change of the chemical compositions of the PDAM/HD coating, seen in a significant decrease in carbon and increase in oxygen and sulfur contents (Table S2†). There was more heparin conjugated on the PDAM/HD coatings than PDAM (Fig. 8C). It was found that the amount of heparin bound to the PDAM/HD coatings was a function of the density of the amine groups of the PDAM/HD coatings (Fig. 8C, and 1C). The highest amount of heparin bound to the PDAM/HD-48h coating reached 900 ng cm−2. The conjugation of heparin on the PDAM/HD coatings prolonged the activated partial thromboplastin time (APTT).49 Meanwhile, the prolonging of APTT was a function of the amount of heparin bound to PDAM/HD coatings. The Hep-PDAM/HD-48h coating had significantly prolonged 16 s of APTT, indicating that the immobilized heparin on the PDAM/HD-48h coating well retained its biological activity (Fig. 8D).
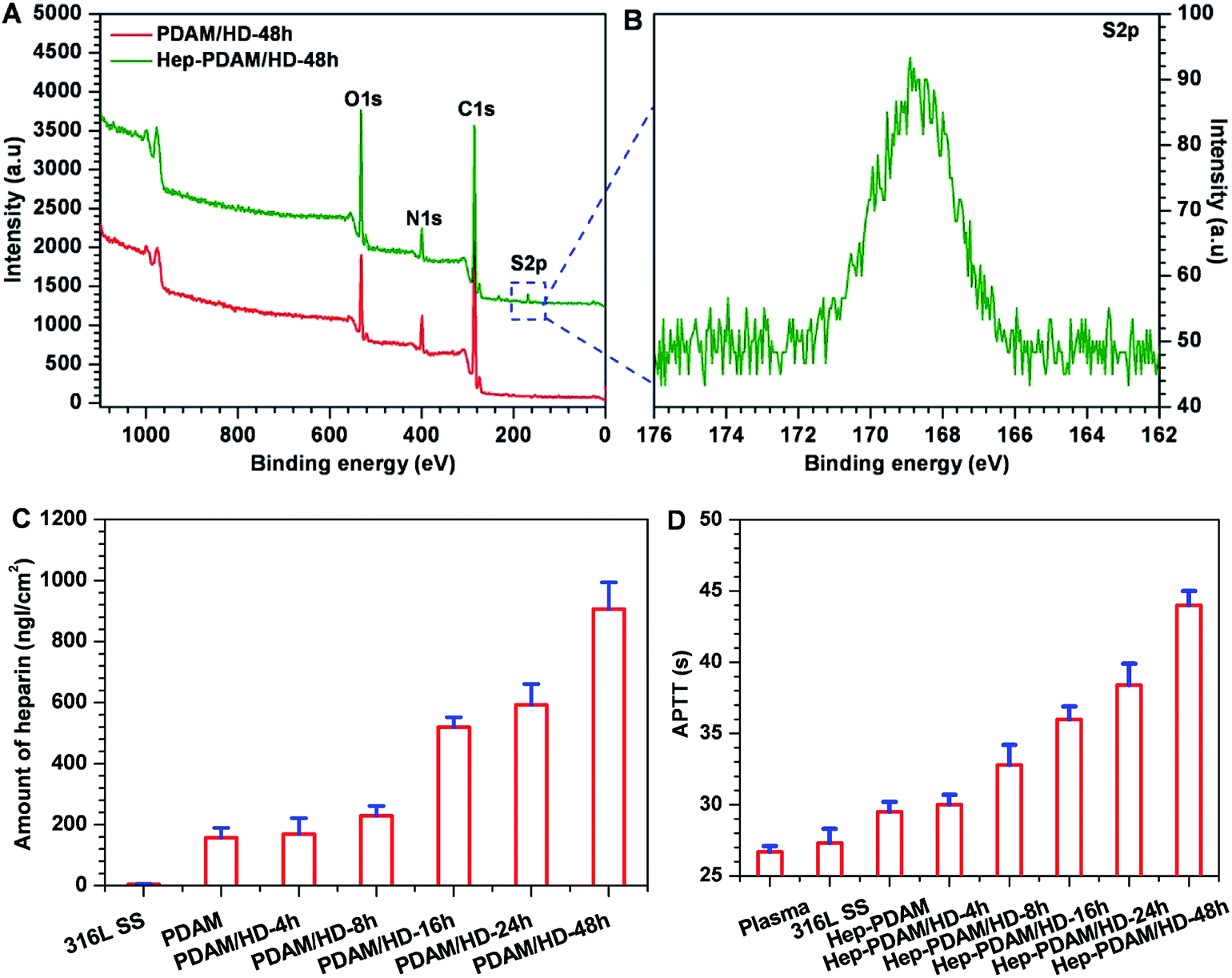 | ||
| Fig. 8 (A) XPS wide scans of the PDAM/HD-48h coating and heparin conjugated PDAM/HD-48h coating; (B) S2p high-resolution spectra of heparin conjugated PDAM/HD-48h coating; (C) the amounts of heparin bound to PDAM and PDAM/HD coatings; (D) APTTs of plasma, 316L SS, PDAM and PDAM/HD coating after heparin conjugation. Data presented as mean ± SD and analyzed using a one-way ANOVA, ***p < 0.001. | ||
4. Conclusions
In this work, a functional adherent amine-rich coating was prepared based on dopamine polymerization. The technique only consisted of a simple one-step dip-coating of the substrates in an aqueous solution of dopamine and HD. Using the technique of PDAM/HD coating, a high density of the amine groups of about 30 nmol cm−2 was obtained on the substrate surface. The PDAM/HD adherent coating not only showed good mechanical properties and stability, but also beneficial biocompatibility for HUVECs. Additionally, the tissue response to PDAM/HD coating was acceptable and attenuated considering the inflammatory response and fibrous encapsulation as compared to 316L SS. More important, the primary amine groups of the PDAM/HD coating can be used to effectively immobilize biomolecules containing carboxylic groups e.g. heparin. These results suggest the promising potential of this PDAM/HD coating for application in the surface modification of biomedical devices.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Project 81271701 and 51173149), the Ministry of Science and Technology of China (Key Basic Research Project no. 2011CB606204), and NSFC Key Program 81330031.Notes and references
- C. D. Hodneland, Y. S. Lee, D. H. Min and M. Mrksich, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5048–5052 CrossRef CAS PubMed.
- D. E. Robinson, A. Marson, R. D. Short, D. J. Buttle, A. J. Day, K. L. Parry, M. Wiles, P. Highfield, A. Mistry and J. D. Whittle, Adv. Mater., 2008, 20, 1166–1169 CrossRef CAS.
- A. B. Sanghvi, K. P. H. Miller, A. M. Belcher and C. E. Schmidt, Nat. Mater., 2005, 4, 496–502 CrossRef CAS PubMed.
- J. S. Lee, A. J. W. Johnson and W. L. Murphy, Adv. Mater., 2010, 22, 5494–5498 CrossRef CAS PubMed.
- K. Alberti, R. E. Davey, K. Onishi, S. George, K. Salchert, F. P. Seib, M. Bornhäuser, T. Pompe, A. Nagy, C. Werner and P. W. Zandstra, Nat. Methods, 2008, 5, 645–650 CrossRef CAS PubMed.
- Z. L. Yang, J. Wang, R. F. Luo, M. F. Maitz, F. J. Jing, H. Sun and N. Huang, Biomaterials, 2010, 31, 2072–2083 CrossRef CAS PubMed.
- Z. L. Yang, Q. F. Tu, J. Wang and N. Huang, Biomaterials, 2012, 33, 6615–6625 CrossRef CAS PubMed.
- H. Kerdjoudj, C. Boura, V. Moby, K. Montagne, P. Schaaf, J. C. Voegel, J. F. Stoltz and P. Menu, Adv. Funct. Mater., 2007, 17, 2667–2673 CrossRef CAS.
- A. Gigout, J. C. Ruiz, M. Wertheimer, M. Jolicoeur and S. Lerouge, Macromol. Biosci., 2011, 11, 1110–1119 CrossRef CAS PubMed.
- S. Pernagallo, O. Tura, M. Wu, K. Samuel, J. J. Diaz-Mochon, A. Hansen, R. Zhang, M. Jackson, G. J. Padfield, P. W. F. Hadoke, N. L. Mills, M. L. Turner, J. P. Iredale and D. C. Hay, Adv. Healthcare Mater., 2012, 5, 646–656 CrossRef PubMed.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- M. C. Xu, Y. F. Zhang, D. Zhai, J. Chang and C. T. Wu, Biomater. Sci., 2013, 1, 933–941 RSC.
- C. T. Wu, W. Fan, J. Chang and Y. Xiao, J. Mater. Chem., 2011, 21, 18300–18307 RSC.
- C. T. Wu, P. P. Han, X. G. Liu, M. C. Xu, T. Tian, J. Chang and Y. Xiao, Acta Biomater., 2014, 10, 428–438 CrossRef CAS PubMed.
- S. M. Kang, N. S. Hwang, J. Yeom, S. Y. Park, P. B. Messersmith, I. S. Choi, R. Langer, D. G. Anderson and H. Lee, Adv. Funct. Mater., 2012, 14, 2949–2955 CrossRef PubMed.
- K. Yang, J. S. Lee, J. Kim, Y. B. Lee, H. Shin, S. H. Um, J. B. Kim, K. I. Park, H. Lee and S. W. Cho, Biomaterials, 2012, 33, 6952–6964 CrossRef CAS PubMed.
- H. Lee, J. Rho and P. B. Messersmith, Adv. Mater., 2008, 20, 1–4 CrossRef.
- I. You, S. M. Kang, Y. Byun and H. Lee, Bioconjugate Chem., 2011, 22, 1264–1269 CrossRef CAS PubMed.
- T. G. Kim, H. Lee, Y. Jang and T. G. Park, Biomacromolecules, 2009, 10, 1532–1539 CrossRef CAS PubMed.
- S. H. Ku and C. B. Park, Biomaterials, 2010, 31, 9431–9437 CrossRef CAS PubMed.
- H. O. Ham, Z. Q. Liu, K. H. A. Lau, H. Lee and P. B. Messersmith, Angew. Chem., Int. Ed., 2011, 50, 732–736 CrossRef CAS PubMed.
- S. H. Ku, J. Ryu, S. K. Hong, H. Lee and C. B. Park, Biomaterials, 2010, 31, 2535–2541 CrossRef CAS PubMed.
- P. L. Bruce, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef PubMed.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef CAS.
- N. F. D. Vecchia, R. Avolio, E. M. E. AlfèM and D. M. Napolitano A, Adv. Funct. Mater., 2013, 23, 1331–1340 CrossRef.
- Y. L. Liu, K. L. Ai and L. H. Lu, Chem. Rev., 2014, 9, 5057–5115 CrossRef PubMed.
- G. C. Li, P. Yang, Y. Z. Liao and N. Huang, Biomacromolecules, 2011, 12, 1155–1168 CrossRef CAS PubMed.
- Z. L. Yang, Q. F. Tu, M. F. Maitz, S. Zhou, J. Wang and N. Huang, Biomaterials, 2012, 33, 7959–7971 CrossRef CAS PubMed.
- Z. L. Yang, Q. F. Tu, Y. Zhu, R. F. Luo, X. Li, Y. C. Xie, M. F. Maitz, J. Wang and N. Huang, Adv. Healthcare Mater., 2012, 1, 548–559 CrossRef CAS PubMed.
- K. Y. Cai, M. Frant, J. Bossert, G. Hildebrand, K. Liefeith and K. D. Jandt, Colloids Surf., B, 2006, 5, 1–8 CrossRef PubMed.
- F. Bernsmann, V. Ball, F. Addiego, A. Ponche, M. Michel, J. J. D. Gracio, V. Toniazzo and D. Ruch, Langmuir, 2011, 27, 2819–2825 CrossRef CAS PubMed.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Langmuir, 2012, 28, 6428–6435 CrossRef CAS PubMed.
- B. D. Ratner, A. S. Hoffman, F. J. Schoen and J. E. Lemons, Biomaterials Science: An Introduction to Materials in Medicine, Academic, San Diego, 1996, 2, pp. 60–64 Search PubMed.
- S. Hong, J. Kim, Y. S. Na, J. Park, S. Kim, K. Singha, G. Im, D. K. Han, W. J. Kim and H. Lee, Angew. Chem., Int. Ed., 2013, 52, 9187–9191 CrossRef CAS PubMed.
- O. C. J. Van, L. Lu, M. K. Chaudhury and R. J. Good, J. Colloid Interface Sci., 1989, 128, 313–319 CrossRef.
- Z. Zhang, Q. Chen, W. Knoll and R. Förch, Surf. Coat. Technol., 2003, 174, 588–590 CrossRef.
- S. Hong, K. Y. Kim, H. J. Wook, S. Y. Park, K. D. Lee, D. Y. Lee and H. Lee, Nanomedicine, 2011, 6, 793–801 CrossRef CAS PubMed.
- Y. H. Ding, Z. L. Yang, C. W. C. Bi, Y. Yang, J. C. Zhang, S. L. Xu, X. Lu, N. Huang, P. B. Huang and Y. Leng, J. Mater. Chem. B, 2014, 2, 3819–3829 RSC.
- M. Tanaka, A. Takayama, E. Ito, H. Sunami, S. Yamamoto and M. Shimomura, J. Nanosci. Nanotechnol., 2007, 7, 763–772 CrossRef CAS PubMed.
- J. L. McGrath, E. A. Osborn, Y. S. Tardy, C. F. Dewey and J. H. Hartwig, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6532–6537 CrossRef CAS PubMed.
- M. A. James, R. Analiz and T. C. David, Semin. Immunol., 2008, 20, 86–100 CrossRef PubMed.
- M. N. Avula, A. N. Rao, L. D. McGill, D. W. Grainger and F. Solzbacher, Acta Biomater., 2014, 10, 1856–1863 CrossRef CAS PubMed.
- J. M. Anderson, Annu. Rev. Mater. Res., 2001, 31, 81–110 CrossRef CAS.
- J. M. Anderson, Curr. Opin. Hematol., 2000, 7, 40–47 CrossRef CAS PubMed.
- C. Gretzer, L. Emanuelsson, E. Liljensten and P. Thomsen, J. Biomater. Sci., Polym. Ed., 2006, 17, 669–687 CrossRef CAS PubMed.
- S. M. Jay, E. A. Skokos, J. Zeng, K. Knox and T. R. Kyriakides, J. Biomed. Mater. Res., Part A, 2010, 93, 189–199 Search PubMed.
- Z. Xia and J. T. Triffitt, Biomed. Mater., 2006, 1, R1 CrossRef CAS PubMed.
- G. C. Li, P. Yang, W. Qin, M. F. Maitz, S. Zhou and N. Huang, Biomaterials, 2011, 32, 4691–4703 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tb01236d |
| This journal is © The Royal Society of Chemistry 2015 |