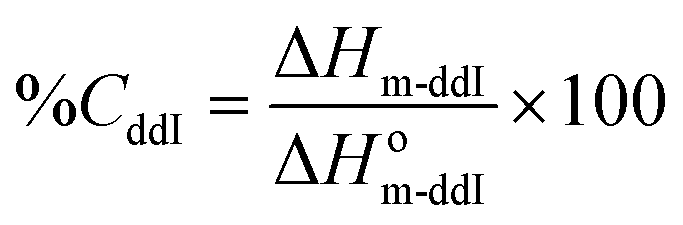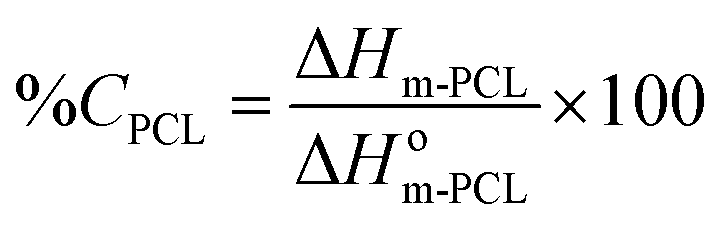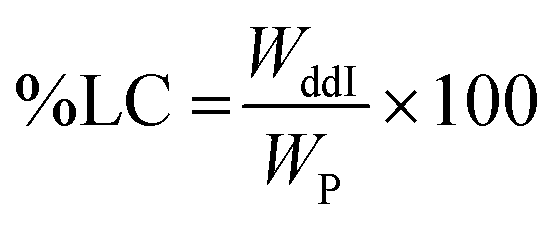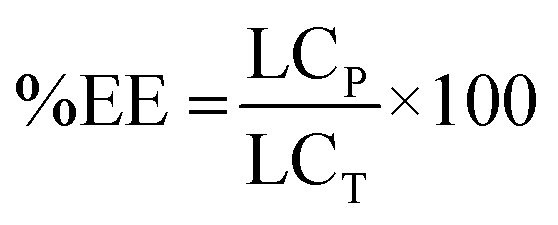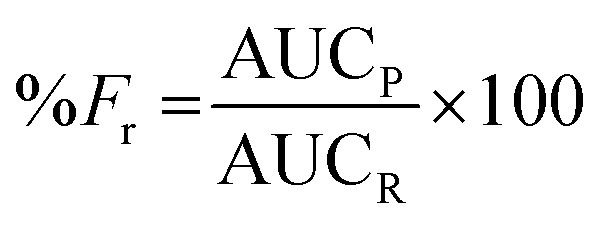Didanosine-loaded poly(epsilon-caprolactone) microparticles by a coaxial electrohydrodynamic atomization (CEHDA) technique†
Received
26th April 2014
, Accepted 12th October 2014
First published on 15th October 2014
Abstract
The goal of this study was to investigate the electrohydrodynamic atomization (EHDA) technology to encapsulate the water-soluble antiretroviral didanosine (ddI) within poly(epsilon-caprolactone) (PCL) particles and stabilize it in the gastric medium where it undergoes fast degradation. A preliminary study employing a one-needle setup enabled the adjustment of the critical process parameters. Then, a configuration of two concentric needles named coaxial electrohydrodynamic atomization (CEHDA) led to the formation of ddI-loaded PCL microcapsules. Scanning electron microscopy analysis showed that the microparticles were spherical and with narrow size distribution. Attenuated total reflectance/Fourier transform infrared spectroscopy confirmed that most of the drug was efficiently encapsulated within the particles, whereas differential scanning calorimetry and X-ray powder diffraction revealed that the drug was preserved mainly in crystalline form. The loading capacity was relatively high (approximately 12% w/w), and the encapsulation efficiency was approximately 100%. In vitro release assays (PBS pH = 7.4) indicated that ddI was released almost completely within 2 h. Moreover, the delayed release was expected to isolate ddI from the biological fluids during the gastric transit. Finally, pharmacokinetics studies in rats showed that ddI-loaded particles lead to a statistically significant increase of the oral bioavailability of almost 4 times and a 2-fold prolongation of the half-life with respect to a ddI aqueous solution, supporting the use of CEHDA as a promising reproducible, scalable and cost-viable technology to encapsulate water-soluble drugs within polymeric particles.
1. Introduction
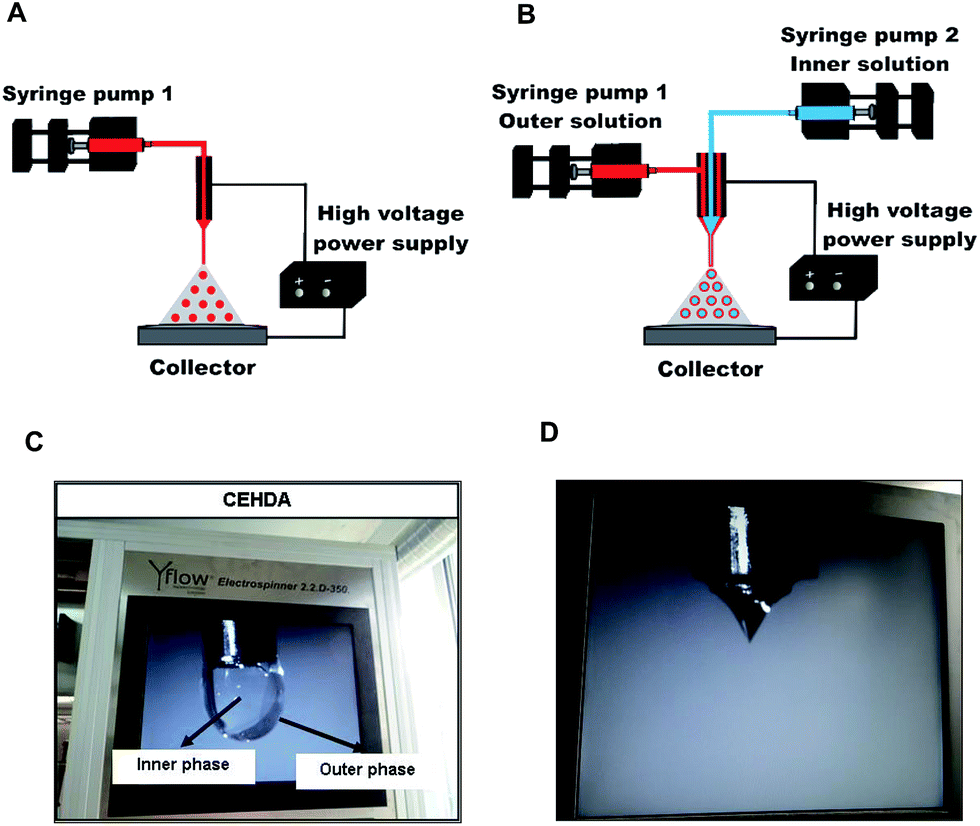
Electrohydrodynamic atomization (EHDA) or electrospraying is a one-step technique with the capability to produce micrometer or sub-micrometer particles with narrow size distribution. EHDA is based on the ability of an electric field to deform the interface of a liquid droplet exiting a capillary.1–4 In a conventional EHDA setup (Fig. 1A), a polymeric liquid is infused at a low flow rate using a programmable syringe pump through a capillary needle connected to a high-voltage supply that provides an electrical potential difference (5–30 kV).2,5–7 When appropriate values of applied electric voltage and flow rate are used, the liquid meniscus at the tip of the needle develops a symmetric conical shape (Taylor cone); the electric charge generates an electrostatic force inside the droplet (Coulomb force) that competes with its surface tension. Thus, a thin jet of liquid bearing a high charge density emerges from the vertex cone and undergoes break up into finer droplets that are subjected to solvent evaporation in their flight to the collector, resulting in the formation of dense solid particles.1,2,8–10 The collector is connected at ground or negative voltage to attract the formed particles.1,2,11 EHDA is affected by a multitude of parameters such as electric field intensity, fluid conductivity, solvent volatility, surface tension, density and viscosity of the sprayed solution, polymer molecular weight and concentration, flow rate, needle diameter and distance to the collector.2,12 Coaxial electrohydrodynamic atomization (CEHDA) is a modification of the original method, in which two electrified concentric needles are used to flow two usually immiscible liquids and produce core/shell structures8,13–15 (Fig. 1B). In the dripping mode, before applying the voltage at the needles, the formation of concentric droplets with the outer liquid surrounding the inner one can be observed (Fig. 1C). After the application of a voltage to both needles and a suitable outer/inner flow rate, a Taylor cone composed of an inner and an outer phase is formed on the needle's tip16,17 (Fig. 1D) that results in the formation of capsules.13,14,18 A remarkable advantage of EHDA and CEHDA over conventional techniques for the production of particles is that droplets repel each other, and therefore, particle aggregation and coalescence can be minimized.6,19,20 Moreover, the process can be conducted at ambient temperature and atmospheric pressure under a continuous production setup; drug-loaded particles with controlled size, nearly monodisperse size distribution and high reproducibility are obtained. Furthermore, these techniques are particularly appealing for the encapsulation of water-soluble drugs because they do not require large aqueous phase volumes (such as in double water-in-oil-in-water emulsions) that favor the migration of the drug. Consequently, a higher drug encapsulation efficiency (%EE) is attained.1,16,21,22
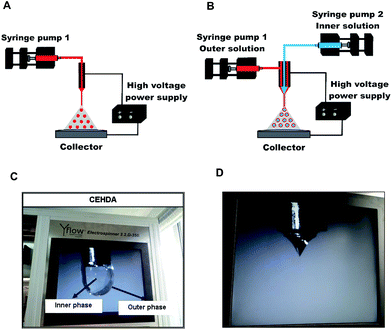 |
| | Fig. 1 (A) EHDA experimental setup, (B) CEHDA experimental setup, (C) dripping mode (core/shell structure) in CEHDA technique, and (D) formation of Taylor cone in CEHDA. | |
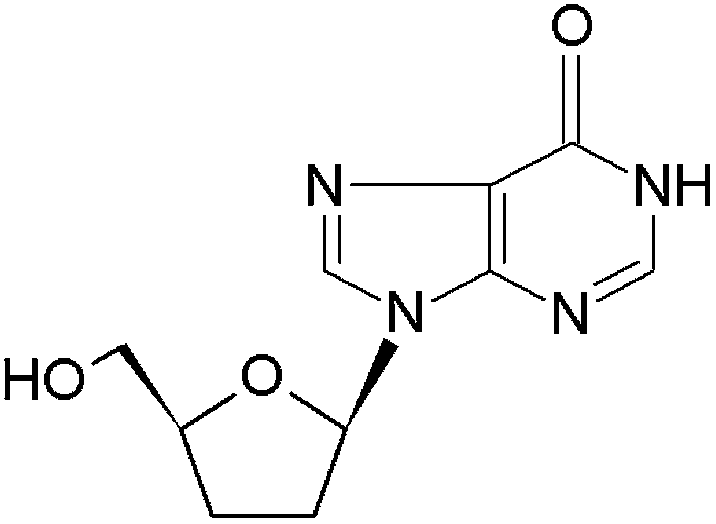
Didanosine (ddI) (Fig. 2) is a water-soluble first-line nucleoside reverse transcriptase inhibitor used in the therapy of the human immunodeficiency virus (HIV) infection.23–26 The most relevant biopharmaceutical drawback of ddI is a low oral bioavailability (20–40%) due to the fast hydrolysis to an inactive hypoxanthine under gastric pH.23,27,28 In addition, ddI displays a short plasma half-life of 1–2 h.23–25 In this context, ddI is administered in buffered formulations containing antacids that partially neutralize the gastric medium and prevent its hydrolysis.25 However, the chronic administration of large doses of buffering salts provokes diarrhea and renal problems and increases the inter-individual variability.29–31 Furthermore, they can reduce the absorption of other antiretrovirals (e.g., protease inhibitors) co-administered with ddI in the therapeutic cocktail.31 Gastro-resistant ddI-loaded granules are commercially available, but the relatively high cost of this product limits patient affordability in constrained-setting countries.32 Therefore, additional efforts are urgently called for to develop innovative dosage forms produced by scalable and cost-effective processes.33,34
 |
| | Fig. 2 Chemical structure of didanosine. | |
Aiming to investigate more efficient methods for the encapsulation of water-soluble drugs, the present work studied for the first time the encapsulation of ddI within polymeric microparticles by a scalable electrospraying method. After the thorough study of the critical parameters of the process, the characterization of the properties of the different systems in vitro and the performance in vivo were assessed with promising results.
2. Materials and methods
2.1. Materials
Poly(epsilon-caprolactone) with molecular weights of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 (PCL14) and 80000 g mol−1 (PCL80), potassium phosphate monobasic, sodium hydroxide, zinc sulfate and sodium acetate were purchased from Sigma-Aldrich (St. Louis, MO, USA). ddI was donated by Richmond Laboratories (Buenos Aires, Argentina). Dichloromethane (DCM), N,N-dimethylacetamide (DMA), methanol (MeOH) and acetonitrile (ACN) were of analytical or HPLC grade and used as received (Sintorgan, Buenos Aires, Argentina).
000 g mol−1 (PCL14) and 80000 g mol−1 (PCL80), potassium phosphate monobasic, sodium hydroxide, zinc sulfate and sodium acetate were purchased from Sigma-Aldrich (St. Louis, MO, USA). ddI was donated by Richmond Laboratories (Buenos Aires, Argentina). Dichloromethane (DCM), N,N-dimethylacetamide (DMA), methanol (MeOH) and acetonitrile (ACN) were of analytical or HPLC grade and used as received (Sintorgan, Buenos Aires, Argentina).
2.2. Preparation of ddI-free and ddI-loaded particles
The particles were produced using an Electrospinner YFLOW 2.2.D-350 (YFLOW Systems and Development S.L., Malaga, Spain). First, a conventional EHDA process was used to assess the critical parameters for the formation of particles instead of fibers: PCL molecular weight, polymer concentration, needle tip-to-collector distance (TTC) and positive and negative voltage. Then, a CEHDA process was used to encapsulate ddI.
2.2.1. Electrohydrodynamic atomization (EHDA).
To assess the effect of the polymer molecular weight and concentration on the formation and the morphology of the particles, a high and low molecular weight PCL were used at the following concentrations: PCL80 (5% and 7.5% w/v) and PCL14 (10%, 12.5% and 15% w/v). Briefly, the corresponding amount of polymer was dissolved in a DCM:DMA (75:25, 10 mL) mixture at room temperature for 12 h. This solution was loaded into a 10 mL syringe and infused at constant flow rate (0.50 mL h−1) using a programmable syringe pump through Teflon tubing (outer/inner diameters of 3.00/1.50 mm) attached to a stainless steel blunt needle (length of 40 mm, outer/inner diameters of 0.70/0.40 mm) that was connected to the positive electrode of a high-voltage power supply. The positive voltage was gradually increased to obtain a stable cone-jet mode and then maintained constant during the entire experiment. A monochrome CCD camera allowed a continuous follow-up of the process. The sample was collected on aluminum foil (10 × 10 cm2) placed on a circular aluminum plate (9 cm in diameter) connected to the negative electrode (−4.00 kV). The collector was placed perpendicular to the needle and at a variable distance from the needle tip (25–30 cm) to evaluate the influence of this parameter on particle formation. Finally, the powders obtained were vacuum-dried (48 h) to ensure the elimination of solvent residues, recovered from the foil with a spatula and stored in sealed glass vials protected from light and moisture until use. The process was conducted inside an acrylic chamber at ambient temperature and atmospheric pressure. The conditions used for EHDA are summarized in Table S1.† Scanning electron microscopy (SEM) analysis (mentioned below) was used to preliminarily determine the formation of particles or fibers and adjust the conditions.
2.2.2. Coaxial electrohydrodynamic atomization (CEHDA).
The coaxial needles system consisted of two concentric stainless steel needles with outer/inner diameters of 1.40/1.10 mm and 0.70/0.40 mm. The outer and inner nozzles were used for feeding of the organic and aqueous solution, respectively. Briefly, PCL14 (1 g) was dissolved in the corresponding solvent (10 mL) as described above, placed in a 10 mL syringe and infused at a constant flow rate through Teflon tubing connected to the outer needle. Concomitantly, the aqueous phase (5 mL) without and with ddI (25 mg mL−1) was infused using a second pump through the inner needle. To improve the formation of core-shell particles, additional organic solvents were evaluated: DCM, DCM:ACN (75:25) and DCM:ACN (50:50). The outer/inner flow rates ratio plays an important role in the stability of the cone-jet and the droplet size.17 Two flow rates for the outer phase (1.00 and 0.50 mL h−1) and three flow rates for the inner phase (0.50, 0.25 and 0.10 mL h−1) were tested. The first was fixed when the second was varied and vice versa. Highly positive voltages were applied to the coaxial system until a stable cone-jet mode was formed, and a negative voltage (−4.00 kV) was applied to the collector. The collector was placed at a distance of 30 cm from the needle tips. The experimental conditions assayed in CEHDA are summarized in Table 1. Considering concentrations of the solutions, processing parameters used and the yield, a production rate of approximately 0.02 g h−1 was obtained.
Table 1 Conditions for the production of ddI-free particles by CEHDAa
| Outer phase solvent |
Outer phase flow rate (mL h−1) |
Inner phase flow rate (mL h−1) |
Outer/inner flow ratio |
Needle positive voltage (kV) |
|
Inner phase solvent: distilled water. Polymer solution: PCL14 10% w/v. Needle tip-to-collector distance: 30 cm. Collector voltage: −4.00 kV.
|
| DCM |
1 |
0.50 |
(2/1) |
15.50 |
| 0.25 |
(4/1) |
15.50 |
| 0.50 |
0.25 |
(2/1) |
14.50 |
| 0.10 |
(5/1) |
13.00 |
| DCM:ACN (75:25) |
1 |
0.50 |
(2/1) |
20.50 |
| 0.25 |
(4/1) |
17.50 |
| 0.50 |
0.25 |
(2/1) |
17.50 |
| 0.10 |
(5/1) |
15.00 |
| DCM:ACN (50:50) |
1 |
0.50 |
(2/1) |
18.50 |
| 0.25 |
(4/1) |
18.00 |
| 0.50 |
0.25 |
(2/1) |
17.00 |
| 0.10 |
(5/1) |
18.00 |
| DCM:DMA (75:25) |
1 |
0.50 |
(2/1) |
20.00 |
| 0.25 |
(4/1) |
16.50 |
| 0.50 |
0.25 |
(2/1) |
18.00 |
| 0.10 |
(5/1) |
13.00 |
2.3. Characterization of the particles
2.3.1. Scanning electron microscopy (SEM).
The morphology and the topography of ddI-free particles and ddI-loaded particles were visualized by SEM using a JEOL JSM-6460LV microscope (JEOL USA Ltd., Peabody, MA, USA). Prior to analysis, samples were metallized with a thin layer of gold (Denton Vacuum Desk II Metallizer, Denton Vacuum LLC, Moorestown, NJ, USA).
2.3.2. Attenuated total reflectance/Fourier transform-infrared spectroscopy (ATR/FT-IR).
To qualitatively assess the encapsulation of ddI within the particles, samples were analyzed by ATR/FT-IR (Nicolet 380 ATR/FT-IR spectrometer, Avatar Combination Kit), Smart Multi-Bounce HATR with ZnSe crystal 45° reflectance (Thermo Scientific, Inc., Waltham, MA, USA) in the wavenumber range between 4000–600 cm−1 (15 scans, spectral resolution of 4.0 cm−1). The following samples were studied: (i) pure PCL14 and free ddI, (ii) a representative PCL14–ddI physical mixture containing the same composition of the particles and (iii) ddI-loaded particles. Spectra were obtained using the OMNIC 8 spectrum software (Thermo Scientific, Inc.).
2.3.3. Differential scanning calorimetry (DSC).
The thermal analysis was performed by DSC (Mettler-Toledo TA-400 differential scanning calorimeter, Columbus, OH, USA). Samples (∼5 mg) were sealed in 40 μL Al crucible-pans (Mettler ME-27331, Greifensee, Switzerland). The thermograms of pure PCL14, ddI-free particles and ddI-loaded particles were obtained in a simple heating temperature ramp between 25 and 210 °C (10 °C min−1) under a dry nitrogen atmosphere. The degree of crystallinity (%C) of ddI and PCL14 in the particles after the process was calculated from eqn (1) and (2), respectively:| | 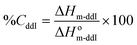 | (1) |
| | 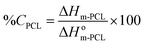 | (2) |
where ΔHm-ddI is the melting enthalpy of ddI, ΔHm-PCL is the melting enthalpy of PCL14 obtained from the thermal analysis of the different samples normalized to the content of each component in that specific sample, and ΔHom-ddI and ΔHom-PCL are the values of melting enthalpy of 100% crystalline ddI and PCL14, respectively. All the enthalpy values are expressed in J g−1.
2.3.4. X-ray powder diffractometry (XRPD).
The crystallinity of different samples was analyzed by XRPD using an X-ray diffractometer (PANalytical X'Pert PRO diffractometer, PANalytical BV, Almelo, The Netherlands), equipped with a PW3710 unit, system of soller slits and monochromator reception. An X-ray generator (Philips PW 1830, PANalytical BV) with a Cu anode, a voltage of 40 kV and a current of 40 mA was used. Diffractograms of pure PCL14, free ddI and ddI-loaded particles were recorded in the 2Θ range 5°–75° with a step angle of 2Θ 0.02° and count time of 1 s at each step.
2.3.5. Loading capacity and encapsulation efficiency.
ddI-loaded particles (10 mg) were suspended in DCM (2 mL) and shaken until complete dissolution of the polymer. Then, phosphate buffer saline (PBS, pH 7.4, 10 mL) was added, and the system was vortexed (30 min) to extract ddI and finally centrifuged (5000 RPM, 1 h) to isolate the aqueous phase that was diluted in PBS to determine the concentration of ddI by UV-visible spectrophotometry (λ = 249 nm, CARY [1E] UV-Visible Spectrophotometer Varian, Varian, Inc., Palo Alto, CA, USA), at 25 °C. A calibration curve was obtained from ddI solutions in PBS with a linearity range between 5 and 30 μg mL−1 (R2 > 0.9997). Measurements were performed in triplicate and results are expressed as mean ± S.D. The statistical method was the unpaired Student's t-test (p < 0.05). The loading capacity (%LC) of the ddI-loaded particles was calculated from eqn (3):| | 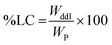 | (3) |
where WddI is the weight of ddI in the particles, and WP is the total weight of particles collected.
The %EE of the ddI-loaded particles was calculated according to eqn (4):
| | 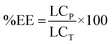 | (4) |
where LC
P is the loading capacity of particles, and LC
T is the theoretical loading capacity of particles.
2.4.
In vitro ddI release
An appropriate amount of particles containing approximately 2.5 mg of ddI was suspended in PBS (pH 7.4, 20 mL) at 37 °C ± 2 and maintained under moderate magnetic stirring (200 RPM) for 120 min. At predetermined time intervals (10 min), an aliquot of the release medium (1 mL) was withdrawn and centrifuged (13000 RPM, 5 min) for ddI analysis and replaced by fresh medium pre-heated at 37 °C. The released ddI amounts were quantified by UV-visible (mentioned above) and corrected by the volume of release medium withdrawn. Assays were carried out in triplicate, and the results are expressed as mean ± S.D.
2.5. Oral pharmacokinetics
Oral bioavailability studies were conducted in male Wistar rats (weighing between 250 and 300 g) under strict controls and according to the guidelines of the “National Institute of Health Guide for the care and use of laboratory animals” publication NIH no. 80-23/96 and national regulations. All efforts were made to prevent the suffering of animals and reduce the number of individuals employed in each experiment. Animals were maintained on 12 h light/dark cycles at 22 ± 2 °C with the air adequately recycled. They received a standard rodent diet and water ad libitum and were fasted overnight (12 h) before the studies. Two types of systems were systematically compared: (i) ddI aqueous solution and (ii) ddI-loaded particles obtained by CEHDA. Each group consisted of four animals and the administered ddI dose and volume were 20 mg kg−1 and 4 mL kg−1, respectively; the ddI concentration in the formulation was 5 mg mL−1. An appropriate amount of particles (containing the amount of the drug according to the required dose per animal weight) was suspended in the corresponding volume of distilled water (4 mL kg−1) and vortexed (5 min) immediately before administration. Formulations were administered to conscious rats through a stomach tube (gavage). At predetermined time intervals (5, 10, 15, 20, 30, 45, 60, 90, 120, 180, 240, 360 and 720 min) blood samples (70 μL) were collected from the tail vein. The total blood volume extracted (approximately 900 μL) was significantly smaller than the maximum recommended (3.5 mL); and therefore, we assume that the volemia decrease did not affect the pharmacokinetic parameters calculated for ddI. The extraction technique of ddI was a modification of a previously reported one.23 Samples were centrifuged to isolate plasma (10000 RPM, 10 min). Then, 20 μL of plasma was deproteinized with MeOH (20 μL) and zinc sulfate (10% w/v, 2 μL) followed by vortex (2 min) and centrifugation (10000 RPM, 10 min). The concentration of the drug in plasma was determined by high performance liquid chromatography (HPLC). For this, an isocratic regime, previously validated, was used with slight modifications24 using a UV detector (λ = 249 nm, UVIS 204 Spectrophotometer, Linear Instruments, Reno, NV, USA) and a Phenomenex Luna 5 μm, C18, 150 mm × 4.60 mm column (Phenomenex® Gemini-NX, Torrance, CA, USA). The mobile phase composed of 0.01 M sodium acetate:MeOH (95:5, pH 6.5) was pumped at a flow rate of 1.2 mL min−1. The calibration curve used to measure the ddI concentration was in a range from 0.16–5 μg mL−1 (R2 > 0.9997).
2.6. Evaluation of in vivo data
Non-compartmental analysis of ddI plasma concentrations was performed using the TOPFIT program (version 2.0, Dr Karl Thomae Gmbh, Schering AG, Gödecke AG, Germany). Parameters were log transformed to reduce heterogeneity of the variance and further compared by unpaired Student's t-test (p < 0.05). Statistical analysis was performed using GraphPad Prism version 6.01 (GraphPad Software, San Diego, CA, USA). The inter-individual variability was assessed by calculating the coefficient of variation (CV%). The following pharmacokinetic parameters were estimated: (i) the maximum plasma concentration (Cmax), (ii) the time to reach the maximum plasma concentration (tmax), (iii) the area-under-the-curve between the administration time and 1 h (AUC0–1), (iv) the area-under-the-curve between the administration time and infinity (AUC0–∞), and (v) the elimination rate constant (ke). The relative oral bioavailability (Fr) was calculated from eqn (5):35| |  | (5) |
where AUCP and AUCR are the AUC0–∞ values of the ddI-loaded particles and the ddI aqueous solution as 100% bioavailability, respectively.
3. Results and discussion
3.1. The rationale
Polymeric microparticles have been shown to protect drug cargos from physicochemical insults in the gastrointestinal tract, modulate their release rate and increase their oral bioavailability.36,37 However, the encapsulation of drugs that are highly water-soluble in a broad pH range, such as ddI (solubility of ∼30 mg mL−1 at pH values below its pKa of 9.12)25,38 within polymeric microparticles employing conventional methods, is usually an inefficient and non-scalable process. Both electrospinning (generation of fibers) and electrospraying (generation of particles) are based on the same principle,12 though the difference between both techniques lies in the density of polymer chains entanglements of the polymer solution.1 Regardless of the great potential of this technique, the production of drug-loaded particles still remains quite an unexplored field when compared to electrospinning.39 PCL is a biodegradable aliphatic polyester that, owing to very good biocompatibility, relatively low cost and easy synthesis, has received a great deal of attention for the production of drug delivery systems40–42 and has been already approved by the US-FDA and the European Medicines Agency for use in pharmaceuticals.43 In this work, we investigated the versatility of electrospraying for the production of ddI-loaded PCL microparticles and assessed the oral bioavailability of the drug. In this framework, we initially explored EHDA to adjust the most critical parameters of the production process. Then, we developed a CEHDA technique that allowed the efficient incorporation of ddI within the core of the polymeric particles.
3.2. Production of ddI-free particles by EHDA
The first step was the selection of a suitable organic solvent or mixture of solvents for the production of particles with the desired morphology. For this, we considered the boiling point (BP, and the evaporation rate) and the electrical conductivity (K) of several organic solvents. Both intrinsic properties have been reported to allow the manipulation of the particle morphology.2,6,44,45 Solvent evaporation from the droplets involves a combination of heat/mass transfer processes.20 A solvent displaying low BP (e.g., DCM, 40 °C) (and fast evaporation rate) would facilitate the formation of particles with porous or hollow structures.11,22,46,47 Conversely, solvents with high BP, such as DMA (166 °C), could result in incomplete evaporation and partial dissolution of semi-solid particles once they reach the collector.1 Regarding K, it is known that an increase in this value leads to a decrease in particle size due to stronger Coulomb repulsion forces and fewer polymer chain entanglements.2,5,6 DCM shows a lower K value (4.3 × 10−11 S cm−1) than ACN (6 × 10−10 S cm−1) and DMA (5.6 × 10−8 S cm−1). Notably, K values that are too low or too high are detrimental in EHDA.2 Therefore, to achieve good solvent evaporation rate as well as an appropriate K, we decided to initially work with a DCM:DMA (75:25) mixture and expected that a decrease of the solvent evaporation rate due to the addition of DMA to DCM would lead to the formation of spherical particles with smooth (and less porous) texture and better release control of the encapsulated cargo.1,12
Polymer concentration is another key parameter to control the process. The formation of particles or fibers depends greatly on the polymer concentration that plays an important role in the chain entanglements regime. When the concentration is high enough, a significant degree of entanglement is achieved, and solid particles are obtained. This regime is denominated, semi-dilute moderately entangled1,2,48 and it was achieved with PCL14 10–15% w/v (Fig. S1†). In contrast, if the degree of entanglement is too high, a semi-dilute highly entangled regime is reached, and beaded fibers or fibers are produced,1,2,48 as exemplified for PCL80 5% and 7.5% w/v in Fig. S2.† Experimental data indicated a strong dependence of the polymer solution viscosity [η] on the polymer molecular weight,48 and these parameters affected the critical chain overlap concentration (Cov). An increase in polymer molecular weight reduces the working window to achieve a reproducible electrospray.2 This phenomenon explains why the use of PCL80, although in a lower concentration than PCL14, did not result in the formation of spherical particles. In advance, PCL14 10% w/v was used to further explore this technology.
An additional parameter that can be used to adjust the extent of solvent evaporation is the TTC distance. A short distance can prevent complete solvent evaporation, resulting in wet particles that collapse and/or coalesce in the collector. In contrast, an increase of the TTC distance leads to particles with more spherical morphology because the polymer chains have enough time to diffuse within the droplet.1,2 In this work, we did not observe substantial differences between TTC distances of 25 and 30 cm. Thus, the greatest TTC distance was selected to ensure a more complete evaporation of the organic solvents, particularly the one with the highest BP. In all the cases, the application of a voltage with opposite polarity on the collector was meant to concentrate the particles after formation. Despite the positive results obtained with drug-free systems, significant differences in the solubility of the hydrophilic drug and the hydrophobic polymer made the adjustment of the solvent system very difficult. Thus, the more versatile CEHDA technique was pursued.
3.3. Production of ddI-free and ddI-loaded particles by CEHDA
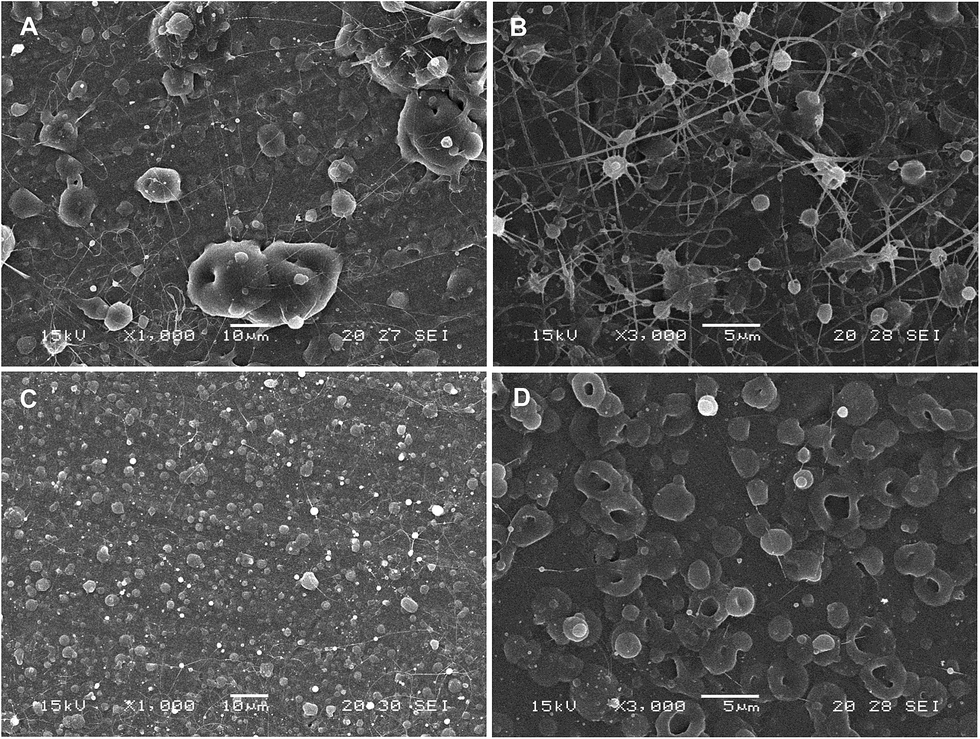
Due to a greater number of critical parameters, CEHDA is a more complex and considerably less popular micro/nanoencapsulation technique than EHDA,9,14,17 and a relatively short history can be tracked in the previously reported scientific studies.39 At the same time, it emerges as a very promising tool to quantitatively encapsulate water-soluble drugs.17 In EHDA feasibility studies, we established the appropriate polymer molecular weight and concentration (PCL14 10% w/v), collector voltage (−4.00 kV) and TTC distance (30 cm). To gain further insight into the versatility of the technique, in CEHDA, other organic solvent systems with different BP and K values were explored. The use of DCM as the outer phase resulted in large porous particles due to the fast evaporation of the solvent and the incomplete diffusion of the polymer within the droplet (Fig. 3A). These results were in agreement with the previously published reports.2,49,50 However, when a DCM:DMA (75:25) mixture was used, particles coalesced, and beaded fibers and fibers were formed (Fig. 3B). This is an interesting observation because the same solvent mixture led to the formation of dense solid particles with narrow size distribution in EHDA. This phenomenon would be associated with the very high K value of DMA that destabilized the CEHDA system containing an internal water phase with the K value (5.5 × 10−8 S cm−1) that was very similar to that of this organic solvent. In this context, we replaced DMA by ACN, a solvent with lower BP and K values, resulting in non-porous spherical particles (Fig. 3C). Conversely, as explained above, a higher K value of the organic phase due to a greater content of ACN made the system instable, as observed for DCM:ACN (50:50) (Fig. 3D). Thus, only when the relative ACN content in the DCM:ACN mixture was reduced from 50% to 25%, spherical and monodisperse particles were obtained (Fig. 3C). In this configuration, water served as the driving liquid and played the dominant role in the formation of the jet and therefore, governed the generation of uniform particles.17,51
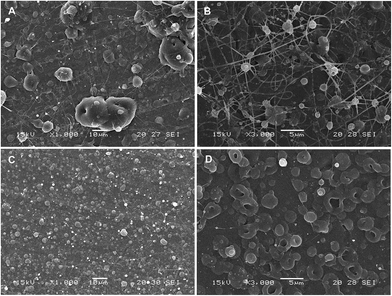 |
| | Fig. 3 SEM micrographs of particles obtained by CEHDA with distilled water as the inner phase and PCL14 10% w/v in (A) DCM, (B) DCM:DMA (75:25), (C) DCM:ACN (75:25) and (D) DCM:ACN (50:50) as the outer phase. Outer/inner flow rate: 0.50/0.25 mL h−1. Scale bar: (A) and (C) = 10 μm; (B) and (D) = 5 μm. | |
In CEHDA, the outer/inner flow rate ratio is another parameter affecting the size and the morphology of the particles.51 To achieve a stable process, usually the flow rate of the inner solution is slower than that of the outer one.52 In general, an intermediate-to-low flow rate that allows the complete evaporation of the solvent, prevents particle coalescence and results in smaller particle sizes is preferred.2,17,53,54 On the other hand, higher flow rates would increase the production rate. Despite the organic solvent in this work, high flow rates of the outer phase (1.00 mL h−1) led to agglomerated and deformed particles. This is a drawback to scale-up the process. In this context, multiplexed configurations have been developed to overcome the low production rate of the single-jet mode.5,22,39
The %yield (ratio between the mass obtained after the process and the total mass of polymer and drug used, considering flow rate and collection time) was 37.1% ± 3.2%. This result was in very good agreement with previous reports and stems from the loss of particles that stick to the chamber walls, and thus do not reach the collector.55 To improve the production yield, modified devices are currently under development.39 This constitutes a fundamental step to enable the process scale-up and the implementation of this new technology under an industrial setting. Based on the morphological analysis (SEM), the following experimental conditions were established for the production of drug-loaded particles with more spherical morphology and nearly monodisperse size distribution: DCM:ACN (75:25) as outer phase and outer/inner flow rate of 0.50/0.25 mL h−1. Subsequently, ddI-loaded particles were produced by incorporating ddI (25 mg mL−1) into the inner phase solution. SEM analysis showed the generation of spherical particles with narrow size distribution and deprivation of ddI crystals on their surface (Fig. 4). These results strongly suggested the effective encapsulation of the drug.
 |
| | Fig. 4 SEM micrograph of ddI-loaded particles obtained by CEHDA using PCL14 10% w/v in DCM:ACN (75:25) as the outer phase. Outer/inner flow rate: 0.50/0.25 mL h−1. Scale bar = 10 μm. | |
3.4. Characterization of ddI-loaded particles
3.4.1. Loading capacity and encapsulation efficiency.
The %LC and %EE were quite high with values of 12.2% ± 0.2% and 96.8% ± 3.2%, respectively. These results confirmed the high potential of CEHDA for the encapsulation of hydrophilic drugs, such as ddI, as opposed to conventional single and double emulsion techniques that resulted in extremely low performances (data not shown). Pertaining to reproducibility, the non-statistically significant differences (p < 0.05) of %LC, %EE and %yield obtained for five independent batches confirmed that once the conditions are established the method is highly reproducible.
3.4.2. ATR/FT-IR analysis.
To reveal the presence of free drug on the surface of the particles, ddI-loaded particles were analyzed by ATR/FT-IR spectroscopy and compared to free ddI, pure PCL14 and a representative PCL14–ddI physical mixture, containing the quantitative composition of the particles. Free ddI showed most of the characteristic bands of hypoxanthine and tetrahydrofurfuryl alcohol: (i) C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1705 cm−1 characteristic of the purine base, (ii) C–N stretching at 1213 cm−1 characteristic of conjugation between the purine base and tetrahydrofurfuryl alcohol and (iii) C–O–C stretching at 1102 and 1062 cm−1 of tetrahydrofurfuryl alcohol.25 Pure PCL14 showed the band of CO at 1725 cm−1.56,57 The physical mixture showed the characteristic bands of both the drug and the polymer, indicating that the drug was detectable by this technique. Spectra of ddI-loaded particles showed only the characteristic bands of the polymer, e.g., the CO stretching was observed without any shifting at 1725 cm−1, indicating that the production process did not alter the degree of crystallinity of PCL14. In addition, the absence of characteristic ddI bands indicated the effective encapsulation. These findings were in full agreement with SEM analysis.
O stretching at 1705 cm−1 characteristic of the purine base, (ii) C–N stretching at 1213 cm−1 characteristic of conjugation between the purine base and tetrahydrofurfuryl alcohol and (iii) C–O–C stretching at 1102 and 1062 cm−1 of tetrahydrofurfuryl alcohol.25 Pure PCL14 showed the band of CO at 1725 cm−1.56,57 The physical mixture showed the characteristic bands of both the drug and the polymer, indicating that the drug was detectable by this technique. Spectra of ddI-loaded particles showed only the characteristic bands of the polymer, e.g., the CO stretching was observed without any shifting at 1725 cm−1, indicating that the production process did not alter the degree of crystallinity of PCL14. In addition, the absence of characteristic ddI bands indicated the effective encapsulation. These findings were in full agreement with SEM analysis.
3.4.3. Thermal analysis.
DSC is an appropriate technique to elucidate the crystalline or amorphous nature of the encapsulated drug.43 ddI showed a sharp endothermic peak at 185 °C (ΔHm = 116.5 J g−1) (Table 2). This %CddI was considered 100% crystallinity.25 Pure PCL14 displayed the typical melting endotherm at 61 °C (ΔHm = 100.3 J g−1) that represented a %CPCL of 71.9% (Table 2); a melting enthalpy value of 139.5 J g−1 was considered for 100% crystalline PCL.56 These results were in agreement with the previously studied reports.56,58,59 ddI-free particles showed the characteristic melting endotherm of PCL14 at 56 °C (ΔHm = 87.1 J g−1), the thermal behavior being similar to that of PCL14 though with a slightly lower crystallinity of 62.4% (Table 2). When the drug-loaded particles were analyzed, PCL14 showed an additional decrease of Tm to 55 °C and of the related enthalpy to 84.0 J g−1 (%C = 60.2%), indicating that ddI partially hindered its crystallization. In the case of ddI, the Tm was identified at 191 °C (ΔHm = 102.4 J g−1), and the %C was 87.9%. The high crystallinity of the encapsulated drug stemmed from the fact that due to the use of CEHDA, it was not encapsulated within a polymer matrix that might prevent its crystallization but within the core of a hollow polymeric capsule.
Table 2 Thermal analysis of ddI-free particles and ddI-loaded particles obtained by CEHDA
| Sample |
T
m (°C) |
ΔHma (J g−1) |
%Ca (%) |
| PCL14 |
ddI |
PCL14 |
ddI |
PCL14 |
ddI |
|
ΔHm and %C values were normalized according to the relative % weight content of drug and polymer in each sample.
The value of ΔHm for 100% crystalline PCL was considered 139.5 J g−1.
|
| ddI |
— |
185.3 |
— |
116.5 |
— |
100.0 |
| PCL14 |
60.6 |
— |
100.3 |
— |
71.9b |
— |
| ddI-free particles |
55.8 |
— |
87.1 |
— |
62.4b |
— |
| ddI-loaded particles |
55.3 |
190.5 |
84.0 |
102.4 |
60.2b |
87.9 |
3.4.4. XRPD.
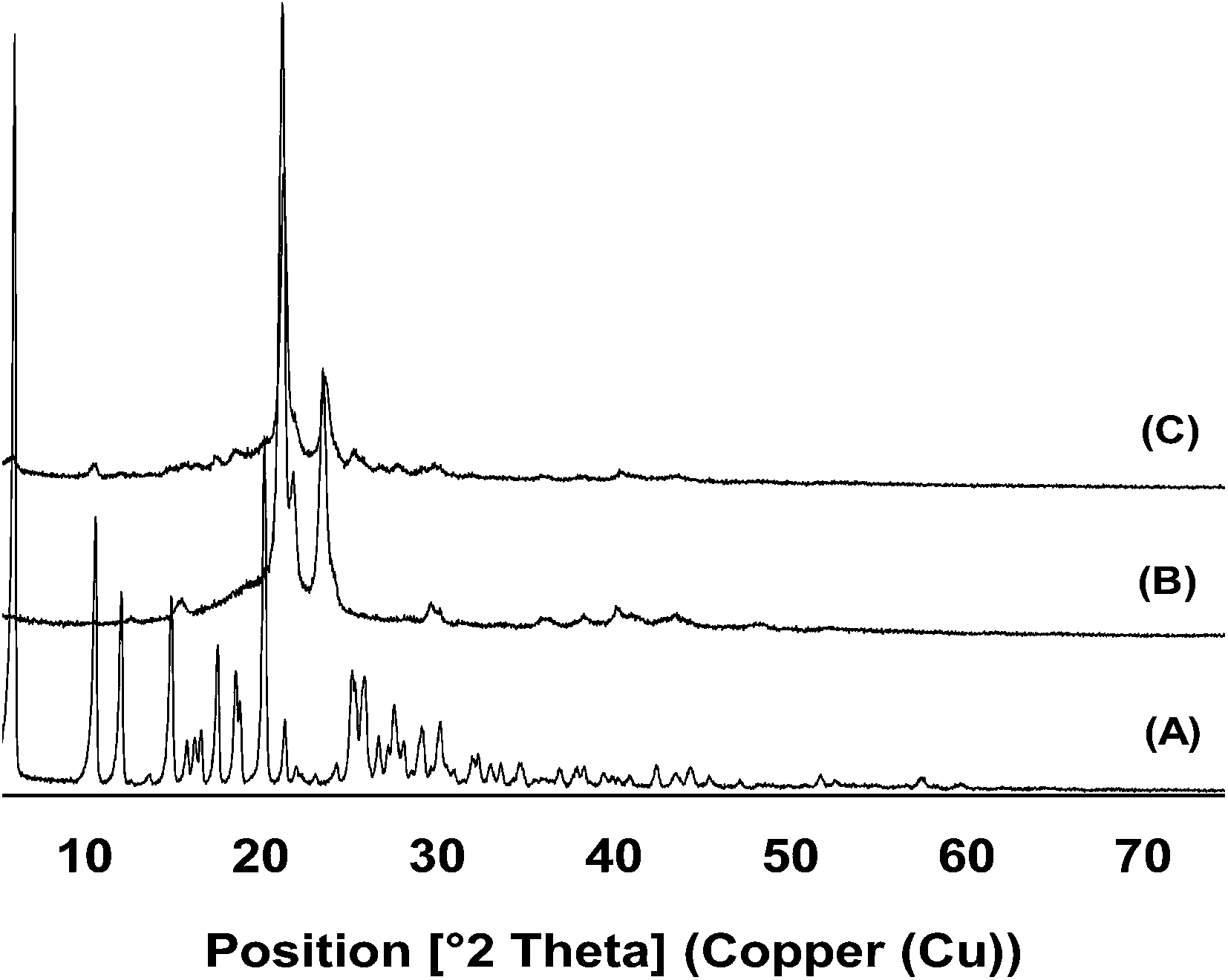
XRPD analysis was used to complement the DSC analysis. The diffractogram of free ddI showed the pattern of a crystalline powder with peaks of characteristic intensities (Fig. 5A). Similarly, PCL14 showed the characteristic pattern of the polymer (Fig. 5B). As expected, ddI-loaded particles showed a diffraction pattern similar to that of pure PCL14, indicating that PCL14 was preserved in a relatively highly crystalline state and that the drug was efficiently encapsulated (Fig. 5C). Otherwise, based on the high %C observed for ddI by DSC, the characteristic peaks of the drug should have also been apparent.
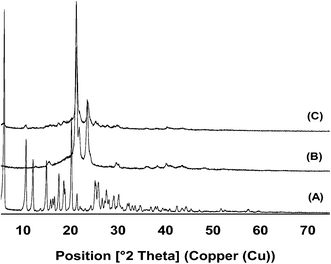 |
| | Fig. 5 XRPD diffractograms of (A) free ddI, (B) pure PCL14 and (C) ddI-loaded particles, obtained by CEHDA. | |
3.5.
In vitro ddI release
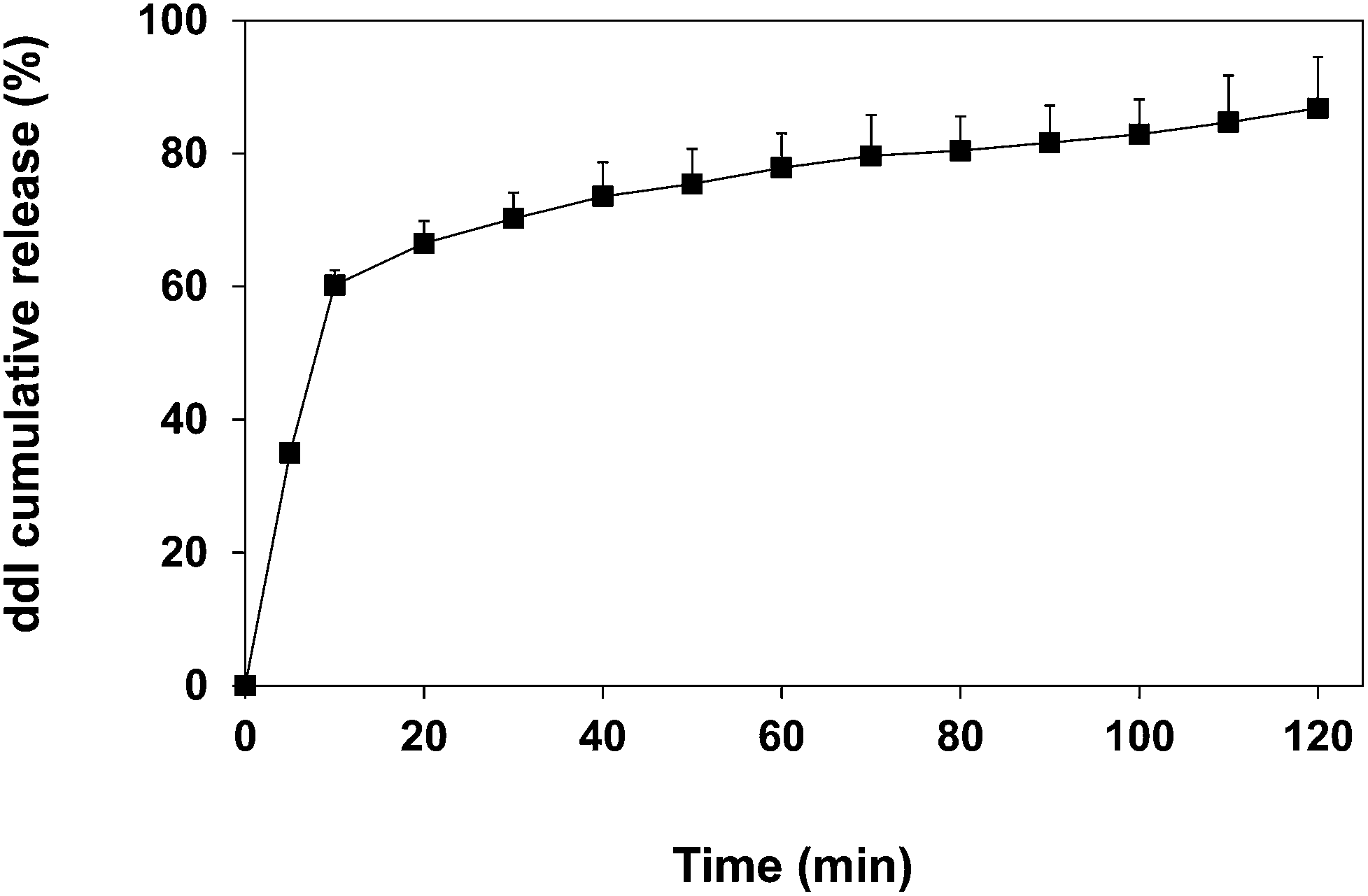
A priori, we envisioned that the protection of the encapsulated cargo from degradation would be intimately associated with the ability of the particles to sustain its release over time and reduce the direct contact with the gastric medium. To assess the ddI release kinetics from ddI-loaded particles obtained by CEHDA, an amount of ddI-loaded particles was suspended in the release medium (PBS, pH 7.4, 37 °C), incubated under moderate magnetic stirring for 120 min, and the released ddI amounts were monitored. The selection of this release medium relied on the fact that the degradation of PCL is very slow under the stomach and the gut conditions, the release mechanism of small drugs from PCL is diffusion,60 and ddI displays similar aqueous solubility (30 mg mL−1) below pH 9.2. Thus, we did not expect a significant impact of the pH on the release rate. Moreover, stability studies in simulated gastric fluid reported elsewhere indicated that ddI concentrations could not be detected after 2 min (t1/2 < 2 min).61 Thus, a degradation profile instead of a release one would have been obtained. Finally, because the tmax of ddI after oral administration under fasted conditions is very short (0.50–1.12 h in humans25 and 0.16–0.25 h in rats61,62), the half-life is 1–2 h in both cases,23,25,63 and we assumed that only the drug that is released will undergo acid degradation, PBS provided appropriate conditions to assess the drug release. Results showed that 60% of the encapsulated drug was released within 10 min, probably due to its highly hydrophilic nature (Fig. 6). It is also known that the burst release of hydrophilic drugs from PCL delivery systems is more pronounced than for lipophilic ones.64 This release profile would ensure the partial protection of the encapsulated drug during the gastric transit.65
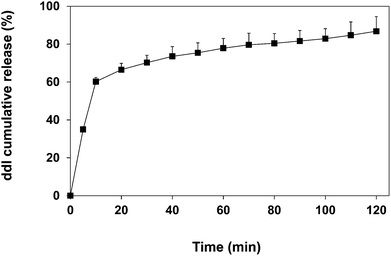 |
| | Fig. 6 Cumulative release of ddI from ddI-loaded particles obtained by CEHDA using distilled water as the inner phase and PCL 10% w/v in DCM:ACN (75:25) as the outer phase. Results are expressed as mean ± S.D. (n = 3). | |
3.6. Oral pharmacokinetics
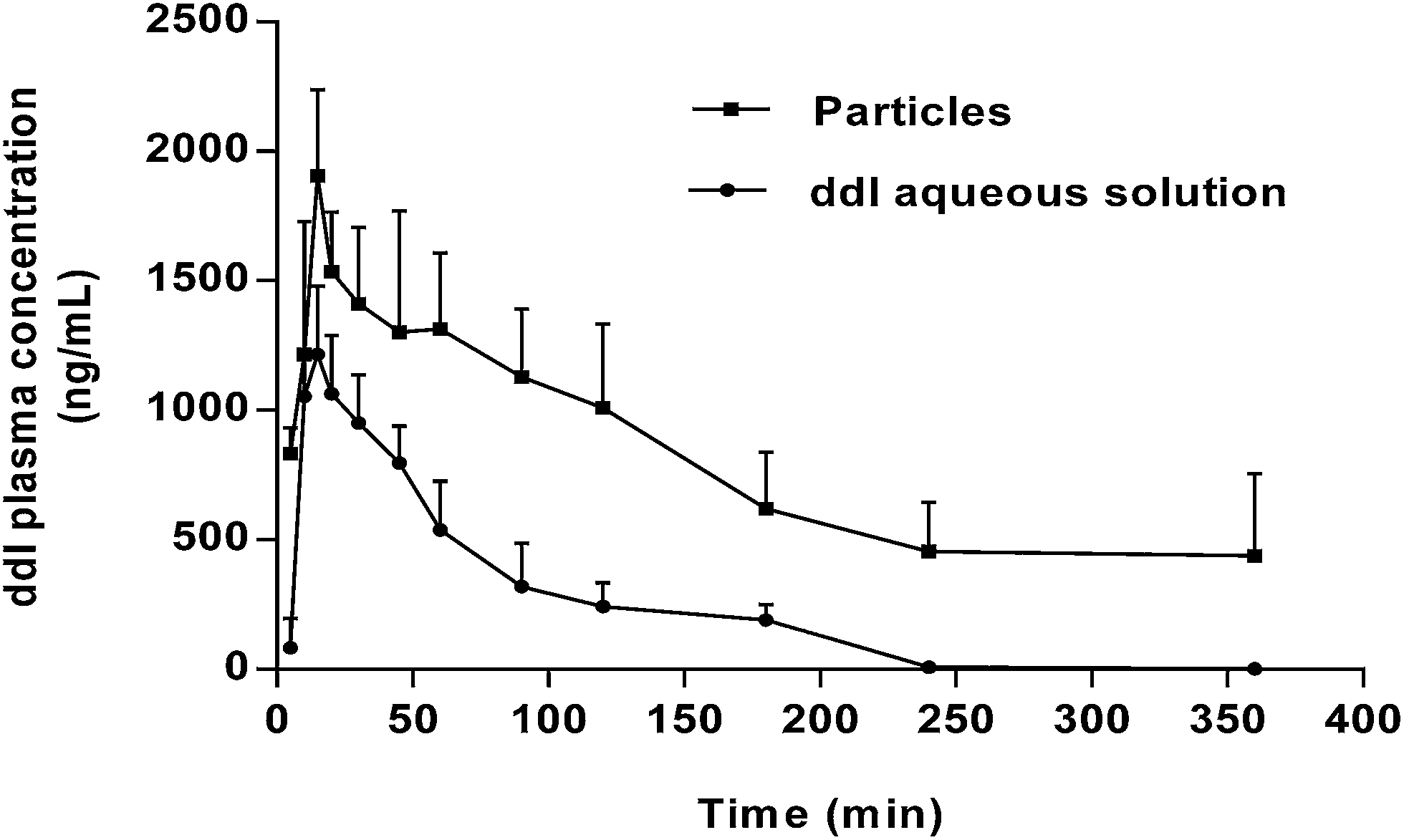
In order to determine if the encapsulation of ddI within PCL microparticles improves its low oral bioavailability, the pharmacokinetics of a ddI aqueous solution and an aqueous suspension of ddI-loaded particles obtained by CEHDA were comparatively assessed. Particles increased the value of Cmax by 1.82 times with respect to the ddI aqueous solution (Fig. 7, Table 3). A similar trend was observed for both AUC values (Table 3), resulting in an Fr (%) value of 375.9%. Differences in tmax were not statistically significant, the values being between 13 and 14 min. Notably, after administration of the ddI solution, the drug was only detected in plasma until 240 min, whereas after administration of the particles, the drug was detected even at a time point of 360 min (Fig. 7). At a later time point (720 min), the drug was not detected in any samples. These results represented a significant decrease of ke (p < 0.05) from 0.51 h−1 for the solution to 0.25 h−1 for the particles and also supported the protection of the encapsulated drug to a relevant extent. At the same time, it should be noted that another mechanism that could contribute to reduce the drug clearance was the significantly higher Cmax attained with the particles. Further studies employing mucoadhesive particles that adhere to the intestinal epithelium would not only protect ddI from gastric degradation but also prolong its release toward drug delivery systems that enable a reduction of the administration frequency, a pending agenda in HIV.66
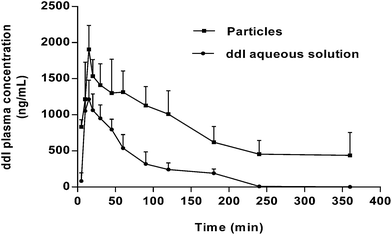 |
| | Fig. 7 ddI plasma concentration (ng mL−1) after administration of ddI aqueous solution and ddI-loaded particles obtained by CEHDA. The ddI dose and concentration were 20 mg kg−1 and 5 mg mL−1, respectively. Results are expressed as mean ± CV% (n = 4). | |
Table 3 Oral pharmacokinetic parameters of ddI. The ddI dose and concentration were 20 mg kg−1 and 5 mg mL−1, respectively (n = 4)a
| Pharmacokinetic parameters |
ddI solution |
ddI-loaded particles |
| Media |
CV% |
Media |
CV% |
|
AUC0–1: area-under-the-curve between 0 and 1 h. AUC0–∞: area-under-the-curve between 0 and infinity. ke: elimination rate constant. * Difference is statistically significant with respect to the ddI solution (p < 0.05). ND:not determined.
|
|
C
max (ng mL−1) |
1279 |
34.19 |
2327* |
3.63 |
|
t
max (min) |
13.75 |
18.18 |
13.33 |
21.65 |
| AUC0–1 (μg mL−1 h−1) |
0.82 |
39.01 |
1.28 |
31.73 |
| AUC0–∞ (μg mL−1 h−1) |
1.91 |
46.19 |
7.18* |
48.22 |
|
F
r (%) |
100.0 |
ND |
375.9 |
ND |
|
k
e (h−1) |
0.51 |
22.79 |
0.25* |
30.59 |
4. Conclusions
In the present work, we developed a CEHDA method for the effective encapsulation of a highly water soluble drug, ddI, within PCL particles. In vitro release assays showed the relatively fast release of the cargo that would be appropriate to ensure the more complete absorption over the gastrointestinal transit. Furthermore, oral administration in Wistar rats of ddI-loaded particles led to a sharp increase of the bioavailability of almost 4 times with respect to a ddI aqueous solution. Overall, these results highlight the potential of this technology for the reproducible and scalable production of micro and nanomedicines. On the other hand, efforts will need to be devoted to improve the process yield and the rate.
Acknowledgements
KPS thanks a Ph.D. scholarship of CONICET. The work was partially supported by grants of UBA (UBACyT 20020090200189) ANPCyT (PICT 224) and CONICET (PIP0220). The RIMADEL network of the CYTED program is also acknowledged.
References
- N. Bock, M. A. Woodruff, D. W. Hutmacher and T. R. Dargaville, Polymers, 2011, 3, 131 CrossRef CAS PubMed.
- N. Bock, T. R. Dargaville and M. A. Woodruff, Prog. Polym. Sci., 2012, 37, 1510 CrossRef CAS PubMed.
- S. D. Nath, S. Son, A. Sadiasa, Y. K. Min and B. T. Lee, Int. J. Pharm., 2013, 443, 87 CrossRef CAS PubMed.
- Y. Wu, S. J. Kennedy and R. L. Clark, J. Biomed. Mater. Res., Part B, 2009, 90, 381 Search PubMed.
-
Y. Wu, A. Duong, L. J. Lee and B. E. Wyslouzil, in The Delivery of Nanoparticles, ed. A. A. Hashim, InTech, Rijeka, Croatia, 2012, ch. 10, pp. 223–242 Search PubMed.
- A. Jaworek and A. T. Sobczyk, J. Electrost., 2008, 66, 197 CrossRef CAS PubMed.
- L. Ding, T. Lee and C.-H. Wang, J. Controlled Release, 2005, 102, 395 CrossRef CAS PubMed.
- R. Pareta and M. J. Edirisinghe, J. R. Soc., Interface, 2006, 3, 573 CrossRef CAS PubMed.
- J. M. López-Herrera, A. Barrero, A. López, I. G. Loscertales and M. Márquez, J. Aerosol Sci., 2003, 34, 535 CrossRef.
- Y. Wu and R. L. Clark, J. Biomater. Sci., Polym. Ed., 2008, 19, 573 CrossRef CAS PubMed.
- B. Almería and A. Gomez, J. Colloid Interface Sci., 2014, 417, 121 CrossRef PubMed.
- S. Chakraborty, I.-C. Liao, A. Adler and K. W. Leong, Adv. Drug Delivery Rev., 2009, 61, 1043 CrossRef CAS PubMed.
- M. Enayati, M.-W. Chang, F. Bragman, M. Edirisinghe and E. Stride, Colloids Surf., A, 2011, 382, 154 CrossRef CAS PubMed.
- Y. Wang, X. Yang, W. Liu, F. Zhang, Q. Cai and X. Deng, J. Microencapsulation, 2013, 30, 490 CrossRef CAS PubMed.
- H. Nie, Y. Fu and C.-H. Wang, Biomaterials, 2010, 31, 8732 CrossRef CAS PubMed.
- S. Hao, B. Wang, Y. Wang and Y. Xu, J. Nanopart. Res., 2014, 16, 2204, DOI:10.1007/s11051-013-2204-2.
- L. Zhang, J. Huang, T. Si and R. X. Xu, Expert Rev. Med. Devices, 2012, 9, 595 CrossRef CAS PubMed.
- S. Ghayempour and S. M. Mortazavi, J. Electrost., 2013, 71, 717 CrossRef PubMed.
- M. A. Tavares Cardoso, M. Talebi, P. A. M. H. Soares, C. U. Yurteri and J. R. van Ommen, Int. J. Pharm., 2011, 414, 1 CrossRef CAS PubMed.
- A. Bohr, J. Kristensen, M. Dyas, M. Edirisinghe and E. Stride, J. R. Soc., Interface, 2012, 9, 2437 CrossRef CAS PubMed.
- A. Bohr, J. Kristensen, E. Stride, M. Dyas and M. Edirisinghe, Int. J. Pharm., 2011, 412, 59 CrossRef CAS PubMed.
- M. Zamani, M. P. Prabhakaran and S. Ramakrishna, Int. J. Nanomed., 2013, 8, 2997 Search PubMed.
- Y. Jin, P. Ai, R. Xin, Y. Tian, J. Dong, D. Chen and W. Wang, Int. J. Pharm., 2009, 368, 207 CrossRef CAS PubMed.
- S. K. Jain, Y. Gupta, A. Jain, A. R. Saxena, P. Khare and A. Jain, Nanomedicine (N. Y., NY, U. S.), 2008, 4, 41 CAS.
-
M. N. Nassar, T. Chen, M. J. Reff and S. N. Agharkar, in Analytical Profiles of Drug Substances and Excipients, ed. H. G. Brittain, Academic Press, Inc, San Diego, California, 1993, vol. 22, pp. 185–227 Search PubMed.
- D. V. N. Srinivasa Rao, N. Srinivas, Ch. Bharathi, Ch. S. Prasad, R. Dandala and A. Naidu, J. Pharm. Biomed. Anal., 2007, 45, 516 CrossRef CAS PubMed.
- S. Høyem, S. Bruheim, G. Maelandsmo, M. Standal, D. E. Olberg, B. Brudeli, A. Asberg, J. Klaveness and P. Rongved, Eur. J. Med. Chem., 2009, 44, 3874 CrossRef PubMed.
- M. Lalanne, K. Andrieux, A. Paci, M. Besnard, M. Ré, C. Bourgaux, M. Ollivon, D. Desmaele and P. Couvreur, Int. J. Pharm., 2007, 344, 62 CrossRef CAS PubMed.
- R. T. Dodge, K. W. Shipp and G. D. Miralles, J. Assoc. Nurses AIDS Care, 1998, 9, 27 CrossRef CAS.
- P. Severino, G. G. G. de Oliveira, H. G. Ferraz, E. B. Souto and M. H. A. Santana, J. Pharm. Anal., 2012, 2, 188 CrossRef CAS PubMed.
- B. D. Damle, V. Mummaneni, S. Kaul and C. Knupp, Antimicrob. Agents Chemother., 2002, 46, 385 CrossRef CAS.
- C. F. da Silva, P. Severino, F. Martins, M. V. Chaud and M. H. A. Santana, J. Microencapsulation, 2009, 26, 523 CrossRef PubMed.
- A. Sosnik, D. A. Chiappetta and A. M. Carcaboso, J. Controlled Release, 2009, 138, 2 CrossRef CAS PubMed.
- A. Kaur, S. Jain and A. K. Tiwary, Acta Pharm., 2008, 58, 61 CrossRef CAS PubMed.
- D. A. Chiappetta, C. Hocht, C. Taira and A. Sosnik, Biomaterials, 2011, 32, 2379 CrossRef CAS PubMed.
- A. C. Hunter, J. Elsom, P. P. Wibroe and S. M. Moghimi, Nanomedicine (N. Y., NY, U. S.), 2012, 8, S5 CAS.
- C. Vilos and L. A. Velasquez, J. Biomed. Biotechnol., 2012, 2012, 672760 CAS.
- B. J. Aungst, Adv. Drug Delivery Rev., 1999, 39, 105 CrossRef CAS.
- A. Sosnik, J. Biomed. Nanotechnol., 2014, 10, 2200 CrossRef CAS PubMed.
- L. Van der Schueren, T. De Meyer, I. Steyaert, Ö. Ceylan, K. Hemelsoet, V. Van Speybroeck and K. De Clerck, Carbohydr. Polym., 2013, 91, 284 CrossRef CAS PubMed.
- D. Paneva, N. Manolova, M. Argirova and I. Rashkov, Int. J. Pharm., 2011, 416, 346 CrossRef CAS PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1 CrossRef CAS PubMed.
- K. P. Seremeta, D. A. Chiappetta and A. Sosnik, Colloids Surf., B, 2013, 102, 441 CrossRef CAS PubMed.
- J. Xie, L. K. Lim, Y. Phua, J. Hua and C.-H. Wang, J. Colloid Interface Sci., 2006, 302, 103 CrossRef CAS PubMed.
- J. Xie, J. C. M. Marijnissen and C.-H. Wang, Biomaterials, 2006, 27, 3321 CrossRef CAS PubMed.
- B. Almería, W. Deng, T. M. Fahmy and A. Gomez, J. Colloid Interface Sci., 2010, 343, 125 CrossRef PubMed.
- S. Hao, Y. Wang, B. Wang, Q. Zou, H. Zeng, X. Chen, X. Liu, J. Liu and S. Yu, Int. J. Pharm., 2014, 463, 10 CrossRef CAS PubMed.
- P. Gupta, C. Elkins, T. E. Long and G. L. Wilkes, Polymer, 2005, 46, 4799 CrossRef CAS PubMed.
- B. Almería, T. M. Fahmy and A. Gomez, J. Controlled Release, 2011, 154, 203 CrossRef PubMed.
- Y. Wu and R. L. Clark, J. Colloid Interface Sci., 2007, 310, 529 CrossRef CAS PubMed.
- S. Gun, M. Edirisinghe and E. Stride, Mater. Sci. Eng., C, 2013, 33, 3129 CrossRef CAS PubMed.
- J. Xie, W. J. Ng, L. Y. Lee and C.-H. Wang, J. Colloid Interface Sci., 2008, 317, 469 CrossRef CAS PubMed.
- A. Rezvanpour and C.-H. Wang, Chem. Eng. J., 2014, 239, 8 CrossRef CAS PubMed.
- H. Ghanbar, C. J. Luo, P. Bakhshi, R. Day and M. Edirisinghe, Mater. Sci. Eng., C, 2013, 33, 2488 CrossRef CAS PubMed.
- D. Grafahrend, P. Jungbecker, G. Seide, H. Leonards, T. Gries, M. Möller and D. Klee, The Open Chemical and Biological Methods Journal, 2010, 3, 1 CrossRef CAS.
- A. Sosnik and D. Cohn, Polymer, 2003, 44, 7033 CrossRef CAS PubMed.
- D. Cohn, G. Lando, A. Sosnik, S. Garty and A. Levi, Biomaterials, 2006, 27, 1718 CrossRef CAS PubMed.
- V. R. Sinha, K. Bansal, R. Kaushik, R. Kumria and A. Trehan, Int. J. Pharm., 2004, 278, 1 CrossRef CAS PubMed.
- M.-J. Choi, A. Soottitantawat, O. Nuchuchua, S.-G. Min and U. Ruktanonchai, Food Res. Int., 2009, 42, 148 CrossRef CAS PubMed.
- F. Barbato, M. I. La Rotonda, G. Maglio, R. Palumbo and F. Quaglia, Biomaterials, 2001, 22, 1371 CrossRef CAS.
- Z. Yan, J. Sun, Y. Chang, Y. Liu, Q. Fu, Y. Xu, Y. Sun, X. Pu, Y. Zhang, Y. Jing, S. Yin, M. Zhu, Y. Wang and Z. He, Mol. Pharmaceutics, 2011, 8, 319 CrossRef CAS PubMed.
- R. Skanji, K. Andrieux, M. Lalanne, J. Caron, C. Bourgaux, J. Degrouard, F. Brisset, C. Gueutin, H. Chacun, N. Dereuddre-Bosquet, A. Paci, G. Vassal, L. Bauduin, S. Garcia-Argote, B. Rousseau, P. Clayette, D. Desmaële and P. Couvreur, Int. J. Pharm., 2011, 414, 285 CrossRef CAS PubMed.
- C. V. Fletcher, R. C. Brundage, R. P. Remmel, L. M. Page, D. Weller, N. R. Calles, C. Simon and M. W. Kline, Antimicrob. Agents Chemother., 2000, 44, 1029 CrossRef CAS.
- T. K. Dash and V. B. Konkimalla, J. Controlled Release, 2012, 158, 15 CrossRef CAS PubMed.
- J. Worsøe, L. Fynne, T. Gregersen, V. Schlageter, L. A. Christensen, J. F. Dahlerup, N. J. M. Rijkhoff, S. Laurberg and K. Krogh, BMC Gastroenterol., 2011, 11, 145 CrossRef PubMed.
- A. Sosnik, J. das Neves and B. Sarmento, Prog. Polym. Sci., 2014 DOI:10.1016/j.progpolymsci.2014.07.010 , in press.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tb00664j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.