DOI:
10.1039/C5TA07254A
(Paper)
J. Mater. Chem. A, 2015,
3, 21733-21743
Insight into quinoxaline containing D–π–A dyes for dye-sensitized solar cells with cobalt and iodine based electrolytes: the effect of π-bridge on the HOMO energy level and photovoltaic performance†
Received
10th September 2015
, Accepted 11th September 2015
First published on 21st September 2015
Abstract
Three new quinoxaline-based organic dyes (AQ201, AQ202, and AQ203), containing thiophene, 3,4-ethylenedioxythiophene (EDOT), and cyclopentadithiophene (CPDT) in the π-system, respectively, have been designed and synthesized for dye-sensitized solar cells. Different from the traditional donor–π-bridge–acceptor (D–π–A) type dyes, the dissymmetric π-bridge on both sides of quinoxaline enables great flexibility in fine-tuning the absorption spectra and energy levels of the resultant molecules. By changing the π-bridge between the bulky triphenylamine donor and quinoxaline group, a negative shift was observed regarding the highest occupied molecular orbital (HOMO) levels of AQ201, AQ202, and AQ203 dyes (0.88, 0.79, and 0.72 V vs. NHE, respectively), while the lowest unoccupied molecular orbital (LUMO) levels of these dyes remained the same (−1.19, −1.20, and −1.20 V vs. NHE, respectively), which, in turn, resulted in a gradual shift of the absorption spectra of AQ dyes. The absorption spectra properties of the dyes are also analysed by density functional theory. The calculated results in combination with the experiments indicate that the absorption bands are mainly dominated by charge transfer transitions from the HOMO and HOMO−1 orbital to the LUMO. In all cases, the [Co(bpy)3]2+/3+ redox-shuttle afforded superior solar cell performance compared to I−/I3−. More importantly, dye AQ202 shows the highest power conversion efficiency (PCE) of 8.37% with the [Co(bpy)3]2+/3+ based electrolyte by maintaining a balance between the spectral absorption range and driving force for dye regeneration. Transient photocurrent decay experiments as well as electrochemical impedance spectroscopy indicate that the lower HOMO levels lead to higher electron lifetime and dye regeneration efficiency.
Introduction
Dye-sensitized solar cells (DSSCs) have attracted considerable attention due to their relatively low production cost, transparency and flexibility.1 It is known that the operating principle of DSSCs usually involves excitation of the dye followed by charge transfer from the dye to the conduction band of TiO2. Thus the oxidized state of the dye is obtained and then regenerated by receiving an electron from a redox mediator in the electrolyte.2 Accordingly, the highest occupied molecular orbital (HOMO) level of the dye should be sufficiently positive compared to the potential of the redox mediator in the electrolyte for efficient dye regeneration. Therefore, dye regeneration depends strongly on both the potential of the redox mediator and the HOMO level of dyes.3 Typically, iodide/triiodide (I−/I3−) and [Co(bpy)3]2+/3+ are most commonly used as redox mediators to obtain highly efficient photovoltaic performances in liquid electrolytes, and their potentials are almost constant.4 According to the Marcus theory, a difference between the dye HOMO level and the redox potential of the electrolyte of 0.2–0.3 eV can spur single-electron transfer, resulting in effective dye regeneration.5 In that case, in order to achieve efficient dye regeneration, it is essential to modulate the HOMO level of the dye to the potential of the redox mediator.
The general design principle of organic sensitizers consists of a donor–π–acceptor (D–π–A) framework that can efficiently facilitate intramolecular charge transfer (ICT) from the ground to excited state and increase light absorption.6 In general, one can tune the HOMO level of the dye by alternating each component in the design. However, by doing that, the LUMO level of the dye also changes significantly, leading to difficulty in independently exploring the effects of the HOMO level on the photovoltaic performance.7 Recently, an auxiliary electron acceptor has been widely introduced into the π-bridge of the D–π–A framework.8 This moiety not only enhances the photovoltaic performance and photostability of sensitizers, but is also beneficial to fine-tune the molecular energy levels.9
It is known that quinoxaline is a strong electron-withdrawing unit due to its high electron affinity, which arises from the two symmetric unsaturated nitrogen atoms.10 Our group recently reported quinoxaline-based sensitizers YA422 (ref. 11) and AQ308,12 which showed outstanding power conversion efficiencies (PCE) of 10.56% and 9.81%, respectively. Those reports inspired us to do some further investigation into the application of quinoxaline as the auxiliary acceptor in DSSCs. In this regard, herein, we have designed and synthesized three new quinoxaline-based organic sensitizers (AQ201, AQ202, and AQ203) by using the dissymmetric π-bridge on both sides of quinoxaline (Scheme 1). The introduction of a phenyl ring between the quinoxaline moiety and the cyanoacrylic acid group stabilizes the LUMO level of the dye.13 And most importantly, an electron-rich unit (thiophene, EDOT, and CPDT, respectively) close to the bulky triphenylamine donor group has been included to tune the HOMO level of the dye.14 As a result, a negative shift was observed in the HOMO levels of AQ201, AQ202, and AQ203 dyes (0.88, 0.79, and 0.72 V vs. NHE, respectively), while the LUMO levels remained the same (−1.19, −1.20, and −1.20 V vs. NHE, respectively). Hence, a red shift and broadening of the absorption spectra were achieved. Those dyes were employed in DSSCs with [Co(bpy)3]2+/3+ and I−/I3− based electrolytes. Overall, the DSSCs using the three dyes with the cobalt-based electrolyte yielded higher PCE compared to the corresponding DSSCs with the iodide-based electrolyte, which is ascribed to the bulky donor group usually required for devices with the cobalt-based electrolyte.11,15 Consequently, the AQ202-based cell with an intermediate HOMO level performed best with a PCE of 8.37% using the [Co(bpy)3]2+/3+ based electrolyte, while the AQ201-based cell with the lowest HOMO level showed the highest PCE of 6.57% with the I−/I3− based electrolyte. Electrochemical impedance spectroscopy (EIS) and transient absorbance spectroscopy (TAS) analyses were performed to understand the influence of the HOMO energy level on the dye regeneration and recombination kinetics of DSSC devices with the two different electrolytes.
 |
| | Scheme 1 The structures of AQ201, AQ202, and AQ203. | |
Results and discussion
Design and synthesis
In this context, molecular engineering was focused on tuning the dye HOMO level and extending the absorption range based on the quinoxaline containing the D–π–A structure, and then improving dye regeneration and photovoltaic performances with [Co(bpy)3]2+/3+ and I−/I3− based electrolytes, respectively. The bulky triphenylamine donor offers the possibility to inhibit charge recombination and block molecular aggregation, especially for the cobalt redox system. As shown in Scheme 2, AQ201, AQ202, and AQ203 were prepared following the same synthetic routes. The synthetic work starts from low-cost, commercially available materials such as 5,8-dibromo-2,3-diphenylquinoxaline and (4-formylphenyl)boronic acid. The three dyes were obtained following a sequence of Suzuki coupling, Stille coupling, bromination, and Knoevenagel condensation. Such a synthetic procedure for AQ201, AQ202, and AQ203 was optimized on the basis of reaction selectivity and product yields. The key intermediates and final dyes were fully characterized with 1H NMR, 13C NMR, and HRMS.
 |
| | Scheme 2 Synthetic procedure of the dyes AQ201, AQ202, and AQ203. | |
Photophysical and electrochemical properties
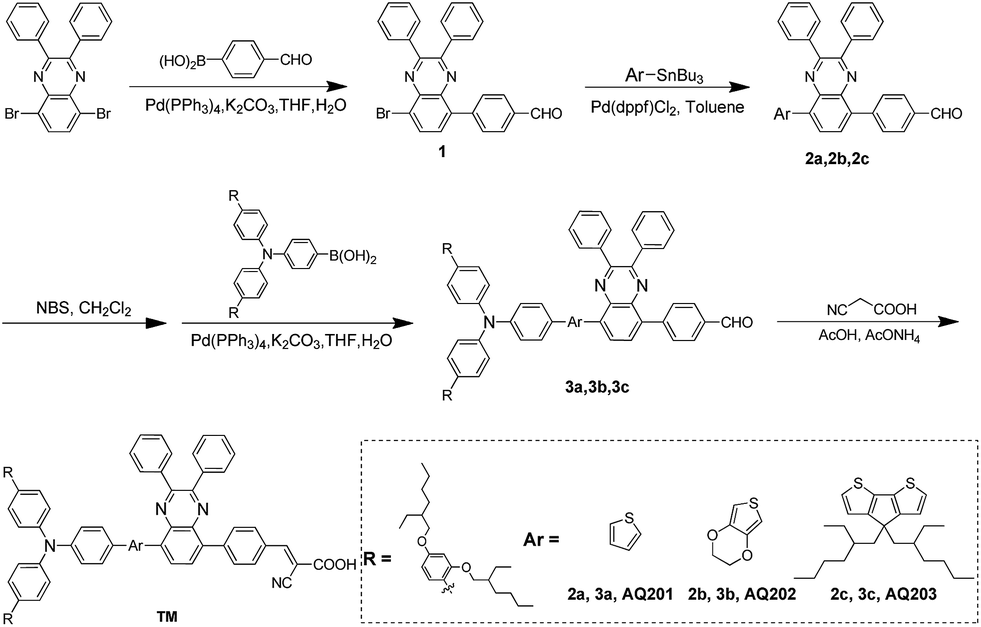
The absorption spectra of AQ201, AQ202, and AQ203 in dichloromethane (CH2Cl2) solution and on 4 μm transparent TiO2 films are shown in Fig. 1 and their relative intensities are tabulated in Table 1. The variation of the π-bridge of the electron-donating moiety leads to significant changes in the position of the lowest energy absorption band. The intramolecular charge transfer (ICT) absorption peak in the visible region is observed at λmax = 491, 515, and 545 nm for AQ201, AQ202, and AQ203, respectively. As listed in Table 1, when replacing the thiophene unit with the EDOT or CPDT unit, the absorption spectra of AQ202 and AQ203 broaden with respect to AQ201 in the visible region, and the absorption peaks (λmax) are red-shifted by 24 and 54 nm, respectively. The red shift in the absorption peak of dye AQ201 sensitized on the 4 μm transparent TiO2 films compared to the absorption spectra in solution might be ascribed to the formation of J-aggregates.16 In addition, when AQ202 and AQ203 dyes are adsorbed on the TiO2 films, the absorption peaks (λmax) blue-shifted to λmax = 513 and 528 nm, respectively. This might be ascribed to deprotonation of the carboxylic acid or H-aggregates.17 In summary, this may be due to the smaller size of the thiophene group than that of EDOT and CPDT groups, leading to different aggregation behavior.
 |
| | Fig. 1 (a) Absorption spectra of dyes AQ201, AQ202, and AQ203 in CH2Cl2; (b) normalized absorption spectra of dyes AQ201, AQ202, and AQ203 on 4 μm TiO2 films. | |
Table 1 Photophysical and electrochemical properties of dyes AQ201, AQ202 and AQ203
| Sample |
λ
max
/nm (ε, M−1 cm−1) |
λ
max
/nm (on TiO2) |
HOMOc/V (vs. NHE) |
E
0–0
/eV |
LUMOe/V (vs. NHE) |
|
Absorption maximum in CH2Cl2 at room temperature.
Absorption maximum on 4 μm transparent TiO2 films.
HOMO was estimated by the ground state oxidation potential (E0ox), and all potentials were obtained in CH2Cl2 with 0.1 M tetrabutylammonium perchlorate (TBAP) as the electrolyte (working electrode: glassy carbon; reference electrode: Ag/AgCl; counter electrode: Pt; calibrated with ferrocene/ferrocenium (Fc/Fc+) as an external reference and converted to NHE by the addition of 0.69 V).
E
0–0 was estimated from the absorption thresholds from the absorption spectra of dyes in solution.
The LUMO was estimated by subtracting E0–0 from the HOMO.
|
|
AQ201
|
491 (32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 898) 898) |
494 |
0.88 |
2.07 |
−1.19 |
|
AQ202
|
515 (25550) |
513 |
0.79 |
1.99 |
−1.20 |
|
AQ203
|
545 (35865) |
528 |
0.72 |
1.92 |
−1.20 |
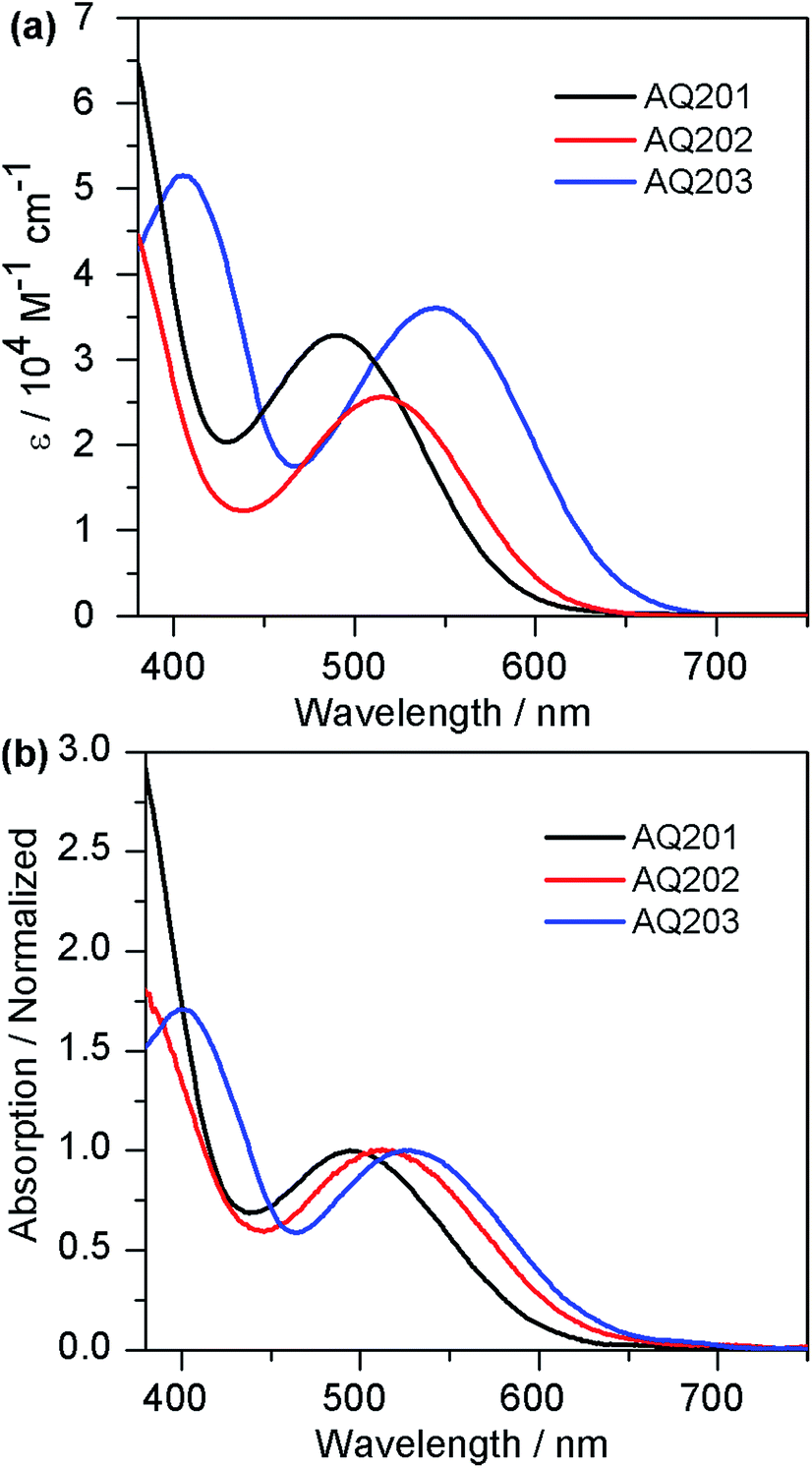
To evaluate the possibility of electron transfer from the excited dye molecule to the conduction band (CB) of TiO2, and from the redox couple in the electrolyte to the oxidized dye molecule, cyclic voltammograms of AQ201, AQ202, and AQ203 dyes were recorded in CH2Cl2 solution (Fig. 2a) to determine the redox potential and the corresponding values are presented in Table 1. The ground state oxidation potentials of AQ201, AQ202, and AQ203 are 0.88, 0.79 and 0.72 V vs. NHE, respectively, which are sufficiently higher than the redox potential of iodide/triiodide (0.45 V vs. NHE), guaranteeing ample driving force for dye regeneration. However, in the presence of [Co(bpy)3]2+/3+ (0.57 V vs. NHE) redox couple, the AQ201 and AQ202 dyes show an advantage over the AQ203 dye for dye regeneration according to the Marcus theory.5 The structural differences in the π-bridge of the electron-donating part of AQ201, AQ202, and AQ203 have a significant influence on their oxidation potentials.
 |
| | Fig. 2 (a) Cyclic voltammograms of dyes AQ201, AQ202, and AQ203; (b) schematic diagram of the energy levels of the TiO2 conduction band, dyes, and [Co(bpy)3]2+/3+ and I−/I3− redox couples. | |
The ground state oxidation potentials (E0ox) of AQ202 and AQ203 are decreased by 90 and 160 mV compared to AQ201via replacing the thiophene with EDOT or CPDT unit. On the other hand, the excited-state oxidation potentials (E*ox) of AQ201, AQ202, and AQ203, derived from the ground state oxidation potential and optical energy gap (E0ox − E0–0), are −1.19, −1.20, and −1.20 V, respectively. E0–0 is estimated from the absorption threshold from the absorption spectra of dyes in solution. The E*ox of these dyes are placed sufficiently above the TiO2 conduction band edge (−0.5 V vs. NHE), ensuring no energetic barriers for electron injection from the excited dyes into the conduction band of TiO2. Herein, the π-bridge on the left side of the quinoxaline unit shows little effect on the LUMO level of D–π–A dyes, which is helpful in molecular designing and HOMO energy level tuning for future novel organic dyes.18
Theoretical approach
Density functional theory (DFT) calculations were carried out using the Gaussian 09 (ref. 19) program to obtain further insight into the electronic distribution of the frontier molecular orbitals and the molecular structures of the dyes AQ201, AQ202, and AQ203. The ground-state structures of those dyes were optimized by DFT at the B3LYP/6-31G(d) level.20 In the calculations, the long alkyl chains were replaced by methyl groups to reduce computational costs without affecting the nature of frontier molecular orbitals. The optimized structures were then used to calculate the 20 lowest singlet electronic transitions using time-dependent DFT calculations (TD-DFT) at the CAM-B3LYP/TZVP level.21 A polarizable continuum model (PCM) was applied using CH2Cl2 as the solvent.22 The TD-DFT results and frontier molecular orbitals of AQ201, AQ202, and AQ203 are shown in Tables 2 and 3, respectively.
Table 2 TD-DFT calculated energies, oscillator strengths (f), and compositions in terms of molecular orbital contributions of AQ201, AQ202, and AQ203
| Dye |
Compositiona |
E (nm) |
f
|
|
H = HOMO, L = LUMO, H−1 = HOMO−1.
|
|
AQ201
|
42% H → L |
463 |
1.5749 |
| 23% H−1 → L |
|
AQ202
|
44% H → L |
475 |
1.5401 |
| 21% H−1 → L |
|
AQ203
|
45% H → L |
507 |
1.9105 |
| 16% H−1 → L |
Table 3 Frontier molecular orbitals of the dyes AQ201, AQ202, and AQ203 calculated at the B3LYP/6-31G level of theory
| Dye |
LUMO |
HOMO |
HOMO−1 |
|
AQ201
|

|

|

|
|
AQ202
|

|
|

|
|
AQ203
|

|

|

|
Table 3 summarizes the orbital distributions of HOMO−1, HOMO, and LUMO. These orbitals were chosen here for discussion because they participate in the lowest singlet electronic transition according to TD-DFT. The three dyes share similar features in the spatial distribution of these frontier molecular orbitals. The HOMO is mainly located on the donor moieties and the first π-bridge on the left side of quinoxaline (Scheme 1), while HOMO−1 extends to the quinoxaline moiety. The LUMO is mainly located on the quinoxaline, phenyl ring and cyanoacrylic acid with a little contribution from the first π-bridge.
The electronic transitions shown by the TD-DFT calculations indicate that the absorption bands of AQ201, AQ202, and AQ203 with the maximum absorption peaks calculated at λ = 463, 475, and 507 nm are dominated by HOMO→LUMO transitions with some contributions from HOMO−1→LUMO. They agree qualitatively well with experimental results, although the energies were overestimated. The insertion of the EDOT and CPDT moiety significantly extends the π-conjugation of the donor in AQ202 and AQ203, which increases the HOMO energy and facilitates its oxidation. Consequently, they exhibit a lower HOMO→LUMO energy gap compared to AQ201, which, in turn, corresponds well with the red shift of the absorption spectra (Fig. 1a).
Photovoltaic performance
The DSSC performances of all the dyes with two different redox electrolytes were tested under AM1.5 G irradiation (100 mW cm−2). Fig. 3a shows the photocurrent density voltage curves (J–V) of DSSCs based on AQ201, AQ202, and AQ203 dyes with the [Co(bpy)3]2+/3+ redox electrolyte, and the photocurrent density voltage curves (J–V) of DSSCs based on the I−/I3− redox electrolyte are shown in Fig. 3b. The detailed photovoltaic parameters are collected in Table 4. Hereby, the influence of altering the π-bridge on the left side of the quinoxaline unit on the DSSC characteristics is apparent. For the devices based on the [Co(bpy)3]2+/3+ redox electrolyte, the AQ202-based cell shows both higher power conversion efficiency (PCE) and short-circuit current density (Jsc), because the AQ202 dye has a wider absorption spectrum as well as matches the Marcus theory requirements of the gap between the HOMO level and [Co(bpy)3]2+/3+redox potential, thus effectively promoting dye regeneration. Clearly, the AQ201-based cell shows the lowest Jsc due to the relatively narrow absorption spectrum range of the three dyes. Although the AQ203 dye exhibits the widest absorption spectrum and highest molar extinction coefficient compared with the two other dyes, the Jsc of the AQ203-based cell with the [Co(bpy)3]2+/3+ redox electrolyte remains lower than that of the AQ202-based cell which is attributed to the smallest driving force for dye regeneration.
 |
| | Fig. 3
J–V curves of DSSCs based on dyes AQ201, AQ202, and AQ203 with (a) the [Co(bpy)3]2+/3+ redox electrolyte and (b) the I−/I3−redox electrolyte. | |
Table 4 Photovoltaic performance of the DSSCs based on AQ201, AQ202 and AQ203 under irradiation of AM 1.5 simulated solar light (100 mW cm−2)a
| Dye |
Electrolyte |
J
sc (mA cm−2) |
V
oc (mV) |
FF |
η (%) |
|
The cobalt electrolyte contains 0.22 M [Co(II) (bpy)3](TFSI)2, 0.06 M [Co(III) (bpy)3](TFSI)3, 0.1 M LiClO4, and 0.5 M 4-tert-butylpyridine in acetonitrile, (bpy = 2,2′-bipyridine, and TFSI = bis(trifluoromethane)sulfonamide); the iodide electrolyte contains 0.1 M LiI, 0.03 M I2, 0.5 M 1-butyl-3-methyl imidazolium iodide (BMII), 0.1 M guanidine isothiocyanate, and 0.5 M 4-tert-butylpyridine (TBP) in acetonitrile:valeronitrile (85:15, v/v).
|
|
AQ201
|
[Co(bpy)3]2+/3+ |
12.52 |
838 |
67.68 |
7.10 |
|
AQ202
|
[Co(bpy)3]2+/3+ |
14.61 |
830 |
69.01 |
8.37 |
|
AQ203
|
[Co(bpy)3]2+/3+ |
14.39 |
797 |
70.27 |
8.06 |
|
AQ201
|
I−/I3− |
12.75 |
756 |
68.16 |
6.57 |
|
AQ202
|
I−/I3− |
13.21 |
720 |
68.08 |
6.48 |
|
AQ203
|
I−/I3− |
13.56 |
692 |
68.08 |
6.39 |
It is informative to compare the results of the devices based on the I−/I3− redox electrolyte with those based on the [Co(bpy)3]2+/3+ redox electrolyte. The AQ203-based cell shows higher Jsc with the I−/I3− redox electrolyte, because the driving force for dye regeneration is sufficient in iodine-based electrolytes. Meanwhile, the Jsc of the devices with the I−/I3− redox electrolyte decreases in the trend of AQ203 > AQ202 > AQ201, which are positively correlated with their absorption spectra range. It should be noted that the AQ201-based cell shows the highest PCE with the I−/I3− redox electrolyte due to the highest open-circuit voltage (Voc) compared with the cells of the other two dyes. Furthermore, the effect of varying the π-bridge is highlighted in terms of the changes in Voc, it is apparent that the Voc of the devices with [Co(bpy)3]2+/3+ or I−/I3−redox electrolyte increases in the order of AQ201 > AQ202 > AQ203. This may be caused by the lowest HOMO level of AQ201 and then the highest dye regeneration efficiency, which reduces the probability of electron recombination from the conduction band of TiO2/excited state dyes to oxidised dyes and improves the Voc.
Fig. 4 shows the IPCE curves of the AQ dye based devices with two different electrolytes. Notably, the IPCE onsets are well consistent with the corresponding electron donating ability, that is, thiophene (AQ201) < EDOT (AQ202) < CPDT (AQ203). In particular, all the AQ dye based devices exhibit small notches around 450 nm, which is attributed to the weak absorbance in this region according to the UV-visible absorption spectra. However, when compared with cobalt-based devices, iodide-based devices displayed a poorer IPCE response with a plateau of around 75% in the visible region, resulting in the lower Jsc. This may be ascribed to the bulky donor group being better suited for the devices with the cobalt-based electrolyte. As a result, the devices using the three dyes with the cobalt-based electrolyte realized higher performances compared to those with the iodide-based electrolyte.
 |
| | Fig. 4 IPCE curves of DSSCs based on dyes AQ201, AQ202, and AQ203 with (a) the [Co(bpy)3]3+/2+ redox electrolyte and (b) the I−/I3− redox electrolyte. | |
Electrochemical impedance spectroscopy (EIS)
Generally, Voc is influenced by the conduction band (Ecb) position and the electron recombination process at the TiO2/dye/electrolyte interface. To obtain a complete analysis of the difference of Voc among these dyes with two different redox electrolytes, EIS was utilized to investigate the interfacial charge transfer and recombination processes of the devices. The EIS measurements were performed in the dark and data were fitted by using the transmission line model.23Fig. 5a depicts the charge transfer resistance (Rct) (between the TiO2 films and the electrolyte) and chemical capacitance (Cchem) of the devices based on AQ201, AQ202, and AQ203 dyes with the [Co(bpy)3]2+/3+ redox electrolyte as a function of the IR (internal resistance) drop corrected potential, and the Rct and Cchem of the devices with the I−/I3− redox electrolyte as a function of the IR drop corrected potential are shown in Fig. 5b. There is no difference in the Rct value trend (AQ201 > AQ202 > AQ203) of the devices when applying either [Co(bpy)3]2+/3+ or I−/I3− as the redox electrolyte. We may attribute these small differences to the small differences in dye structures leading to slightly different effectiveness as a blocking layer.
 |
| | Fig. 5 Charge transfer resistance (Rct) and chemical capacitance (Cchem) plotted against the IR drop corrected potential for DSSCs based on dyes AQ201, AQ202, and AQ203 with (a) the [Co(bpy)3]2+/3+ redox electrolyte and (b) the I−/I3− redox electrolyte. | |
Chemical capacitance, as the capacitive response of the TiO2 photoanode, depends on the density of electronic state distribution below the Ecb, which can be used as an indirect measure of the Fermi level (EFn). Overall, there is a minor difference in the Cchem of the devices based on AQ201, AQ202, and AQ203 dyes with either [Co(bpy)3]2+/3+ or I−/I3− redox electrolyte, indicating only a small shift of the TiO2EFn position. Accordingly, with both [Co(bpy)3]2+/3+ and I−/I3− as redox electrolytes, the increase in the Voc mainly results from an increase in the electron lifetime (τn), because τn, positively correlated with Voc, is calculated by the equation of τn = Rct × Cchem (Fig. 6). Clearly, there is also no difference in the τn value trend (AQ201 > AQ202 > AQ203) of the devices using either [Co(bpy)3]3+/2+ or I2/I3− as the redox electrolyte, which is in good agreement with the observed Voc trend of the devices. In conclusion, the differences in Voc arise only from the structural difference in the π-bridge between the bulky triphenylamine donor group and quinoxaline, demonstrating that, similar to the π-bridge, the thiophene unit is beneficial for achieving higher Voc, while the CPDT unit contributes negatively in this system.
 |
| | Fig. 6 Electron lifetime (τn) plotted against chemical capacitance (Cchem) of DSSCs based on dyes AQ201, AQ202, and AQ203 with (a) the [Co(bpy)3]2+/3+ redox electrolyte and (b) the I−/I3− redox electrolyte. | |
Photoinduced charge-transfer dynamics
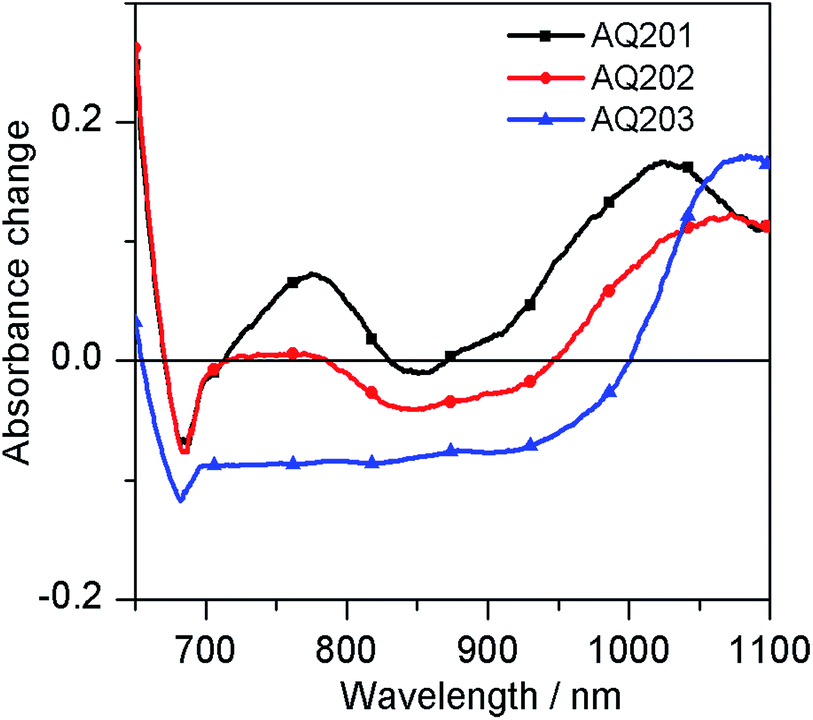
The above results show that the three dyes experience enough driving force for dye regeneration in the iodide-based electrolyte, while the devices with the cobalt-based electrolyte result in higher PCE. Accordingly, micro-to-millisecond transient absorption spectroscopy (TAS) measurements were focused on the cobalt-based electrolyte to gain insight into the influence of the π-bridge on the oxidized dye lifetime and its regeneration in the [Co(bpy)3]2+/3+ redox system. Photoinduced absorption spectroscopy (PIA) is used to locate spectral features corresponding to the oxidized dye absorbance. All dyes exhibit comparable spectral features in their oxidized states, which are measured under 510 nm excitation and absorbance changes are reported in Fig. 7. An isosbestic point between the absorbance of the dye ground and oxidized states is found for these dyes at around 700 nm. It is observed that replacing the thiophene with EDOT or CPDT group not only affects the ground state electronic devices, but also decreases the oxidized state absorbance, with a modification in its absorbance shape. For all the dyes, we observe a positive transient absorbance of around 1000–1100 nm. Hence, following the decay kinetics of the transient absorption signal measured at 1050 nm allowed for the analysis of the charge recombination and dye regeneration rate constants.
 |
| | Fig. 7 PIA spectra measured upon laser excitation (λexc = 510 nm) of AQ201, AQ202 and AQ203 dyes adsorbed on 4 μm mesoporous TiO2 films in the absence of redox electrolytes. | |
The electron transfer and dye regeneration lifetimes of AQ201 and AQ202 were extracted by fitting the spectra with mono-exponential decays (acetonitrile) and bi-exponential decays (cobalt). However, the kinetic fit of AQ203 was extracted by fitting the spectra with tri-exponential decays (acetonitrile and cobalt). This may be due to the larger size and more donating nature of the CPDT group than those of thiophene and EDOT groups, or even to different aggregation behavior. It is likely that not only triphenylamine but also the CPDT unit is acting as a charge donor. And then, AQ203 showed kinetically complicated behavior under the same conditions, which probably refers to several complicated recombination processes. Fig. 8 shows the TAS signals of AQ201, AQ202, and AQ203 in the absence and presence of the cobalt redox electrolyte under similar conditions, and their fitting curves. In the absence of a redox mediator in pure acetonitrile solvent, the decay of the absorption signal presents the recombination process of the oxidized dye and the electrons injected in TiO2. Oxidized AQ201 has a lifetime of 478 μs. Elevating the HOMO level leads to a decrease in the lifetime of AQ202 and AQ203 to 316 and 262 μs, respectively, confirming hole stabilization by the lower HOMO levels. In the presence of the cobalt redox couple, regeneration of the oxidized dye occurs and significantly accelerates the decay of the oxidized dye signal. The lifetime is 13.4 μs for AQ201, 67.4 μs for AQ202 and 81.3 μs for AQ203. Assuming a first-order rate for both dye regeneration and back electron transfer reactions, we can estimate regeneration yields for all the dyes according to ηreg = kreg/(kreg + krec), ki = 1/τi. The summary of the lifetime and the dye regeneration yields (ηreg) is shown in Table 5. We conclude from these results that regeneration of the three dyes will be the limiting step in the operation of devices with the cobalt electrolyte. However, dye regeneration is certainly governed by the chemical structures of the dyes, especially with respect to its impact on the HOMO energy level.
 |
| | Fig. 8 Microsecond normalized transient absorption decays of AQ201, AQ202 and AQ203 dyes at 1050 nm (a) in the absence (no iodide and cobalt electrolytes added) and (b) in the presence of the cobalt redox electrolyte under similar conditions. | |
Table 5 Time constants extracted from the fits of transient absorption and calculated regeneration yields
| Dye |
τ
rec (μs) |
τ
reg cobalt (μs) |
η
reg cobalt
|
|
AQ201
|
478 |
13.4 |
0.97 |
|
AQ202
|
316 |
67.4 |
0.83 |
|
AQ203
|
262 |
81.3 |
0.76 |
Conclusion
In summary, we have synthesized three new quinoxaline-based organic dyes and studied the effect of the different π-bridges on the HOMO levels of the dyes and the performance of DSSCs with [Co(bpy)3]2+/3+ and I−/I3− based electrolytes. The cyclic voltammograms show that the HOMO levels of the dyes increase in the trend of AQ203 > AQ202 > AQ201, while the LUMO levels of these dyes remained the same. It is interesting to note that the AQ201-based DSSC with the lowest HOMO level showed the poorest PCE of 7.10% with the cobalt-based electrolyte, but the best PCE of 6.57% with the iodide-based electrolyte. Moreover, the AQ202-based cell shows the highest PCE of 8.37% with the cobalt-based electrolyte, due to a wider absorption spectrum as well as a higher driving force for dye regeneration. Last but not least, the AQ203-based cells are not the best with either [Co(bpy)3]2+/3+ or I−/I3− based electrolyte despite the widest absorption spectrum and highest molar extinction coefficient, due to the smallest driving force for dye regeneration. Electrochemical impedance spectroscopy (EIS) and transient absorbance spectroscopy (TAS) indicate that the lower HOMO level leads to higher electron lifetime and dye regeneration efficiency. This work emphasizes the significance of the HOMO level of the dye in the trade-off between the spectral absorption range and energy level matching, which are responsible for determining the device PCE. Designing rational metal-free organic dyes will be focused in further work towards molecular energy level tuning, giving possibilities to increase the device performance.
Experimental
Materials
All chemicals and reagents were from commercial sources without purification. Solvents for chemical synthesis were purified by distillation. All chemical reactions were carried out under an argon atmosphere. The starting materials [4-bis(2′,4′-bis((2-ethylhexyl)oxy)biphenyl-4-yl)amino]phenylborate14b and 5,8-dibromo-2,3-diphenylquinoxaline11 were synthesized according to the published procedures.
Characterization
1H NMR and 13C NMR spectra were recorded on Bruker AM-400 MHz instruments with tetramethylsilane (TMS) as the internal standard. HRMS was performed by using a Waters LCT Premier XE spectrometer. The absorption spectra of the sensitizer dyes in solution and those adsorbed on TiO2 films were measured with a Varian Cary 500 spectrophotometer. The stability and redox behaviors of dye molecules were analyzed by cyclic voltammetry (CV) in CH2Cl2 solutions without deoxidization. The cyclic voltammograms (CV) were determined by using a CHI660C electrochemical workstation (Chenhua Co. Ltd, Shanghai, China) in a three-electrode cell. The working electrode was glassy carbon, used in conjunction with a Pt auxiliary electrode and an Ag/AgCl wire pseudo-reference electrode. All the experiments were performed in CH2Cl2 solutions containing 0.1 M tetrabutylammonium perchlorate (TBAP) at a scan rate of 100 mV s−1. The ferrocenium/ferrocene (Fc/Fc+) redox couple was used as an external potential reference.
Fabrication and photovoltaic measurements
4 or 8 μm TiO2 films composed of 20 nm nanoparticles, α-terpineol and ethyl cellulose, and a 4 μm thick scattering layer (60% nanoparticles of 20 nm and 40% large particles of 100 nm) was prepared according to the established method.10 These nanocrystal titania films were treated with 40 mM TiCl4 aqueous solution at room temperature overnight, following by sintering at 450 °C for 30 min. After cooling down to room temperature, these films were dipped into a 3 × 10−4 M solution of dyes in chloroform/ethanol (v/v: 3/7) mixed solvent with 5 mM CDCA for 3 h. After this, the electrodes were rinsed with CH2Cl2 and ethanol and then dried. The size of the electrodes was 0.12 cm2. The counter electrode (Pt-coated TCO) and the working electrode (dye-loaded TiO2 film) were sealed together by a hot-melt 30 μm thick spacer under heating. The Pt-counter electrodes were prepared following the literature.10 The iodide electrolyte contained 0.1 M LiI, 0.03 M I2, 0.5 M 1-butyl-3-methyl imidazolium iodide (BMII), 0.1 M guanidine isothiocyanate, and 0.5 M 4-tert-butylpyridine (TBP) in acetonitrile:valeronitrile (85:15, v/v), and the cobalt electrolyte contained 0.22 M [Co(II) (bpy)3](TFSI)2, 0.06 M [Co(III) (bpy)3](TFSI)3, 0.1 M LiClO4, and 0.5 M 4-tert-butylpyridine in acetonitrile, (bpy = 2,2′-bipyridine, and TFSI = bis(trifluoromethane)sulfonamide).
The DSSCs were evaluated by recording the J–V curves with a Keithley 2400 source meter under the illumination of Air Mass 1.5 G simulated solar light coming from a solar simulator (Oriel-91160 equipped with a 300 W Xe lamp). Incident monochromatic IPCE spectra were obtained with an Oriel-74125 system and the intensity of incident monochromatic light was measured with a Si detector (Oriel-71640). Electrical impedance experiments (EIS) were performed in the dark. The spectra were scanned in a frequency range from 0.1 Hz to 100 kHz at room temperature with the applied potential set at open circuit. The alternate potential amplitude was set at 10 mV.
Photoinduced charge-transfer dynamics
The nanosecond laser flash photolysis technique was applied to the dye-sensitized, 4 μm thick, transparent TiO2 mesoporous films deposited on normal flint glass. The samples were excited by pulses produced at a repetition rate of 30 Hz by a broad-band optical parametric oscillator (GWU OPO-355) pumped by a Powerlite 7030 frequency-tripled Q-switched Nd:YAG laser (Continuum, Santa Clara, California, USA). The output excitation wavelength was tuned at λexc = 530 nm with a pulse width of 7 ns (fwhm). The laser beam was expanded by a planoconcave lens to irradiate a large cross-section of the sample, whose surface was kept at a 45° angle to the excitation beam. The probe light, produced by a continuous wave xenon arc lamp, was first passed through a monochromator tuned in 20 nm steps throughout the 420–740 nm range of the VIS light spectrum. The probe light passed various optical elements, the sample, and then a second monochromator, before being detected by a fast photomultiplier tube (Hamamatsu, R9110). Data waves were recorded on a DSA 602A digital signal analyzer (Tektronix, Beaverton, Oregon, USA). Satisfactory signal-to-noise ratios were typically obtained by averaging over 1500 laser shots. The transient absorption measurements were performed on the previously described dye-sensitized, 4 μm-thick, transparent TiO2 films either immersed in [Co(bpy)3]2+/3+ or I−/I3− based electrolyte.
Synthesis
Synthesis of 2a.
Under an argon atmosphere, compound 1 (439 mg, 1.0 mmol), K2CO3 (5 mL, 2 M aqueous solution) and Pd(PPh3)4 (42 mg, 0.036 mmol) were dissolved in 20 mL of THF. After stirring for half an hour, the mixture was heated to 50 °C and a solution of (4-formylphenyl)boronic acid (1.0 mmol) in THF (5 mL) was added slowly. The mixture was then refluxed for 12 h before cooling to room temperature and extracted with CH2Cl2. The organic layers were washed with brine water and dried over anhydrous Na2SO4. After removing the solvent under reduced pressure, the residue was chromatographed on a silica gel column with PE/CH2Cl2 (3/1–1/1, v/v) as eluent to give compound 2 (260 mg, 56% yield). 1H NMR (400 MHz, CDCl3, ppm): δ = 10.15 (s, 1H), 8.18 (d, J = 8.4 Hz, 1H), 8.06 (d, J = 8.4 Hz, 2H), 7.99 (d, J = 8.8 Hz, 2H), 7.75–7.71 (m, 3H), 7.56 (d, J = 8.8 Hz, 2H), and 7.44–7.32 (m, 6H). 13C NMR (100 MHz, CDCl3, ppm): δ = 192.19, 153.26, 153.01, 143.81, 139.03, 138.74, 138.68, 138.29, 138.23, 135.47, 132.92, 131.44, 130.34, 130.19, 130.00, 129.46, 129.37, 129.33, 128.38, 128.29, and 124.51. HRMS (m/z): [M + H+] calcd for C27H18N2OBr, 465.0602; found: 465.0538.
Synthesis of 3a.
Under an argon atmosphere, 2-(tributylstannyl)thiophene (193 mg, 0.52 mmol), compound 2 (200 mg, 0.43 mmol) and Pd(dppf)Cl2 (7.3 mg, 0.01 mmol) were dissolved in anhydrous toluene (20 mL) and refluxed for 12 h. The organic phase was extracted with CH2Cl2, washed with HCl (1 M), dried over Na2SO4 and concentrated under reduced pressure. The crude solid was purified by chromatography on silica gel using PE/CH2Cl2 (3/1–1/1, v/v) as eluent to give a yellow solid 3a (180 mg, 89% yield). 1H NMR (400 MHz, CDCl3, ppm): δ = 10.13 (s, 1H), 8.23 (d, J = 8.8 Hz, 1H), 8.04 (s, 4H), 7.93 (d, J = 4.4 Hz, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.76–7.74 (m, 2H), 7.59–7.56 (m, 3H), 7.41–7.29 (m, 6H), and 7.22 (t, J = 4.2 Hz, 1H). 13C NMR (10 MHz, CDCl3, ppm): δ = 192.27, 152.05, 151.79, 144.45, 138.69, 138.63, 138.53, 138.51, 137.21, 137.10, 135.27, 133.07, 131.48, 130.44, 130.27, 130.03, 129.75, 129.36, 129.14, 128.88, 128.31, 128.28, 126.90, 126.84, and 126.72. HRMS (m/z): [M + H+] calcd for C31H21N2OS, 469.1375; found: 469.1388.
Synthesis of 4a.
Compound 3a (160 mg, 0.34 mmol) and N-bromosuccinimide (NBS) (73 mg, 0.41 mmol) were dissolved in anhydrous CH2Cl2 in an ice bath for 2 h. Then, a Suzuki coupling reaction was performed with [4-bis(2′,4′-bis((2-ethylhexyl)oxy)bi-phenyl-4-yl)amino]phenylborate by using a similar procedure to that of compound 2 to give an orange solid 4a (210 mg, 45% yield). 1H NMR (400 MHz, CDCl3, ppm): δ = 10.12 (s, 1H), 8.23 (d, J = 8.8 Hz, 1H), 8.04 (s, 4H), 7.83 (d, J = 4.4 Hz, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.80–7.78 (m, 2H), 7.61–7.59 (m, 4H), 7.45 (d, J = 8.2 Hz, 4H), 7.41–7.39 (m, 3H), 7.36–7.27 (m, 6H), 7.22–7.18 (m, 6H), 6.57–6.55 (m, 4H), 3.89–3.86 (m, 8H), 1.76–1.68 (m, 4H), 1.45–1.26 (m, 32H), and 0.97–0.87 (m, 24H). 13C NMR (100 MHz, CDCl3, ppm): δ = 192.26, 159.93, 157.27, 152.02, 151.56, 147.80, 147.67, 145.61, 144.51, 138.75, 138.66, 138.59, 137.04, 136.71, 135.23, 133.46, 133.12, 131.48, 130.77, 130.55, 130.48, 130.34, 130.08, 129.37, 129.21, 129.17, 128.30, 128.12, 126.42, 126.08, 123.96, 123.44, 123.17, 122.03, 105.22, 100.35, 70.75, 70.56, 39.54, 39.44, 30.70, 30.64, 29.20, 29.07, 24.13, 23.97, 23.16, 23.14, 14.21, 11.27, and 11.25. HRMS (m/z): [M + H+] calcd for C93H106N3O5S, 1376.7853; found: 1376.7836.
Synthesis of AQ201.
Under an argon atmosphere, a mixture of 4a (170 mg, 0.12 mmol), cyanoacetic acid (85 mg, 1 mmol), and ammonium acetate (120 mg) in acetic acid (25 mL) and THF (5 mL) was heated at reflux for 12 h. After cooling down, the mixture was poured into water. The precipitates were collected by filtration and washed with water. The residue was purified by column chromatography on silica gel with CH2Cl2/ethanol (10:1, v/v) to yield a red solid AQ201 (120 mg, 69% yield). 1H NMR (400 MHz, acetone-d6, ppm): 8.45–8.43 (m, 2H), 8.26 (d, J = 8.8 Hz, 2H), 8.14 (d, J = 8.2 Hz, 2H), 8.08–8.04 (m, 2H), 7.83 (d, J = 8.4 Hz, 2H), 7.71 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 8.8 Hz, 2H), 7.51–7.49 (m, 5H), 7.45–7.44 (m, 3H), 7.40–7.35 (m, 3H), 7.28 (d, J = 8.4 Hz, 2H), 7.17 (d, J = 8.8 Hz, 6H), 6.69 (s, 2H), 7.64 (d, J = 8.2 Hz, 2H), 396–3.94 (m, 8H), 1.76–1.68 (m, 4H), 1.50–1.30 (m, 32H), and 0.98–0.86 (m, 24H). 13C NMR (100 MHz, acetone-d6, ppm): δ = 161.01, 158.17, 149.46, 146.41, 143.98, 134.76, 132.50, 131.43, 131.35, 130.92, 129.07, 129.02, 127.16, 124.71, 124.13, 123.68, 123.05, 106.71, 101.08, 71.35, 71.04, 40.43, 40.35, 31.51, 31.34, 24.61, 23.78, 23.76, 14.46, 14.39, 11.64, 11.48, and 5.45. HRMS (m/z): [M + H+] calcd for C96H107N4O6S, 1443.7911; found: 1443.7953.
Synthesis of 3b.
Compound 3b was obtained by using a similar procedure to that of 3a (150 mg, 75% yield). 1H NMR (400 MHz, CDCl3, ppm): δ = 10.05 (s, 1H), 8.63 (d, J = 8.8 Hz, 1H), 7.96 (s, 4H), 7.82 (d, J = 4.4 Hz, 1H), 7.69 (d, J = 8.4 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 7.32–7.23 (m, 6H), and 7.55 (s, 1H). 13C NMR (100 MHz, CDCl3, ppm): δ = 191.25, 150.66, 150.06, 143.61, 140.35, 139.74, 137.71, 137.46, 137.25, 136.09, 134.83, 134.07, 130.63, 130.41, 129.48, 129.37, 128.98, 128.28, 127.98, 127.19, 126.64, 111.86, 102.97, 64.02, and 63.27. HRMS (m/z): [M + H+] calcd for C33H23N2O3S, 527.1429; found: 527.1428.
Synthesis of 4b.
Compound 4b was obtained by using a similar procedure to that of 4a (190 mg, 80% yield).1H NMR (400 MHz, CDCl3, ppm): δ = 10.11 (s, 1H), 8.74 (d, J = 8.8 Hz, 1H), 8.03 (s, 4H), 7.88 (d, J = 8.4 Hz, 1H), 7.84–7.82 (m, 2H), 7.75 (d, J = 8.4 Hz, 2H), 7.60 (d, J = 8.4 Hz, 2H), 7.45 (d, J = 8.8 Hz, 4H), 7.39–7.35 (m, 4H), 7.33–7.23 (m, 6H), 7.20–7.18 (m, 4H), 6.57–6.56 (m, 4H), 4.48–4.47 (m, 2H), 4.42–4.41 (m, 2H), 390–3.87 (m, 8H), 1.77–1.70 (m, 4H), 1.45–1.27 (m, 32H), and 0.98–0.87 (m, 24H). 13C NMR (100 MHz, CDCl3, ppm): δ = 192.31, 159.86, 157.23, 151.67, 150.82, 138.81, 138.49, 135.78, 135.08, 131.46, 130.67, 130.34, 130.06, 129.36, 129.14, 129.05, 128.26, 128.18, 125.55, 123.73, 123.19, 121.37, 105.18, 100.31, 70.72, 70.55, 64.94, 64.39, 39.50, 39.40, 34.25, 30.64, 30.59, 30.35, 29.73, 29.47, 29.15, 29.02, 24.08, 23.92, 23.11, 23.08, 14.15, 11.20, and 11.19. HRMS (m/z): [M + H+] calcd for C95H108N3O7S, 1434.7908; found: 1437.7910.
Synthesis of AQ202.
Compound AQ202 was obtained by using a similar procedure to that of AQ201 (90 mg, 78% yield). 1H NMR (400 MHz, acetone-d6, ppm): δ = 8.73 (s, 1H), δ = 8.37 (s, 1H), 8.18 (d, J = 8.8 Hz, 1H), 8.06 (d, J = 8.8 Hz, 1H), 7.88–7.72 (m, 5H), 7.59 (d, J = 8.4 Hz, 2H), 7.45 (d, J = 4.2 Hz, 4H), 7.36–7.28 (m, 6H), 7.23 (d, J = 8.4 Hz, 2H), 7.12–7.18 (m, 6H), 6.66–6.56 (m, 4H), 4.43–4.36 (m, 4H), 3.92–3.91 (m, 8H), 1.73–1.66 (m, 4H), 1.44–1.28 (m, 32H), and 0.97–0.84 (m, 24H). 13C NMR (100 MHz, acetone-d6, ppm): δ = 160.92, 158.14, 155.10, 146.51, 139.87, 139.47, 134.39, 132.43, 131.58, 131.45, 131.32, 131.25, 130.97, 129.74, 128.93, 127.75, 124.41, 123.74, 116.75, 106.65, 103.72, 101.13, 71.38, 71.04, 65.16, 40.40, 40.35, 31.50, 31.34, 24.84, 24.61, 23.77, 14.49, 14.42, 11.65, and 11.50. HRMS (m/z): [M + H+] calcd for C114H140N3O5S2, 1501.7966; found: 1501.7733.
Synthesis of 3c.
Compound 3c was obtained by using a similar procedure to that of 3a (320 mg, 47% yield). 1H NMR (400 MHz, CDCl3, ppm): δ = 10.12 (s, 1H), 8.22–8.20 (m, 1H), 8.03 (s, 4H), 7.88–7.86 (m, 2H), 7.79–7.77 (m, 2H), 7.58 (d, J = 4.4 Hz, 2H), 7.45–7.44 (m, 3H), 7.35–7.29 (m, 3H), 7.20 (d, J = 4.2 Hz, 1H), 6.99–6.97 (m, 1H), 1.97–1.93 (m, 4H), 1.29–1.22 (m, 2H), 1.01–0.88 (m, 16H), and 0.78–0.60 (m, 12H).
Synthesis of 4c.
Compound 4c was obtained by using a similar procedure to that of 4a (250 mg, 65% yield).1H NMR (400 MHz, CDCl3, ppm): δ = 10.05 (s, 1H), 8.13 (s, 1H), 7.97 (s, 4H), 7.80 (d, J = 8.4 Hz, 2H), 7.72–7.70 (m, 2H), 7.51 (d, J = 8.8 Hz, 2H), 7.42–7.35 (m, 9H), 7.28–7.21 (m, 5H), 7.10 (d, J = 8.2 Hz, 7H), 6.49–6.48 (m, 4H), 3.82–3.78 (m, 8H), 1.94–1.86 (m, 4H), 1.69–1.61 (m, 4H), 1.35–1.18 (m, 34H), 0.95–0.80 (m, 40H), and 0.68–0.55 (m, 12H). 13C NMR (100 MHz, CDCl3): δ = 192.29, 159.86, 157.21, 138.79, 138.65, 135.12, 133.28, 131.42, 130.69, 130.49, 130.38, 130.05, 129.39, 129.07, 128.37, 128.26, 125.92, 123.73, 123.15, 105.15, 100.27, 70.69, 70.52, 43.43, 39.48, 39.39, 35.21, 34.19, 30.63, 30.57, 29.14, 29.01, 28.61, 27.45, 24.04, 23.90, 23.10, 23.07, 22.84, 14.14, 11.17, and 10.70. HRMS (m/z): [M + H+] calcd for C114H140N3O5S2, 1695.0234; found: 1695.0217.
Synthesis of AQ203.
Compound AQ203 was obtained by using a similar procedure to that of AQ201 (85 mg, 63% yield). 1H NMR (400 MHz, acetone-d6, ppm): δ = 8.35 (d, J = 8.4 Hz, 1H), 8.29 (s, 1H), 8.16–8.12 (m, 3H), 8.03 (d, J = 8.8 Hz, 2H), 7.93 (d, J = 8.8 Hz, 1H), 7.69–7.67 (m, 2H), 7.54–7.50 (m, 4H),7.37–7.35 (m, 7H), 7.24–7.22 (m, 3H), 7.15–7.13 (m, 3H), 7.04–7.00 (m, 6H), 6.56 (s, 2H), 6.48 (d, J = 8.8 Hz, 2H), 3.83–3.81 (m, 8H), 1.64–1.53 (m, 4H), 1.24–1.16 (m, 34H), 0.85–0.74 (m, 40H), and 0.60–0.51 (m, 12H). 13C NMR (100 MHz, acetone-d6) δ = 160.98, 158.16, 146.42, 139.96, 134.61, 132.43, 131.43, 131.35, 130.92, 129.13, 129.00, 124.54, 123.69, 106.69, 101.09, 71.35, 71.05, 54.79, 43.91, 40.43, 40.34, 36.11, 35.02, 32.64, 31.51, 31.34, 24.82, 24.61, 23.78, 23.76, 23.57, 23.51, 23.34, 14.49, 14.41, 11.65, 11.50, and 11.18. HRMS (m/z): [M + H+] calcd for C117H141N4O6S2, 1762.0293; found: 1762.0281.
Acknowledgements
For financial support of this research, we thank the Science Fund for Creative Research Groups (21421004), the National Basic Research 973 Program (2013CB733700), and NSFC/China (21172073, 21372082, 21572062 and 91233207). Yue Hu thanks the China Scholarship Council and the University of Edinburgh for a studentship. This work has made use of the resources provided by the Edinburgh Compute and Data Facility (ECDF) (http://www.ecdf.ed.ac.uk/).
Notes and references
-
(a) B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef;
(b) M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed;
(c) B. E. Hardin, H. J. Snaith and M. D. McGehee, Nat. Photonics, 2012, 6, 162–169 CrossRef CAS;
(d) S. F. Zhang, X. D. Yang, Y. Numata and L. Y. Han, Energy Environ. Sci., 2013, 6, 1443–1464 RSC;
(e) Y.-S. Yen, H.-H. Chou, Y.-C. Chen, C.-Y. Hsu and J. T. Lin, J. Mater. Chem., 2012, 22, 8734–8747 RSC.
-
(a) A. Hagfeldt, G. Boschloo, L. C. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed;
(b) Y. Ooyama and Y. Harima, ChemPhysChem, 2012, 13, 4032–4080 CrossRef CAS PubMed;
(c) Z. J. Ning, Y. Fu and H. Tian, Energy Environ. Sci., 2010, 3, 1170–1181 RSC.
-
(a) J. Y. Cong, X. C. Yang, L. Kloo and L. C. Sun, Energy Environ. Sci., 2012, 5, 9180–9194 RSC;
(b) J. W. Ondersma and T. W. Hamann, Coord. Chem. Rev., 2013, 257, 1533–1543 CrossRef CAS;
(c) J. N. Clifford, E. Palomares, M. K. Nazeeruddin, M. Grätzel and J. R. Durrant, J. Phys. Chem. C, 2007, 111, 6561–6567 CrossRef CAS.
-
(a) G. Boschloo and A. Hagfeldt, Acc. Chem. Res., 2009, 42, 1819–1826 CrossRef CAS PubMed;
(b) M. Wang, C. Grätzel, S. M. Zakeeruddin and M. Grätzel, Energy Environ. Sci., 2012, 5, 9394–9405 RSC;
(c) T. W. Hamann, Dalton Trans., 2012, 41, 3111–3115 RSC;
(d) A. Yella, H.-W. Lee, H. N. Tsao, C. Y. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed;
(e) S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
-
(a) A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed;
(b) B.-G. Kim, K. Chung and J. Kim, Chem.–Eur. J., 2013, 19, 5220–5230 CrossRef CAS PubMed;
(c) J. N. Clifford, E. Martĺnez-Ferrero, A. Viterisi and E. Palomares, Chem. Soc. Rev., 2011, 40, 1635–1646 RSC.
-
(a) C. Teng, X. C. Yang, S. F. Li, M. Cheng, A. Hagfeldt, L.-Z. Wu and L. C. Sun, Chem.–Eur. J., 2010, 16, 13127–13138 CrossRef CAS PubMed;
(b) J. Massin, L. Ducasse, M. Abbas, L. Hirsch, T. Toupance and C. Olivier, Dyes Pigm., 2015, 118, 76–87 CrossRef CAS;
(c) D. P. Hagberg, T. Marinado, K. Martin Karlsson, K. Nonomura, P. Qin, G. Boschloo, T. Brinck, A. Hagfeldt and L. C. Sun, J. Org. Chem., 2007, 72, 9550–9556 CrossRef CAS PubMed;
(d) X. F. Lu, Q. Y. Feng, T. Lan, G. Zhou and Z.-S. Wang, Chem. Mater., 2012, 24, 3179–3187 CrossRef CAS;
(e) A. Yella, R. Humphry-Baker, B. F. E. Curchod, N. A. Astani, J. Teuscher, L. E. Polander, S. Mathew, J.-E. Moser, I. Tavernelli, U. Rothlisberger, M. Grätzel, M. K. Nazeeruddin and J. Frey, Chem. Mater., 2013, 25, 2733–2739 CrossRef CAS.
-
(a) M. Velusamy, K. R. J. Thomas, J. T. Lin, Y.-C. Hsu and K.-C. Ho, Org. Lett., 2005, 7, 1899–1902 CrossRef CAS PubMed;
(b) W. H. Zhu, Y. Z. Wu, S. T. Wang, W. Q. Li, X. Li, J. Chen, Z.-S. Wang and H. Tian, Adv. Funct. Mater., 2011, 21, 756–763 CrossRef CAS.
-
(a) Y. Z. Wu, W. H. Zhu, S. M. Zakeeruddin and M. Grätzel, ACS Appl. Mater. Interfaces, 2015, 7, 9307–9318 CrossRef CAS PubMed;
(b) W. J. Ying, J. B. Yang, M. Wielopolskic, T. Moehl, J.-E. Moser, P. Comte, J. L. Hua, S. M. Zakeeruddin, H. Tian and M. Grätzel, Chem. Sci., 2014, 5, 206–214 RSC;
(c) Y. Z. Wu and W. H. Zhu, Chem. Soc. Rev., 2013, 42, 2039–2058 RSC.
- X. Li, S. C. Cui, D. Wang, Y. Zhou, H. Zhou, Y. Hu, J.-G. Liu, Y. T. Long, W. J. Wu, J. L. Hua and H. Tian, ChemSusChem, 2014, 7, 2879–2888 CrossRef CAS PubMed.
- J. B. Yang, P. Ganesan, J. Teuscher, T. Moehl, Y. J. Kim, C. Y. Yi, P. Comte, K. Pei, T. W. Holcombe, M. K. Nazeeruddin, J. L. Hua, S. M. Zakeeruddin, H. Tian and M. Grätzel, J. Am. Chem. Soc., 2014, 136, 5722–5730 CrossRef CAS PubMed.
- X. Li, Y. Zhou, J. Chen, J. B. Yang, Z. W. Zheng, W. J. Wu, J. L. Hua and H. Tian, Chem. Commun., 2015, 51, 10349–10352 RSC.
-
(a) D. Joly, L. Pelleja, S. Narbey, F. Oswald, J. Chiron, J. N. Clifford, E. Palomares and R. Demadrille, Sci. Rep., 2014, 4, 4033 CrossRef CAS PubMed;
(b) S. Haid, M. Marszalek, A. Mishra, M. Wielopolski, J. Teuscher, J.-E. Moser, R. Humphry-Baker, S. M. Zakeeruddin, M. Grätzel and P. Bäuerle, Adv. Funct. Mater., 2012, 22, 1291–1302 CrossRef CAS;
(c) Z. Y. Yao, M. Zhang, H. Wu, L. Yang, R. Z. Li and P. Wang, J. Am. Chem. Soc., 2015, 137, 3799–3802 CrossRef CAS PubMed.
-
(a) M.-W. Lee, J.-Y. Kim, D.-H. Lee and M. J. Ko, ACS Appl. Mater. Interfaces, 2014, 6, 4102–4108 CrossRef CAS PubMed;
(b) H. N. Tsao, C. Y. Yi, T. Moehl, J.-H. Yum, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, ChemSusChem, 2011, 4, 591–594 CrossRef CAS PubMed.
- S. M. Feldt, E. A. Gibson, E. Gabrielsson, L. C. Sun, G. Boschloo and A. Hagfeldt, J. Am. Chem. Soc., 2010, 132, 16714–16724 CrossRef CAS PubMed.
- S.-L. Li, K.-J. Jiang, K.-F. Shao and L.-M. Yang, Chem. Commun., 2006, 2792–2794 RSC.
- Z.-S. Wang, Y. Cui, Y. Dan-oh, C. Kasada, A. Shinpo and K. Hara, J. Phys. Chem. C, 2007, 111, 7224–7230 CrossRef CAS.
- Y. Z. Wu, X. Zhang, W. Q. Li, Z.-S. Wang, H. Tian and W. H. Zhu, Adv. Energy Mater., 2012, 2, 149–156 CrossRef CAS.
-
M. J. Frisch, et al., Gaussian, Inc, Wallingford CT, 2009 Search PubMed.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS;
(b) T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- A. Schaefer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef CAS.
- M. Cossi and V. Barone, J. Chem. Phys., 2001, 115, 4708–4717 CrossRef CAS.
-
(a) F. Fabregat-Santiago, J. Bisquert, G. Garcia-Belmonte, G. Boschloo and A. Hagfeldt, Sol. Energy Mater. Sol. Cells, 2005, 87, 117–131 CrossRef CAS;
(b) F. Fabregat-Santiago, G. Garcia-Belmonte, I. Mora-Sero and J. Bisquert, Phys. Chem. Chem. Phys., 2011, 13, 9083–9118 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta07254a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a