
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of highly electrochemically active Li2S nanoparticles for lithium–sulfur-batteries†
M.
Kohl
ab,
J.
Brückner
ab,
I.
Bauer
ab,
H.
Althues
a and
S.
Kaskel
*ab
aFraunhofer Institute for Material and Beam Technology (IWS), Winterbergstraße 28, D-01277 Dresden, Germany
bDepartment of Inorganic Chemistry, Dresden University of Technology, Bergstraße 66, D-01062 Dresden, Germany. E-mail: Stefan.Kaskel@chemie.tu-dresden.de
First published on 3rd July 2015
Abstract
Carbothermal reduction of lithium sulfate below its melting point was used to produce sub-micron sized lithium sulfide particles which retain the morphology of the source particle. Using a lithium polysulfide-doped ether electrolyte, significantly enhanced activation of lithium sulfide could be accomplished in battery cells, achieving high discharge capacities up to 1360 mA h gsulfur−1 at a 0.1C rate. Showing an economically viable and scalable reaction routine for future lithium–sulfur-batteries.
Introduction
The lithium sulfur battery has the potential to significantly surpass current Li-ion battery limitations related to low intrinsic discharge capacities of intercalation cathode materials. At room temperature, sulfur has the highest known specific electrochemical capacity of solid materials (1672 mA h g−1), compared to about 250 mA h g−1 for LiCoO2.1 On a system cell level, energy densities exceeding 400 W h kg−1 are expected, rendering the lithium sulfur battery system as a highly attractive next generation battery with substantially increasing interest in recent years. Nevertheless many open questions, especially in regards to a possible entry into the field of consumer electronics and automotive, remain. The major challenge is to achieve both: long-life cycle reversibility and high energy densities.Recent work indicates that the high reactivity of the lithium metal anode limits the cycling reversibility through decomposition of electrolyte.2 Because of the dendritic growth of lithium during plating, metallic lithium without SEI-protection is formed in each cycle leading to undesired byproducts and a rapid depletion of liquid electrolytes. Due to this fact stabilization of the solid electrolyte interface is very difficult and prolonged cycle life is only observed at an excess of electrolyte.3–5 To overcome this problem alternative anode materials have been developed. Intercalation materials such as hard carbons6 or conversion electrodes such as silicon2,7 and tin8 do not form dendrites during charging and can form more stable SEIs. Improved cycling stability exceeding 1000 cycles has been reported.2,6 However, in order to use these materials as anodes, prelithiation as an additional and time consuming process step is necessary prior to final cell assembly. Another promising alternative is to use lithium sulfide (Li2S) as cathode active material. In recent years research of lithium sulfide as cathode material was not as advanced as it was for sulfur. However, lithium sulfur battery researchers are more and more addressing new strategies to use lithium sulfide in Li–S batteries, because no prelithiation of anodes is required when using lithium sulfide. The latter would allow processing schemes, analogue to Li-ion battery cathode active materials, which are also fully lithiated and do not need a lithium containing anode.9
Recently Li2S nanoparticles were reported to have enhanced electrochemical activity of over 1000 mA h g−1 of respective mass of sulfur. They use commercially available lithium sulfide, which is then processed by grinding or solution based precipitation.10–14 Either way, working with commercially available lithium sulfide, is not practicable for consumer oriented batteries, since prices are ≈400 times higher than for elemental sulfur. Also Li2S is usually delivered with other lithium salt impurities such as lithium oxide, -hydroxide and -carbonate. In addition, commercially available lithium sulfide consists of 30–100 μm sized particles and has no electrochemical activity due to the large particle size.15
In this work a novel approach for the synthesis of submicron-sized and highly electrochemically active Li2S-particles, based on the carbothermal reduction of lithium sulfate is reported. Ball milling is described as an effective method to reduce the particle size of the lithium sulfate starting material. Due to a tailored temperature program for the carbothermic reduction the small particle size of Li2SO4 could be preserved and transferred to the resulting Li2S particles. Moreover the addition of lithium polysulfide to the electrolyte will be demonstrated to have significant impact for enhanced lithium sulfide particle activation. Composite cathodes thereby achieve high lithium sulfide loadings of 3.5–4 mg cm−2 and a high active material utilization up to 83%.
Results and discussion
Lithium sulfide can be synthesized through carbothermic reduction of lithium sulfate according to the following equation:| Li2SO4 + xC → Li2S + xCOy (x = 1–4; y = 1–2) |
Corresponding to the chemical enthalpies and entropies of the reactants and products the Gibbs energies ΔGR of the carbothermic reduction reactions can be calculated in order to estimate the most likely reaction scheme at a given temperature. However, this does not include activation energies that need to be met and volatile products leaving the reaction zone immediately after synthesis.
According to the Gibbs free energies the reaction of four mole carbon with one mole lithium sulfate to lithium sulfide and carbon monoxide is most favored at temperatures above 725 °C (Fig. 1). The carbothermic reduction of lithium sulfate to lithium sulfide has been published since the 1950s however all published patents are referring to operation temperatures of over 845 °C, the melting point of lithium sulfate.16–18 However, according to our studies, lithium sulfide synthesized at this temperature has a discharge capacity of only 20 mA h gsulfur−1, (Fig. S1†) due to large particles size of the resulting lithium sulfide from the lithium sulfate melt in the Li2SO4/carbon black blend.
 | ||
| Fig. 1 Ellingham diagram for different carbothermal reductions. Numbers at the end of the lines are ΔGR values at 820 °C. | ||
To achieve smaller lithium sulfide particles a new approach of the carbothermic reduction was developed. Instead of heating above the melting point of lithium sulfate, lithium sulfate was heated and reduced below its melting point. A temperature of 820 °C, slightly below the melting point, was found to produce high performance Li2S-materials. The advantage is that lithium sulfide prepared in this way retains the morphology from the lithium sulfate with even smaller particle diameters than the parent sulfate. The latter is a consequence of volume reduction to 45% of the original lithium sulfate particle volume due to the lower molar volume of lithium sulfide compared to lithium sulfate. At this temperature, the reaction of lithium sulfate to lithium sulfide and carbon monoxide is favored, according to Fig. 1, which was also confirmed by the mass decrease from the reactants to solid products to 33.3 and 30.3% for samples with 66 and 69 wt% Li2SO4·H2O, respectively.
In order to evaluate a successful carbothermic reduction, SEM and X-ray diffraction (XRD) were performed. SEM images (Fig. 2) demonstrate a change in crystallinity from monocline (Li2SO4·H2O) to an octahedral crystal, which is an indication for Li2S particles. XRD measurements (Fig. 3) reveal the conversion of lithium sulfate monohydrate to lithium sulfide showing no other crystalline impurities.
 | ||
| Fig. 2 SEM images from a Li2SO4·H2O sample before (a) and after (b) the heat treatment at 820 °C under argon atmosphere. | ||
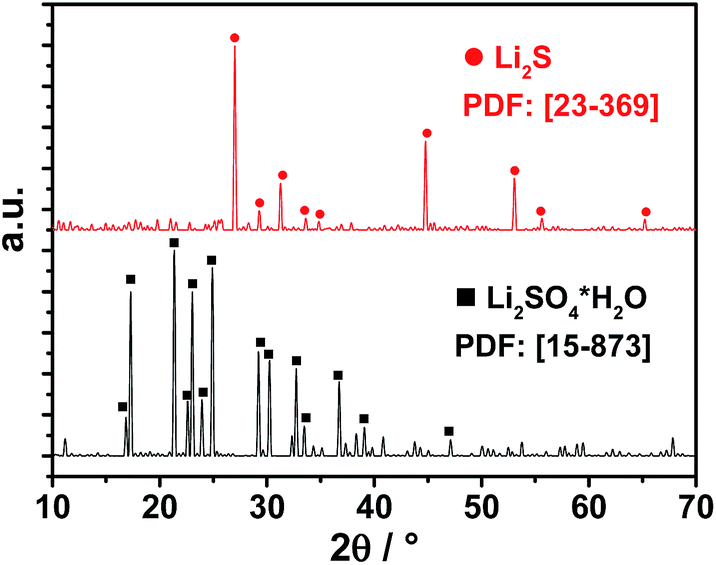 | ||
| Fig. 3 XRD powder patterns showing the conversion from Li2SO4·H2O (black lines) to Li2S (red lines) after carbothermic reduction at 820 °C. | ||
A commercially available carbon black (KB EC-600JD) with a high specific pore volume and surface area (Fig. 4) was employed to serve as both, the porous carbon host structure for the active species in the cathode, as well as the reductant for the carbothermic reduction.
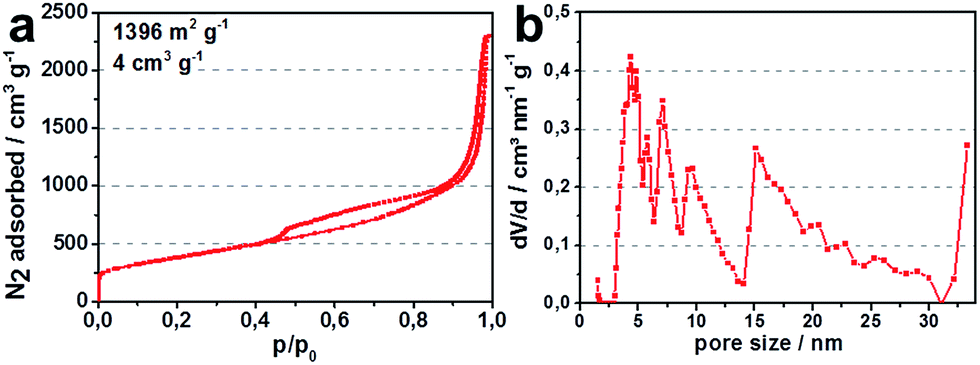 | ||
| Fig. 4 (a) N2-isotherm (77 K) for carbon black KB EC-600JD; (b) pore size distribution for KB EC-600JD. | ||
Fig. 5a illustrates discharge capacities, for lithium sulfide from 4 h ball milled lithium sulfate. High specific surface area enhances the amount of active reaction sites of the conductive carbon host matrix for the active species in the cell, lithium sulfide, polysulfides and sulfur. Therefore, more of the active species are able to participate in the cell reaction which increases the degree of utilization of active material.19 This is why commercially available KB EC-600JD with a total pore volume of 4 cm3 g−1 and surface area of 1396 m2 g−1 was chosen for the experiments. The high total pore volume, especially the mesopore volume of KB EC-600JD enables a high loading of active species infiltrated into the porous carbon host during cycling, leading to a sufficient distribution of active species within the pores. This contributes to good rate capability, high discharge potentials and only a slight decrease of discharge capacity during cycling of 0.15% per cycle between cycles 15 and 90 (Fig. 5a, capacity in mA h gcathode−1 is based on all in the cathode film existing materials. Meaning Li2S-corresponding mass of sulfur active material, carbon black, PTFE binder and spherical coles).
 | ||
| Fig. 5 (a) Galvanostatic discharge capacity over cycle number and (b) voltage profiles during charge and discharge for chosen cycles at a C-rate of 0.1 for Li2S based cathodes made from 4 h ball milled Li2SO4 with KB EC-600JD. Li2S activated in first charge up to a voltage of 3.5 V. | ||
To investigate the influence of the particle size on the specific capacity, Li2SO4 samples were ball milled for 12 and 60 h.
Prolonged dispersion ball milling of lithium sulfate leads to smaller particle sizes. Bulk Li2SO4 consists of ∼300 μm large particles, after 4 h of ball milling it decreases to ∼500 nm, after 12 h to ∼300 nm and after 60 h it decreases to ∼150 nm. The resulting lithium sulfide is about 50 to 100 nm in size (Fig. 6). Increasing the milling time further with the same parameters does not reduce the size of lithium sulfate particles further (Fig. S2†).
 | ||
| Fig. 6 SEM image of (a) bulk Li2SO4·H2O; (b) Li2SO4·H2O ball milled for 4 h; (c) Li2SO4·H2O ball milled for 60 h, TEM offset is including KB EC-600JD; (d) TEM image of Li2S and carbon black KB EC-600JD of Li2SO4·H2O ball milled for 60 h. | ||
With decreasing particle size of the lithium sulfate and of the resulting lithium sulfide, discharge capacities are significantly increasing. For 4 h and 60 h ball milling the difference in discharge capacity is ∼170 mA h gsulfur−1 or 30% (Fig. 7). The cycle stability before cell breakdown is more than 50 cycles with a linear decrease in capacity by 0.19% per cycle between cycle 15 and 40. The improved discharge capacity of smaller lithium sulfide particles results from the larger specific surface area of small particles compared to bigger counterparts. The reason is that kinetic limitation of charge-transfer between lithium sulfide particles and the electrolyte during the first charging, which converts lithium sulfide to polysulfides and sulfur, is faster for smaller particles.15 Therefore more lithium sulfide is activated and converted to sulfur at a given temperature, current rate and electrolyte amount. This results in a higher degree of utilization of lithium sulfide for smaller particles.
 | ||
| Fig. 7 Galvanostatic discharge capacity over cycle number at a C-rate of 0.1 for Li2S based cathodes made from 4 h and 60 h ball milled Li2SO4 with KB EC-600JD as porous carbon host structure in the cathode. | ||
We also found that the addition of lithium polysulfide to the electrolyte can promote charge-transfer between lithium sulfide particles and the electrolyte to overcome the kinetic limitations. Due to the presence of polysulfide as a catalyst, the extraction of S-species from Li2S is facilitated as compared to the direct formation of polysulfide from Li2S which requires the simultaneous oxidation and recombination of S-species on the Li2S surface.
A comparable process is the oxidation of lithium sulfide to polysulfides with the help of sulfur.20,21
When applied to the lithium sulfide containing battery cells an addition of only 0.125 M Li2S6 in the electrolyte increases the discharge capacity by ∼200 mA h gsulfur−1 at the beginning of the cycling process and 120 mA h gsulfur−1 at cycle 50 (Fig. 8a), because lithium polysulfides are oxidized to sulfur during the first charge, directly after cell assembly. The amount of lithium polysulfides, when regarded as active material contributing to the cell capacity, should increase the discharge capacity only by an amount of 67 mA h gsulfur−1. Thus, the addition of lithium polysulfides enhances the utilization of lithium sulfide in the cathode, presumably by promoting charge transfer effects at the surface of lithium sulfide particles.
 | ||
Fig. 8 (a) Galvanostatic discharge capacity over cycle number at a C-rate of 0.1 for Li2S based cathodes made from 60 h ball milled Li2SO4 with KB EC-600JD as porous carbon host structure in the cathode and 1 M LiTFSI, 0.25 M LiNO3 in DME/DOL (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) (filled symbols) or 1 M LiTFSI, 0.125 M LiNO3 and 0.125 M Li2S6 polysulfide in DME/DOL (1:1 v/v) (empty symbols), respectively, as electrolyte; (b) comparing the discharge capacities of cathodes consisting of 78 and 68 wt% Li2S with 0.125 M Li2S6 polysulfide containing electrolyte; voltage profiles during charge and discharge for chosen cycles at a C-rate of 0.1 for Li2S based cathodes made from 60 h ball milled Li2SO4, (c) without and (d) with 0.125 M Li2S6 addition to the electrolyte. :1 v/v) (filled symbols) or 1 M LiTFSI, 0.125 M LiNO3 and 0.125 M Li2S6 polysulfide in DME/DOL (1:1 v/v) (empty symbols), respectively, as electrolyte; (b) comparing the discharge capacities of cathodes consisting of 78 and 68 wt% Li2S with 0.125 M Li2S6 polysulfide containing electrolyte; voltage profiles during charge and discharge for chosen cycles at a C-rate of 0.1 for Li2S based cathodes made from 60 h ball milled Li2SO4, (c) without and (d) with 0.125 M Li2S6 addition to the electrolyte. | ||
The significant difference in discharge capacity between 68 wt% and 78 wt% lithium sulfide containing cathodes, especially in the first ten cycles, is based on the lower amount of electrochemical active species in the 68 wt% cell, leading to a better distribution along the porous carbon black host material (Fig. 8b). The continual decline of discharge capacities is based on the effect of the polysulfide shuttle, which is present especially after a cycle number of over 50, despite the addition of LiNO3 to the electrolyte solution. The polysulfide shuttle leads to a loss of electrochemical active material at the anode side. Due to an high active material loading of 3.5–4 mg cm−2 of Li2S and resulting high areal discharge capacities of about 2.9 to 3.4 mA h cm−2, the 0.125 M LiNO3 gets readily depleted from the electrolyte to due lithium formation during charging. Especially in the presented case, because only an amount of 15 μl of electrolyte was used in coin cell preparation, in order to be comparable to realistic cell manufacturing. This is the minimum amount which the electrodes need for sufficient wetting and reversible cycling. The depletion of nitrate during cycling is one of the main challenges in using this additive.22 Due to the reason that the discharge capacity degradation is mainly based on depleting nitrate, this effect can be delayed by giving more LiNO3 to the electrolyte solution. However by adding more lithium nitrate, especially in the case of the polysulfide containing electrolyte, the electrolyte gains in density and both used electrolytes, the one with and the one without polysulfide doping, are not directly comparable anymore.
In Fig. 9a the cyclic voltammograms for lithium sulfide cathodes with an electrolyte containing only conducting salt (LiTFSI) and lithium SEI former (LiNO3) in DME/DOL show in the 1st cycle a charge peak at 3.33 V vs. Li/Li+ in which lithium sulfide is oxidized to polysulfides and sulfur. For lithium sulfide embedded in micro- or mesopores of the porous carbon black this oxidation occurs normally at a voltage of 2.35 V. Therefore, all lithium sulfide particles exhibit an overpotential of ≈1 V for the conversion into polysulfides and sulfur during the 1st charge. The cathodic discharge peaks at 2.35 V and 2.06 V represent the electrochemical reduction of lithium polysulfides Sx2− (x = 5–8) to short chain polysulfide S42− and the reduction of S42− to S22− and Li2S, respectively.23 Consequently, after activation during the first charge, the synthesized lithium sulfide behaves like normal sulfur – which it gets oxidized to in the first charge. By adding lithium polysulfides Li2S6 to the electrolyte, the cyclic voltammogram of the 1st charge shows two separate peaks at 2.27 and 2.40 V vs. Li/Li+ corresponding to the oxidation of Li2S and polysulfides to sulfur. This sulfur on the other hand helps to oxidize the remaining Li2S particles at a voltage of 3.25 V, a lower voltage than it is needed for the polysulfide-free cells (Fig. 9). The following cycle displays exactly the same behavior as the cell without addition of polysulfides to the electrolyte, illustrating that after the initial activation of lithium sulfide particles the entire electrochemical active lithium sulfide is now contributing to the cell reaction during subsequent cycling and no further activation is needed.
 | ||
| Fig. 9 (a and b) Cyclic voltammograms for the two first cycles at a scan speed of 20 μV s−1, for Li2S based cathodes made from 60 h ball milled Li2SO4 with KB EC-600JD as porous carbon host structure in the cathode. (a) 1 M LiTFSI, 0.25 M LiNO3 in DME/DOL (1:1 v/v) and (b) 1 M LiTFSI, 0.125 M LiNO3 and 0.125 M Li2S6 polysulfide in DME/DOL (1:1 v/v) as electrolyte. | ||
The additional anodic peak at a voltage of 3.26 V vs. Li/Li+, in the 2nd cycle for either cells with or without polysulfide supplement can be attributed to the decomposition of LiTFSI conducting salt in the electrolyte. This is a common problem for high voltage Li-ion batteries, because decomposed LiTFSI is able to corrode aluminum based current collectors.24 For the cell without polysulfide addition this decomposition is visible as a shoulder at 3.25 V for the 1st cycle (Fig. 9b). The decomposition peak is below and also detected at somewhat lower voltages than the single oxidation peak for the first charge in the cell without polysulfides addition to the electrolyte (Fig. 9a). Thus, polysulfides or sulfur cannot be formed from lithium sulfide without any LiTFSI salt decomposition in the cell without polysulfide addition to the electrolyte. This can develop into a serious problem for the long term cycling stability, even if LiTFSI is only decomposed during the first charge.
It can be presumed, that even for the 1st cycle after cell assembly, no special formation is needed and cells only need to be activated to a voltage of 2.6 V vs. Li/Li+, the normal charge cut-off for lithium sulfur batteries,1 if polysulfides are added to the electrolyte. This can be also confirmed by the differences in the voltage profiles of the first charge between 4 h and 60 h ball milled samples (Fig. 5b and 8c, red line) and the 60 h ball milled sample with polysulfide addition to the electrolyte (Fig. 8d, red line). The initial formation of lithium polysulfides from lithium sulfide particles, which happens during the first overpotential peak,15 is 0.15 V lower for Li2S from 60 h ball milled Li2SO4 (3.23 V) than for 4 h ball milled one (3.38 V), due to faster activation kinetics of smaller lithium sulfide particles. When adding polysulfides to the electrolyte, the initial kinetic limitation and overpotential does not exist. Also the subsequent voltage plateau at 3.18 V and 2.90 V for the 4 h and 60 h ball milled samples, respectively, are lower for cells with polysulfide containing electrolyte with 2.45 V, demonstrating that initial charging to 2.6 V can be sufficient to activate sub-micron sized lithium sulfide particles in polysulfide containing electrolyte.
Conclusions
In summary, we have presented a novel approach for synthesizing nanosized lithium sulfide (Li2S) using an economically viable and scalable carbothermal reduction. Ball milled lithium sulfate was reduced at temperatures below its melting point to retain the submicron particle size. In addition it was found that smaller lithium sulfide particles exhibit significantly enhanced discharge capacities than larger particles. Furthermore, the crucial impact of lithium polysulfides as an electrolyte additive on the activation barriers of lithium sulfide was demonstrated. In particular, charging directly after cell assembly to high cut-off voltages of 3.4 V and above is not needed for 150 nm particle sized lithium sulfide when polysulfide is used as an additive.Experimental
In order to reduce the particle size and to ensure sufficient mixing, lithium sulfate monohydrate (Carl Roth, >99% p.a.) and Ketjen Black (KB EC-600JD) porous carbon black were ball milled for 4 h and 60 h respectively at 400 rpm with 10 min interval in a zirconia lined grinding beaker with 2 mm sized zirconia balls within a dispersion of ethanol. Subsequent carbothermal reduction of lithium sulfate monohydrate/Ketjen Black powder, with 66 or 69 wt% Li2SO4·H2O, was executed at 820 °C for 3 h with an argon gas flow of 1200 sccm in a 40 mm diameter tube furnace (HTM Reetz). Such treated samples with Li2S contents of 71 or 81 wt% were transferred into an argon filled glove box (MBraun) with monitored oxygen value of <0.1 ppm and moisture level of <0.1 ppm.Electrode sheets were prepared with a dry-process approach in an argon filled glove box as described elsewhere.25 Lithium sulfide/Ketjen Black powder is grinded with 3 wt% PTFE binder (Sigma Aldrich) and 1 wt% spherical coal as additive for better processing. Grinding of the powders leads to sheets with thicknesses of ≈100 μm. These sheets were then laminated onto carbon coated aluminum foil current collector. Circular electrode discs with a diameter of 10 mm (corresponding to 3.5–4 mg cm−2 lithium sulfide loading with a Li2S content of 68 and 78 wt%, respectively) were punched from the electrode sheets.
Coin cells (MTI Corp., CR2016) were assembled in a glove box. Metallic lithium (Pi-Kem, 99.0%, diameter 15.6 mm, thickness 250 μm) was used as anode. Porous polypropylene (Celgard 2500) was used as separator. The standard electrolyte was composed of 1 M lithium bis(trifluoromethylsulfonyl) imide (LiTFSI, Sigma Aldrich, 99.95%), 0.25 M lithium nitrate (LiNO3, Alfa Aesar, 99.98%, anhydrous) in 1,2-dimethoxyethane (DME, Sigma Aldrich, 99.5%, anhydrous) and 1,3-dioxolane (DOL, Sigma Aldrich, 99.8%, anhydrous) with a mixing ration of 1:1 v/v. The polysulfide containing electrolyte consisted of 1 M LiTFSI, 0.125 M LiNO3 and 0.125 M Li2S6 polysulfide in DME/DOL (1:1 v/v). The amount of electrolyte was fixed at 15 μL, as this is the minimum amount needed to fill the dead volume of the coin cell. Chemicals were used as received, except for LiTFSI, DME, and DOL. In order to remove residual water, LiTFSI and LiNO3 were dried at 120 °C under vacuum for 24 h before use and DME and DOL were dried and stored over a 3 Å molecular sieve.
Scanning electron microscope (SEM) images were acquired with a JEOL JSM-6610LV, TEM (transmission electron microscope) images with a Jeol JEM-2100, cyclic voltammetry measurements were performed with an Ivium-n-stat, X-ray diffraction powder patterns were recorded using a Siemens D5005, N2-isothermes were measured at 77 K on a Quantachrome QuadraSorb and galvanostatic charge/discharge cycling tests were performed with a BaSyTec Cell Test System (CTS) at room temperature. Cut-off voltages for cycling were 1.8 and 2.6 V or 3.4 V for the activation step, if not mentioned otherwise. Current rate during charge activation of lithium sulfide in the 1st cycle was at a C-rate of 0.01 if not mentioned otherwise. For cycling a C-rate of 0.1 (167 mA gsulfur−1) was chosen.
Acknowledgements
This research was financed by the Fraunhofer project LiScell. We are grateful for the support. TEM measurements were performed by Dr Joerg Kaspar from Fraunhofer IWS Dresden.Notes and references
- X. Ji and L. Nazar, J. Mater. Chem., 2010, 20, 9821–9826 RSC.
- Z. Wei Seh, W. Li, J. J. Cha, G. Zheng, Y. Yang, M. T. McDowell, P.-C. Hsu and Y. Cui, Nat. Commun., 2013, 4, 1331 CrossRef PubMed.
- Y. Yang, G. Zheng and Y. Cui, Energy Environ. Sci., 2013, 6, 1552 CAS.
- W. Li, G. Zheng, Y. Yang, Z. W. Seh, N. Liu and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7148 CrossRef CAS PubMed.
- S. Lu, Y. Cheng, X. Wu and J. Liu, Nano Lett., 2013, 13, 2485 CrossRef CAS PubMed.
- J. Brückner, S. Thieme, F. Böttger-Hiller, I. Bauer, H. T. Grossmann, P. Strubel, H. Althues, S. Spange and S. Kaskel, Adv. Funct. Mater., 2014, 24, 1284–1289 CrossRef PubMed.
- J. Hassoun, J. Kim, D.-J. Lee, H.-G. Jung, S.-M. Lee, Y.-K. Sun and B. Scrosati, J. Power Sources, 2012, 202, 308–313 CrossRef CAS PubMed.
- J. Hassoun and B. Scrosati, Angew. Chem., Int. Ed. Engl., 2010, 49, 2371–2374 CrossRef CAS PubMed.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- Y. Yang, M. T. McDowell, A. Jackson, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2010, 10, 1486–1491 CrossRef CAS PubMed.
- F. Wu, H. Kim, A. Magasinski, J. T. Lee, H.-T. Lin and G. Yushin, Adv. Energy Mater., 2014, 4, 1400196 Search PubMed.
- F. Wu, A. Magasinski and G. Yushin, J. Mater. Chem. A, 2014, 2, 6064 CAS.
- S. Jeong, D. Bresser, D. Buchholz, M. Winter and S. Passerini, J. Power Sources, 2013, 235, 220–225 CrossRef CAS PubMed.
- J. Guo, Z. Yang, Y. Yu, H. D. Abruña and L. A. Archer, J. Am. Chem. Soc., 2013, 135, 763–767 CrossRef CAS PubMed.
- Y. Yang, G. Zheng, S. Misra, J. Nelson, M. F. Toney and Y. Cui, J. Am. Chem. Soc., 2012, 134, 15387–15394 CrossRef CAS PubMed.
- E. Tor, United States Pat. Trademark Off., USPO395813, 1958, pp. 1–8.
- J. Hml, Eur. Pat. Off., EP0994071B, 2004, pp. 1–14.
- A. Grahame, Eur. Pat. Off., EP1187790B, 2004, pp. 3–7.
- J. Zheng, M. Gu, M. J. Wagner, K. A. Hays, X. Li, P. Zuo, C. Wang, J.-G. Zhang, J. Liu and J. Xiao, J. Electrochem. Soc., 2013, 160, A1624–A1628 CrossRef CAS PubMed.
- R. D. Rauh, F. S. Shuker, J. M. Marston and S. B. Brummer, J. Inorg. Nucl. Chem., 1977, 39, 1761–1766 CrossRef CAS.
- R. D. Rauh, K. M. Abraham, G. F. Pearson, J. K. Surprenant, S. B. Brummer and E. I. C. Corporation, J. Electrochem. Soc., 1979, 126, 523–527 CrossRef CAS PubMed.
- S. S. Zhang, Electrochim. Acta, 2012, 70, 344–348 CrossRef CAS PubMed.
- M. Gao, X. Xiong, W. Wang, S. Zhao, C. Li, H. Zhang, Z. Yu and Y. Huang, J. Power Sources, 2014, 248, 1149–1155 CrossRef CAS PubMed.
- H. Louis, Y.-G. Lee, K. M. Kim, W. I. Cho and J. M. Ko, Bull. Korean Chem. Soc., 2013, 34, 1795–1799 CrossRef CAS.
- S. Thieme, J. Brückner, I. Bauer, M. Oschatz, L. Borchardt, H. Althues and S. Kaskel, J. Mater. Chem. A, 2013, 1, 9225 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: S1: discharge capacity of cathodes from pristine Li2SO4·H2O reduced at 900 °C. S2: SEM image of 84 h ball milled Li2SO4·H2O. See DOI: 10.1039/c5ta04504e |
| This journal is © The Royal Society of Chemistry 2015 |