
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A triazine–resorcinol based porous polymer with polar pores and exceptional surface hydrophobicity showing CO2 uptake under humid conditions†
Shyamapada
Nandi
a,
Ulrike
Werner-Zwanziger
b and
Ramanathan
Vaidhyanathan
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research, Dr. Homi Bhabha Road, Pashan, Pune-411008, Maharashtra, India. E-mail: vaidhya@iiserpune.ac.in
bDepartment of Chemistry, IRM, NMR-3, Dalhousie University, 6274 Coburg Road, PO BOX 15000, Halifax, NS B3H4R2, Canada
First published on 8th September 2015
Abstract
Several applications including post-combustion carbon capture require capturing CO2 under humid conditions. To obtain a material capable of interacting more strongly with CO2 than water, surface hydrophobicity and polarizing pores have been incorporated simultaneously into an ultra-microporous Bakelite-type polymer comprising of triazine–triresorcinol building units. Being built from C–C bonds, it exhibits exceptional chemical stability (survives conc. HNO3(g) + SO3(g) without losing any porosity). Triazine–phenol lined channels enable adsorption of CO2 (2.8 mmol g−1 with a good selectivity of 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (85% N2:15% CO2) at 303 K, 1 bar) and the inherent surface hydrophobicity amply minimizes the affinity for H2O. When the adsorption was carried out using a humid CO2 stream (∼50% RH) the material loses only about 5% of its capacity. In a steam-conditioning experiment, the sample was exposed to high humidity (∼75% RH) for a day, and without any further activation, was tested for CO2 adsorption. It retains more than 85% of its CO2 capacity. And this capacity was intact even after 48 h of steam conditioning. The role of phenol in contributing to the surface hydrophobicity is exemplified by the fact that a ∼17% lithiation of the phenolic sites nearly removes all of the surface hydrophobicity. The local structure of the polymer has been modeled using tight-binding DFT methods (Accelrys) and three low energy conformers were identified. Only the CO2 isotherm simulated using the lowest energy conformer matches the experimental isotherm quite well. The triazine–phenol polymer presented here has good hydrophilic–hydrophobic balance, where the basic triazine units and the phenol groups seem to co-operatively assist the CO2 capture under humid conditions. These properties along with its excellent acid stability make the material a suitable candidate for post-combustion CO2 capture. Also, the study presents a new approach for simultaneously introducing polarizing character and surface hydrophobicity into a porous material.
:1 (85% N2:15% CO2) at 303 K, 1 bar) and the inherent surface hydrophobicity amply minimizes the affinity for H2O. When the adsorption was carried out using a humid CO2 stream (∼50% RH) the material loses only about 5% of its capacity. In a steam-conditioning experiment, the sample was exposed to high humidity (∼75% RH) for a day, and without any further activation, was tested for CO2 adsorption. It retains more than 85% of its CO2 capacity. And this capacity was intact even after 48 h of steam conditioning. The role of phenol in contributing to the surface hydrophobicity is exemplified by the fact that a ∼17% lithiation of the phenolic sites nearly removes all of the surface hydrophobicity. The local structure of the polymer has been modeled using tight-binding DFT methods (Accelrys) and three low energy conformers were identified. Only the CO2 isotherm simulated using the lowest energy conformer matches the experimental isotherm quite well. The triazine–phenol polymer presented here has good hydrophilic–hydrophobic balance, where the basic triazine units and the phenol groups seem to co-operatively assist the CO2 capture under humid conditions. These properties along with its excellent acid stability make the material a suitable candidate for post-combustion CO2 capture. Also, the study presents a new approach for simultaneously introducing polarizing character and surface hydrophobicity into a porous material.
Introduction
High surface area Metal Organic Frameworks (MOFs), Covalent Organic Frameworks (COFs), porous polymers, traditional zeolites and inorganic mesoporous materials are being investigated steadily for their applications in gas capture and separation.1–10 In many of the metal–organic based sorbents there are still severe concerns regarding their stability to extremely harsh environments which constitute the majority of industrial effluents (steam, acidic vapors, basic conditions, particulates, etc.).1–4,11–14 To address water stability, recently, modification in terms of the choice of the metal (Zr, Al, Cr, and Ni) or hydrophobic side chains or protecting groups on the ligand backbone have been carried out to improve the stability.15–19 Here we have taken a slightly different approach, wherein we have made metal-free organic frameworks built from exceptionally strong C–C bonds to address the stability issues. Of course, a similar approach has been adopted before, resulting in several porous polymers and composites.6,20–34 Some of them have shown excellent stabilities and CO2 capture capabilities.27–34 Recently, a family of porous covalent triazine frameworks (PCTF)35 have been synthesized under ionothermal conditions. They have nano- to microporous frameworks and some show exceptionally high surface areas (2235 m2 g−1)36 and exhibit good selectivities for CO2 over N2 and CH4.35,36 Melamine based microporous organic polymers,37 polymer nanosieve membranes,38 and polyethylene type porous organic polymers have also been investigated for gas adsorption.39 However, very few polymers have a CO2 uptake above 2 mmol g−1 under ambient conditions (Table S1†) with reasonable selectivity (>100). For example, some of the Porous Aromatic Framework (PAF) series show uptakes in excess of 2 mmol g−1 under ambient conditions.40 A series of porous polymer networks (PPNs) with exceptionally high surface areas and high pressure CO2, CH4 and H2 capacities have been reported by Yuan et al.31 But the compounds from both the PAF and PPN series do not have sufficiently high CO2 selectivities. However, loading of polyamines into PAF-5 gave excellent CO2 adsorbents, but such sorbents requiring a guest loading could have issues with obtaining consistent guest loading across multiple batches and during scale up, which has been realized also by the authors.41 We believe that it is advantageous to have active sites as a part of the framework. The advantage of using porous organic polymers was exemplified in a recent report wherein a humidity swing was employed to carry out CO2 capture.42 Also, the inherent ability of polymer derived porous carbons to capture CO2 under humid conditions was demonstrated by Lu and co-workers.43 In addition to the consideration of the stability of the material to humidity, its impact on CO2/N2 separation is also important.9,27,44In a typical post-combustion CO2 capture by solid adsorbents the effluent or flue gas at 90–100 °C is cooled down to 50–30 °C, stripped of acidic vapors (NOx and SOx) and water vapor and then the relatively dry stream (85% N2:15% CO2) is fed to the CO2 recovery/adsorption unit.3,11,45 The extent to which the flue gas stream is stripped of acidic fumes and dried has a direct impact on the cost and design complexity involved in the capture process itself. Developing solid sorbents which exhibit very good stability towards heat, steam and acidic fumes, with good CO2 uptake at room temperature and 1 bar and high CO2/N2 selectivity could minimize or take away stripping of humidity and reactive vapors and thereby could bring down the parasitic load on the process. Cross-linked organic oligomers and polymers are known for their exceptional chemical and thermal stability.24–31,46,47 When made from bulky monomers devoid of highly polar acidic or basic functionalities these cross-linked polymers occur as an insoluble powder, however, they generally do not have sufficient functionality to polarize gas molecules or to interact with them via other weak forces. Many of them show N2 uptake at 77 K while their CO2 capture abilities have not been explained.24,25,39 In microporous polymers wherein CO2 capture has been observed,25,48 majority of the CO2 capture happens via gas trapping in the micropores, and such pores could be amenable to water molecules as well. Very recently, CO2 capture using benzimidazole and triazine based polymers was reported,49,50 but they were not evaluated for CO2 capture under humid conditions. To capture CO2 under the humid flue gas conditions, functionalizing a pore with polarizing and basic groups favoring interactions with CO2 and simultaneously providing hydrophobicity to the pores would be effective, but this is quite challenging. In fact, MOFs made of highly polar carboxylates, phosphonates, sulfonates tend to interact with CO2via dispersive, electrostatic and quadrupolar forces which make them as excellent CO2 sorbents.51 Similarly, zeolites used in CO2 scrubbing, Zeolite-13x, has polarizing pore walls.52,53 Unfortunately, most of these polar MOFs adhere to water as much as they adhere to CO2. When such polar groups are sheathed by protective groups, a drastic decrease in CO2-framework interactions results,54 this makes tuning the material for CO2 over water a very tricky task. In an attempt to address the stability issues and capture of CO2 under humid conditions, here, we report a triazine–triresorcinol based ultra-microporous polymer having a highly hydrophobic surface and polar pore walls combining to form a Hydrophobic Polar Framework (HPF-1). Furthermore, we have used Tight-Binding Density Functional Theory (DFT-TB) calculations to simulate the structure of HPF-1, something which is being employed increasingly in recent times to obtain meaningful, structural insights on amorphous polymers.55,56
Experimental
The polymer is prepared using simple Bakelite chemistry by reacting triazine–triresorcinol with terephthaldehyde under solvothermal conditions. A reaction between 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tris(benzene-1,3-diol) (0.203 g; 0.5 mmol) and terephthaldehyde (0.100 g; 0.75 mmol) in a solution containing 5 ml 1,4-dioxane + 3 ml tetrahydrofuran was carried out by heating at 200 °C for 96 h (Fig. 1). The product, a yellowish brown color powder was isolated by filtration and was washed with dimethylformamide (15 ml), tetrahydrofuran (10 ml) and finally with methanol and acetone. The powder was found to be amorphous from powder X-ray diffraction (Fig. S1†).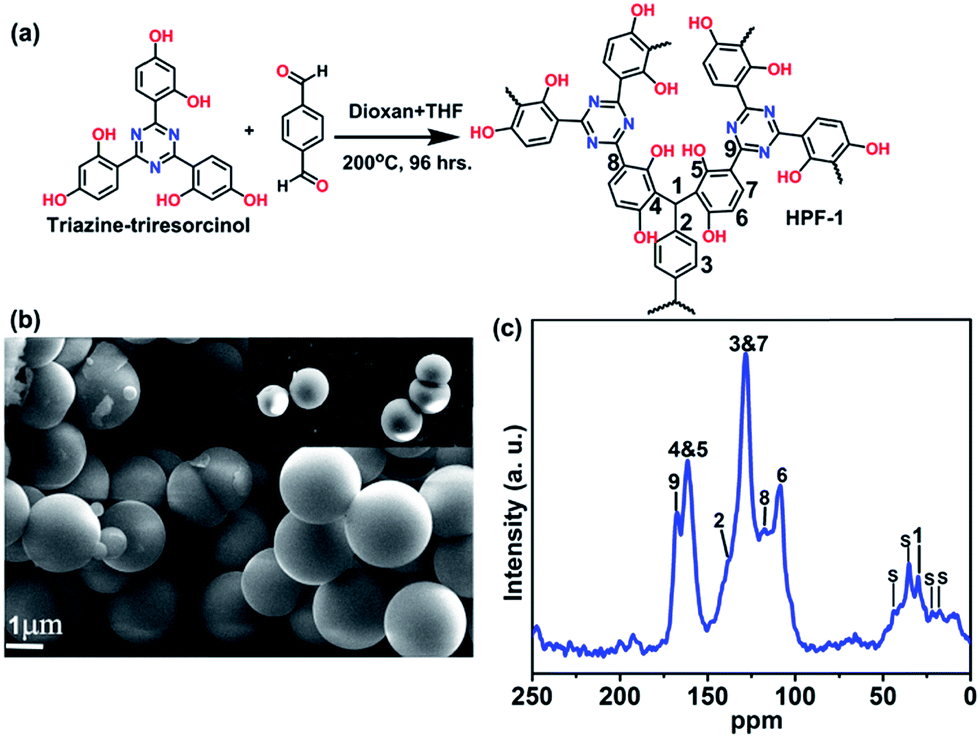 | ||
| Fig. 1 (a) The reaction involved in the synthesis of HPF-1. (b) Microspheres formed by HPF-1 as seen from the FE-SEM image. (c) Shows the 13C-MASNMR (400 MHz) of HPF-1, the peaks corresponding to the aromatic backbone and triazine groups can be observed. Few of the peaks in the aliphatic region correspond to the occluded THF and dioxan molecule in the polymer labelled as ‘s’. | ||
The field emission-SEM image indicated the sample to be highly homogeneous microspheres (Fig. 1b and S2†). Thermogravimetric analysis revealed exceptional thermal stability of up to 380 °C (Fig. S3†). We attribute the stability of HPF-1 to the strong C–C bond formed between the monomers, characteristic of Bakelite.48,57 Solid state NMR indicated the presence of Bakelite type couplings (Fig. 1c), along with some unsubstituted sites on the resorcinol rings and very few terminal aldehyde groups, which agreed well with the stretching frequencies observed in the infrared spectra (Fig. S4†).
Results and discussion
The porosity of HPF-1 was established using N2 adsorption carried out at 77 K (Fig. 2a). HPF-1 has a Brunauer–Emmett–Teller (BET) surface area of 576 m2 g−1 and a Langmuir surface area of 777 m2 g−1. A density functional theory based model fitted to the adsorption branch of the 77 K N2 isotherm shows the majority of the pores being 5.5 Å in size (inset of Fig. 2a). Different preparation batches were screened and they gave the same uptake indicating HPF-1 forming as a pure phase with good reproducibility. At 195 K, the material showed a CO2 uptake of 9.35 mmol g−1, which represents the saturation CO2 uptake capacity of the material (Fig. 2b). The material showed a moderate CO2 uptake of 2.8 mmol g−1 at 303 K, which is the one of the highest uptake reported for a porous polymer under ambient conditions (Table S1,† selected porous polymers have been compared in this table). Encouraged by seeing a very low uptake of N2 at room temperature, we carried out CO2 and N2 adsorptions at different temperatures.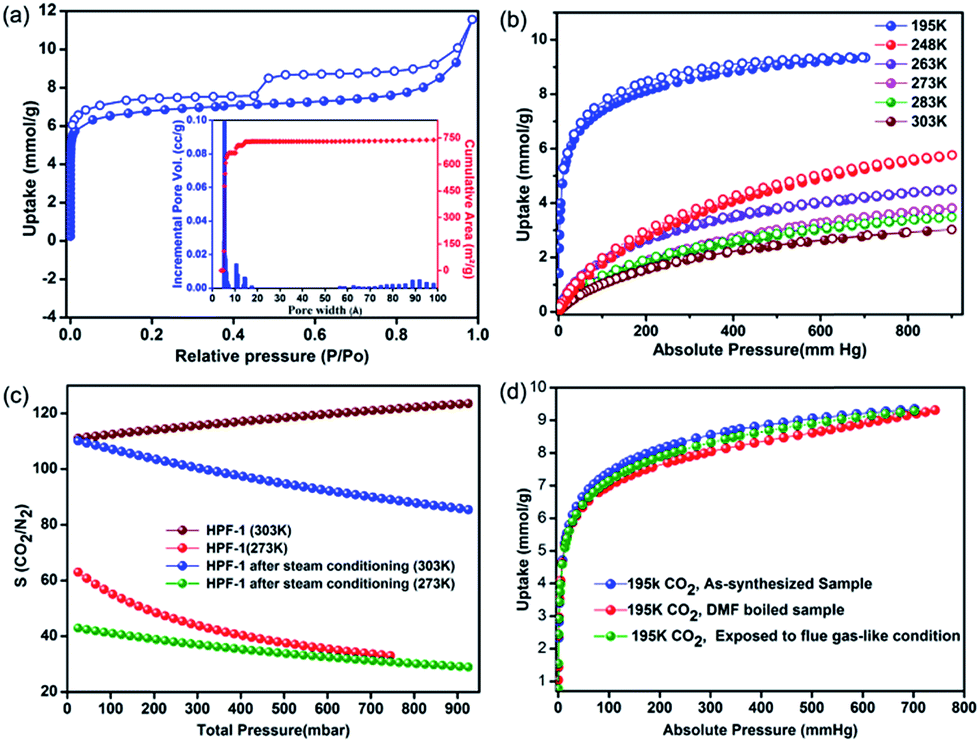 | ||
| Fig. 2 (a) The 77 K N2 adsorption isotherm of HPF-1. The inset shows the pore size distribution with a high concentration of ultra-micropores (5.5 Å) and a hierarchy of pores in the mesoporous regime but in extremely small relative concentrations. (b) Variable temperature CO2 isotherm with a saturation uptake of ∼9.5 mmol g−1 at 195 K and a 2.8 mmol g−1 at 303 K, 1 bar. (c) The IAST based CO2/N2 selectivity, it drops down in the presence of humidity, however, is still quite high (90) at 303 K. (d) Comparison of the 195 K saturation CO2 isotherms of the acid (conc. HNO3 + SO3(g) + boiling water) and solvent (DMF, 150 °C) treated HPF-1. | ||
The Ideal Adsorption Solution Theory (IAST) model was used to calculate the selectivity for CO2 over N2 for 85% N2:15% CO2 composition. It turns out that HPF-1 has a selectivity of 120:1 for CO2 at 303 K and 1 bar pressure (Fig. 2c). This is quite high (Table S1†) and can be attributed to the inherent molecular sieving effect of the ultra-micropores. Interestingly, the selectivity increases with increasing temperature, this is quite rare in porous polymers.20 This arises due to the CO2 capacity dropping down much gradually compared to the N2 with increasing temperatures. This could be due to the stronger interaction of weakly acidic CO2 with the triazine lined framework compared to N2. Such interactions would definitely be exaggerated in small micropores as those present in the polymer.
Chemical stability under harsh conditions such as acidic vapors and hydrolytic stability under steam and boiling water for such porous materials could make them potential candidates for a variety of application including flue gas capture. A typical gas-fired flue gas consists of the composition 7.4–7.7% CO2, 14.6% H2O, ∼4.45% O2, 200–300 ppm CO, 60–70 ppm NOx, and 73–74% N2.58 If a material stable to these harsh conditions can exhibit good low-pressure CO2 uptake and recyclability, they could make an apt candidate for vacuum swing CO2 separation applications.59,60 With this aim, we subjected HPF-1 to a steady stream of acidic vapors generated by heating a solution containing sulfur trioxide, SO3(g) (generated from chlorosulfonic acid) + conc. HNO3 + H2O for about 48 h. Considering the organic nature of HPF-1, in separate tests, we boiled it in water, DMF, water/DMF, DMF/DMSO, n-butanol and toluene. All the above harsh treatments did not result in any product decomposition or any drop in the porosity of the material, as evidenced from their saturation CO2 uptakes at 195 K (Fig. 2d). Demonstration of stability under such harsh chemical environments has been identified as crucial and a few studies on porous organic frameworks and metal organic frameworks have been reported recently.18,61
To understand the nature of the pore surface in this polymer, as it is functionalized with polar phenolic groups, we carried out vapor sorption studies using water and toluene as probes. It could be seen that the water sorption isotherm had type III behavior indicating weak adsorbate–adsorbent interactions (Fig. 3a). The heat of adsorption (HOA) calculated using virial analysis showed a value of 43 kJ mol−1 at zero-loading (Fig. 3b), which is lower than what was reported for a hydrophobic material and is just below the heat of vaporization of water.54 The toluene adsorption, however, shows type I behavior. This is reflected in the HOA for toluene with a value of 35 kJ mol−1 at zero-loading, which builds up to a value as high as 62 kJ mol−1 at 2.2 mmol g−1 loading. This could be due to strong adsorbate–adsorbate interactions which can be expected for toluene molecules trapped in confined pores. Thus, HPF-1 clearly has stronger interactions with toluene than water. A contact angle measurement on a sample that was prepared by spreading the as-synthesized HPF-1 powder on a glass substrate showed a contact angle of 153° for water (Fig. 3c), which confirms its highly hydrophobic surface. Some of the super-hydrophobic polymers touted for their hydrophobicity have this value at 164°.62 In spite of such a high contact angle, there is not much selectivity towards toluene in the sorption measurements suggesting that the interior of the material, comprising the pores, are not as hydrophobic as the surface.
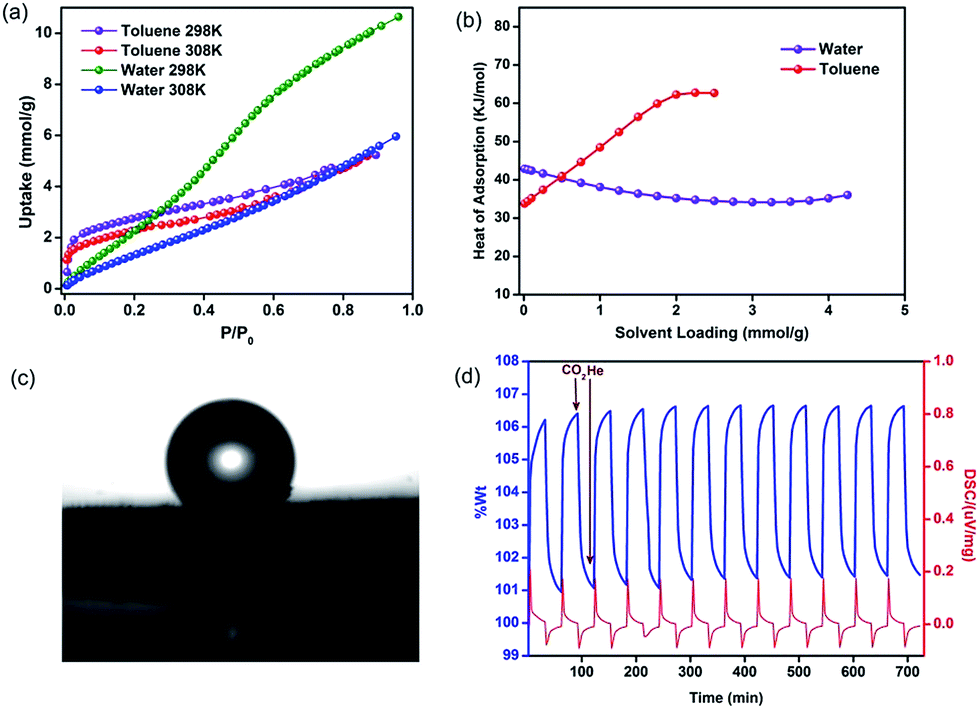 | ||
| Fig. 3 (a) Vapor sorption isotherms of HPF-1 carried out at different temperatures. The toluene shows type-I behavior while water shows hardly any adhesion, as indicated by a near-linear isotherm. (b) The HOA is consistent with the shapes of the isotherms indicating that water interacts very weakly with a zero-loading HOA of 43 kJ mol−1 which is near its vaporization point. (c) Contact angle measurement of HPF-1 showing a highly hydrophobic surface (153°). (d) TGA cycling experiments indicating a facile removal of adsorbed CO2 by a He purge. | ||
A zero-loading heat of adsorption for CO2 in the range of 25–35 kJ mol−1 is indicative of a facile CO2 recovery.63 In HPF-1, the HOA of CO2, estimated from a virial/DFT model, had a moderate value of 26 kJ mol−1, implying the possibility of a facile CO2 regeneration. We have demonstrated this experimentally through a TGA cycling experiment. A near 100% recovery of adsorbed CO2 by a He sweep at 303 K was obtained on the TGA and fifteen such adsorption/desorption cycles have been carried out without any loss in capacity (Fig. 3d). From the water sorption studies it is clear that HPF-1 has relatively weak interactions with water, but the uptake of water is still appreciable. This makes it imperative to demonstrate the ability of the material to adsorb CO2 even after being exposed to sufficient water or in other words the selectivity of CO2 over water. For this purpose we have carried out a steam conditioning study wherein other porous materials, with comparable CO2 uptakes, carbon molecular sieves, ZnAtzOx,64 zeolite 4A and the title material were activated and then exposed to a flow of humid N2 (100 ml min−1 over a 75% RH obtained from a saturated NaCl solution maintained at 60 °C) for a period of 24 h. This wet material was tested for CO2 adsorption without any further activation. As can be seen from Fig. 4, a microporous carbon molecular sieve (3kType-172CMS) loses 39% of its CO2 capacity and ZnAtzOx loses 55%, zeolite 4A loses 83%, while HPF-1 loses 20% of its CO2 capacity. Even a hydrophobic standard, silica-alumina (P/N 004/16821/00), loses 40% of its CO2 capacity (see Section 9 of the ESI†). As can be seen from Fig. 2c, HPF-1's CO2/N2 selectivity drops down in the presence of humidity, however, even for this steam conditioned phase, the selectivity is still quite high (90) at 303 K. And this capacity and selectivity were intact even after 48 h of steam conditioning.
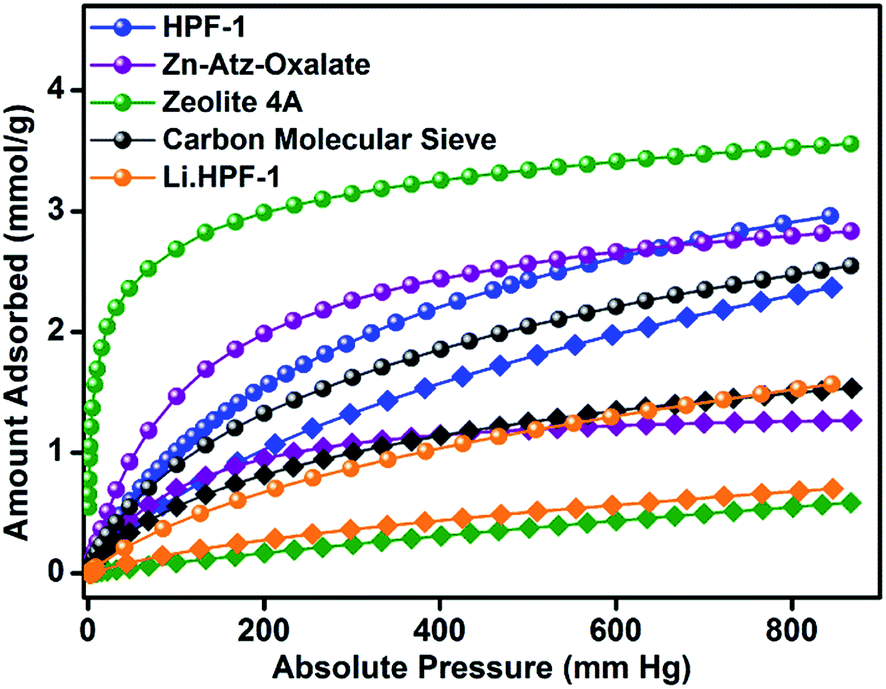 | ||
| Fig. 4 Comparison of the effect of humidity on the CO2 adsorption behavior of selected porous materials with varying hydrophobic–hydrophilic character and similar CO2 uptakes. All isotherms were carried out at 303 K. Important: after the steam conditioning the material was not subjected to any activation. Note: spheres represent the activated phase; squares represent the steam conditioned phase. | ||
To quantify the CO2 adsorption of HPF-1 from a dynamic humid CO2 stream, in a separate and simple experiment, we activated (evacuated at 160 °C for 12 h under 10−4 Torr) about 1 g of the sample and then exposed it to a flow of humid CO2 (100 ml min−1 over a 75% RH obtained from a saturated NaCl solution maintained at 60 °C) for 24 h. This sample was then cut off from the CO2 stream and was exposed to ambient conditions to release any CO2 filling the vessel. The adsorbed CO2 was desorbed by heating the sample at 60 °C and the evolved CO2 was captured on to a pre-treated solution of CaO (see ESI†). Brisk bubbling was observed followed by the occurrence of a white crystalline precipitate of CaCO3, which was extracted by filtration and dried with an acetone wash. Following this, a mass balance was carried out, which indicated the formation of 0.266 g of CaCO3 from 1 g of HPF-1. (CO2)x, which translates to 2.66 mmol g−1 of CO2 per gram of HPF-1. This is 5% less than the capacity obtained from a single component CO2 isotherm. The humidity to which the sample has been exposed during this experiment is much higher than the maximal humidity (15%) expected in a flue gas. Furthermore, the CaCO3 formed was characterized to be anhydrous CaCO3 (ICSD: 18165) using PXRD and the TGA indicated a weight loss agreeing extremely well with what was expected for a 2.66 mmol of CO2 per gram of HPF-1 (Fig. S16†).
During our investigation on porous hydrophobic materials, we made an interesting observation that a MOF made up of an alternating fluorine and amine lining did not show any CO2 uptake.65 Thus, it could be possible that just the presence of hydrophobic sites proximal to a strongly CO2 interacting site is not sufficient to obtain good CO2-framework interactions over water. Or in other words, there has to be an optimal hydrophobic–hydrophilic balance provided by the adjacently positioned functional groups giving rise to adsorption pockets that could favor a less polar CO2 over water. Here the phenol groups could be providing such hydrophobic-polar environments, as they are known to act as partitioning agents due to their ability to tune their hydrophilic–hydrophobic character depending on the type of substituent on the ring or the environment it is suspended in.66 To demonstrate the role of the phenolic groups in contributing to the material's hydrophobicity, in another experiment, we loaded the material with ∼17% of Li (quantified from Microwave Plasma Atomic Emission Spectrometry) and this lithiated sample was subjected to the same steam conditioning treatment. The CO2 adsorption studies on this humidified lithiated sample show a drastic 56% drop in CO2 capacity (Fig. 4). A video has been presented in the ESI† to demonstrate the complete loss of surface hydrophobicity with this ∼17% Li loading, clearly expressing the critical role of phenol groups in providing a hydrophobic texture to the material.
In recent times, a simulation based approach has been shown to bring significant structural insights on porous polymers.47,55,56 Experimental structure determination is almost impossible owing to the lack of solubility in these highly cross-linked polymers preventing the use of techniques such as gel permeation chromatography or routine solution NMR. To obtain a probable structure of HPF-1, we created a small oligomer by combining the monomers in a 2:3 ratio and minimized its structure using DFT methods with Materials Studio. Then we carried out random polymerization with it to obtain a larger oligomer (polymer). The polymer thus generated could take different configurations depending on the slight differences in the geometry of the initial energy minimized smaller oligomer. However, the geometric constraints in terms of acceptable bond lengths, angles and van der Waals distances applied during these operations avoided the generation of too many metastable structures with shallow local minima. Three low energy polymer configurations were chosen based on the final energy (alpha-, beta and gamma, Fig. S16†). These were then minimized again using tight-binding DFT methods (DFT-TB). Following this, an amorphous cell was created independently for the alpha, beta and gamma phases. Again, a DFT tight-binding calculation was done to optimize the geometry, lattice parameters and the energy. This yielded a structure with a triclinic cell: P1; a = 37.8895 Å, b = 35.1144 Å, c = 23.5303 Å, α = 89.768(2)°, β = 91.638(4)°, and γ = 100.648(4)° (Fig. 5). During the entire process complete rotational and torsional freedom was maintained. We remark that only when the initial oligomer was properly geometry optimized, could the larger oligomer be formed with acceptable bonds and favorable van der Waals requirements. The lowest energy polymer configuration, alpha, had relative energies two and three times lower than the beta and gamma, respectively (Fig. 5 and S18†). The structure of the alpha phase had highly corrugated ultra-micropores, which are lined by the phenol groups from the resorcinol unit and the nitrogens of the triazine groups protrude along the walls making them accessible. A Connolly representation shows the presence of three-dimensional access via small ultra-microporous openings (Fig. 5d). The surface area calculations done using Materials Studio yielded a theoretical surface area of 570 m2 g−1 (exptl BET and 77 K N2: 576 m2 g−1) and a pore volume of 0.24 cm3 g−1 (exptl, 77 K N2: 0.27 cm3 g−1). We simulated the pure-component (0–1 bar) isotherms at 303 K for both CO2 and N2 for all the three configurations using a Monte-Carlo method (Accelrys).64 The simulated isotherm matched exceptionally well with the experimental one (Fig. 6a), but only for the alpha form. From the simulations, the average heat of adsorption was estimated to be 30 kJ mol−1 which is very close to the experimentally determined 26 kJ mol−1 (inset of Fig. 6a).
 | ||
| Fig. 5 Three dimensional packing of the lowest energy configuration, the α-phase, of HPF-1 formed using a random polymerization of monomeric units with terephthaldehyde (DFT-TB minimized). A view along (a) the C-axis showing the small cavities (4 × 8.0 Å), hydrogens have been removed for clarity; (b) the B-axis showing the ultra-micropores (7.5 × 7.0 Å, not factoring the van der Waals radii); (c) representation differentiating single oligomers of specific site symmetry via different color coding. (d) The Connolly representation of HPF-1 showing the presence of highly corrugated channels running along all three-directions (blue – opening to the pores). | ||
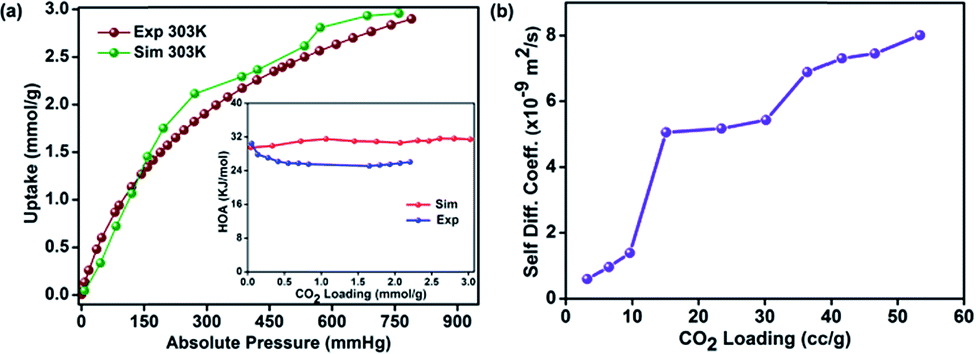 | ||
| Fig. 6 (a) Comparison of the CO2 isotherm simulated using the α-phase and the experimental isotherm at 303 K. The inset shows the comparison of the HOA plots. (b) The self-diffusion coefficients of CO2 in HPF-1 fitted using a spherical pore model. | ||
The complex pore structure of HPF-1 comprising of extremely corrugated channels could pose possible diffusion limitations to the movement of CO2 molecules across the polymer. To address this, the diffusion of CO2 within the hydrophobic-polar channels of the HPF-1 was measured using the rate of adsorption studies. The equilibration kinetics associated with 10 different pressure points were extracted and the data were fitted to a spherical pore model (Fig. S19†).67
A CO2 self-diffusivity coefficient of 10−9 m2 s−1 was calculated, (Fig. 6b) which is higher than the value obtained for zeolites and is comparable to some of the MOFs.68–70 In fact, these diffusivity values are similar to those obtained for HTFP, a highly hydrophobic MOF, built from ligands rich in aromatic groups.68 Also, the CO2 diffusion in HPF-1 appears considerably facile compared to a fluorine lined ultra-microporous MOF.71 This could be due to the relatively weaker framework-CO2 interactions in HPF-1. The CO2 self-diffusivities used in these comparisons are for materials that are being used in industrial CO2 capture and the ones that have been identified as potential candidates. However, there is a marked difference in the pathways that CO2 would be travelling through, this difference arises from the pore sizes and also from the structure of the pore walls.4 Given this, the MOFs and porous organic polymers with larger pores and with soft or flexible pore walls are always expected to have an advantage over zeolites with narrow windows and rigid pore walls. The jumps found in the diffusivity values with increasing CO2 loadings (Fig. 6b) could be due to the corrugated pores making the structure digress from the spherical model. Yet, other models (linear or slab) did not fit well. To the best of our knowledge, till date there are no reports of CO2 kinetics in porous polymers.
Conclusions
Here, we have synthesized a porous polymer by employing a tri-resorcinol with a triazine core as the building unit. The emphasis of the material includes its ability to selectively capture CO2 over N2, and its exceptional stability under harsh conditions (NOx, SOx, and steam) that mimic flue gas environments. Such stability from a porous material is highly desirable and we attribute it to the lack of hydrolyzable groups and the polymer backbone being built up from C–C bonds. When the adsorption was carried out using a humid CO2 stream the material loses only about 5% of its capacity, still having a selectivity of 90:1. Converting 17% of the phenol groups into –OLi results in a complete loss of the surface hydrophobicity exemplifying the role of phenolic groups in providing hydrophobicity. To obtain structural insights that could explain the hydrophobic–hydrophilic character of the material, we have proposed a structure based on amorphous cell simulation. HPF-1 has corrugated ultra-microporous channels copiously lined with phenol groups and basic triazine units which agrees extremely well with our expectations of the pore surface based on solvent sorption studies and preferential CO2 sorption. The 303 K CO2 adsorption isotherms and the associated HOA profiles simulated based on this structure seem to match well with the experimentally observed ones, which add to the confidence of the proposed structure. In fact, HPF-1 brings out a phenol–triazine–aldehyde based chemistry which enables us to develop porous polymers with a fine balance between hydrophobicity and polar character. The catalyst-free and easily up-scalable synthesis and cheap ingredients make this class of materials an attractive target.
Acknowledgements
We gratefully acknowledged IISER Pune and MHRD, Govt. of India for financial support. S. Nandi thanks DST, CII, SERB and Enovex for financial support through the Prime Minister's Fellowship.Notes and references
- H.-C. Joe-Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415 RSC.
- J. M. Huck, L.-C. Lin, A. H. Berger, M. N. Shahrak, R. L. Martin, A. S. Bhown, M. Haranczyk, K. Reuter and B. Smit, Energy Environ. Sci., 2014, 7, 4132 CAS.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. M. Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yaoa and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130 CAS.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308 RSC.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed.
- M. J. Bojdys, J. Jeromenok, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 2202 CrossRef CAS PubMed.
- A. I. Cooper, CrystEngComm, 2013, 15, 1483 RSC.
- Z. Zhang, Z.-Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868 CAS.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhao, Q. Gong, Z. Lib and J. Li, Chem. Commun., 2013, 49, 653 RSC.
- Q. Wang, J. Luo, Z. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42 CAS.
- T. C. Drage, C. E. Snape, L. A. Stevens, J. Wood, J. Wang, A. I. Cooper, R. Dawson, X. Guo, C. Satterley and R. Irons, J. Mater. Chem., 2012, 22, 2815 RSC.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O'Hare and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478 CAS.
- C. Mottillo and T. Friscic, Angew. Chem., Int. Ed., 2014, 53, 7471 CrossRef CAS PubMed.
- D. Feng, K. Wang, J. Su, T.-F. Liu, J. Park, Z. Wei, M. Bosch, A. Yakovenko, X. Zou and H.-C. Zhou, Angew. Chem., Int. Ed., 2015, 54, 149 CrossRef CAS PubMed.
- S. B. Kalidindi, S. Nayak, M. E. Briggs, S. Jansat, A. P. Katsoulidis, G. J. Miller, J. E. Warren, D. Antypov, F. Cor, B. Slater, M. R. Prestly, C. M. Gastaldo and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2015, 54, 221 CrossRef CAS PubMed.
- T. A. Makal, X. Wang and H.-C. Zhou, Cryst. Growth Des., 2013, 13, 4760 CAS.
- Q. Liu, L. Ning, S. Zheng, M. Tao, Y. Shi and Y. He, Sci. Rep., 2013, 3, 2916 Search PubMed.
- Y. Liu, Z. U. Wang and H.-C. Zhou, Greenhouse Gases: Sci. Technol., 2012, 2, 239 CrossRef CAS PubMed.
- H. A. Patel, S. H. Je, J. Park, D. P. Chen, Y. Jung, C. T. Yavuz and A. Coskun, Nat. Commun., 2013, 4, 1357 CrossRef PubMed.
- H. A. Patel, F. Karadas, A. Canlier, J. Park, E. Deniz, Y. Jung, M. Atilhan and C. T. Yavuz, J. Mater. Chem., 2012, 22, 8431 RSC.
- X. Zhu, S. M. Mahurin, S.-H. An, C.-L. Do-Thanh, C. Tian, Y. Li, L. W. Gill, E. W. Hagaman, Z. Bian, J.-H. Zhou, J. Hu, H. Liu and S. Da, Chem. Commun., 2014, 50, 7933–7936 RSC.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268 CrossRef CAS PubMed.
- Y. Luo, B. Li, W. Wang, K. Wu and B. Tan, Adv. Mater., 2012, 24, 5703 CrossRef CAS PubMed.
- P. Arab, M. G. Rabbani, A. K. Sekizkardes, T. İslamoğlu and H. M. El-Kaderi, Chem. Mater., 2014, 26, 1385 CrossRef CAS.
- J. Lua and J. Zhang, J. Mater. Chem. A, 2014, 2, 13831 Search PubMed.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457 CrossRef CAS PubMed.
- Y. Li, T. Ben, B. Zhang, Y. Fu and S. Qiu, Sci. Rep., 2013, 3, 2420 Search PubMed.
- A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328 CrossRef CAS PubMed.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291 CrossRef CAS PubMed.
- D. Q. Yuan, W. G. Lu, D. Zhao and H. C. Zhou, Adv. Mater., 2011, 23, 3723 CrossRef CAS PubMed.
- G. Qi, Y. Wang, L. Estevez, X. Duan, N. Anako, A.-H. A. Park, W. Li, C. W. Jones and E. P. Giannelis, Energy Environ. Sci., 2011, 4, 444 CAS.
- M. Rose, W. Bohlmann, M. Sabo and S. Kaskel, Chem. Commun., 2008, 2462 RSC.
- C. F. Martín, E. Stöckel, R. Clowes, D. J. Adams, A. I. Cooper, J. J. Pis, F. Rubiera and C. Pevida, J. Mater. Chem., 2011, 21, 5475 RSC.
- A. Bhunia, I. Boldog, A. Möller and C. Janiak, J. Mater. Chem. A, 2013, 1, 14990 CAS.
- A. Bhunia, V. Vasylyeva and C. Janiak, Chem. Commun., 2013, 49, 3961 RSC.
- M. G. Schwab, B. Fassbender, H. W. Spiess, A. Thomas, X. Feng and K. Müllen, J. Am. Chem. Soc., 2009, 131, 7216 CrossRef CAS PubMed.
- N. Du, H. B. Park, G. P. Robertson, M. M. Dal-Cin, T. Visser, L. Scoles and M. D. Guiver, Nat. Mater., 2011, 10, 372 CrossRef CAS PubMed.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574 CrossRef CAS PubMed.
- R. Dawson, E. Stöckel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239 CAS.
- S. Sung and M. P. Suh, J. Mater. Chem. A, 2014, 2, 13245 CAS.
- H. He, W. Li, M. Zhong, D. Konkolewicz, D. Wu, K. Yaccato, T. Rappold, G. Sugar, N. E. David and K. Matyjaszewski, Energy Environ. Sci., 2013, 6, 488 CAS.
- G. P. Hao, W.-C. Li, D. Qian, G.-H. Wang, W.-P. Zhang, T. Zhang, A.-Q. Wang, F. Schüth, H.-J. Bongard and A.-H. Lu, J. Am. Chem. Soc., 2011, 133, 11378 CrossRef CAS PubMed.
- J. Yu and P. B. Balbuena, J. Phys. Chem. C, 2013, 117, 3383 CAS.
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreibera and T. E. Muller, Energy Environ. Sci., 2012, 5, 7281 CAS.
- J.-H. Zhu, Q. Chen, Z.-Y. Sui, L. Pan, J. Yu and B.-H. Han, J. Mater. Chem. A, 2014, 2, 16181 CAS.
- B. Li, R. Gong, W. Wang, X. Huang, W. Zhang, H. Li, C. Hu and B. Tan, Macromolecules, 2011, 44, 2410 CrossRef CAS.
- A. P. Katsoulidis and M. G. Kanatzidis, Chem. Mater., 2011, 23, 1818 CrossRef CAS.
- A. K. Sekizkardes, S. Altarawneh, Z. Kahveci, T. İslamoğlu and H. M. El-Kaderi, Macromolecules, 2014, 47, 8328 CrossRef CAS.
- M. Saleh, S. B. Baek, H. M. Lee and K. S. Kim, J. Phys. Chem. C, 2015, 119, 5395 CAS.
- G. K. H. Shimizu, R. Vaidhyanathan and J. M. Taylor, Chem. Soc. Rev., 2009, 38, 1430 RSC.
- F. Sua and C. Lu, Energy Environ. Sci., 2012, 5, 9021 Search PubMed.
- Z. Zhang, W. Zhang, X. Chen, Q. Xia and Z. Li, Sep. Sci. Technol., 2010, 45, 710 CrossRef CAS PubMed.
- J. M. Taylor, R. Vaidhyanathan, S. S. Iremonger and G. K. H. Shimizu, J. Am. Chem. Soc., 2012, 134, 14338 CrossRef CAS PubMed.
- C. Reece, D. J. Willock and A. Trewin, Phys. Chem. Chem. Phys., 2015, 17, 817 RSC.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710 CrossRef CAS PubMed.
- A. P. Katsoulidis, J. He and M. G. Kanatzidis, Chem. Mater., 2012, 24, 1937 CrossRef CAS.
- X. Xu, C. Song, R. Wincek, J. M. Andresen, B. G. Miller and A. W. Scaroni, Fuel Chemistry Division Preprints, 2003, 48, 162 CAS.
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954 CrossRef PubMed.
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030 CAS.
- R. T. Woodward, L. A. Stevens, R. Dawson, M. Vijayaraghavan, T. Hasell, I. P. Silverwood, A. V. Ewing, T. Ratvijitvech, J. D. Exley, S. Y. Chong, F. Blanc, D. J. Adams, S. G. Kazarian, C. E. Snape, C. T. Drage and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 9028 CrossRef CAS PubMed.
- K. P. Rao, M. Higuchi, K. Sumida, S. Furukawa, J. Duan and S. Kitagawa, Angew. Chem., Int. Ed., 2014, 126, 8364 CrossRef PubMed.
- J. M. Simmons, H. Wu, W. Zhou and T. Yildirim, Energy Environ. Sci., 2011, 4, 2177 CAS.
- R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650 CrossRef CAS PubMed.
- C. Y. Su, A. M. Goforth, M. D. Smith, P. J. Pellechia and H. C. zur Loye, J. Am. Chem. Soc., 2004, 126, 3576 CrossRef CAS PubMed.
- S. Katsuta, K. Nakamura, Y. Kudo, Y. Takeda and H. Kato, J. Chem. Eng. Data, 2011, 56, 4083 CrossRef CAS.
- K. Malek and M.-O. Coppens, J. Chem. Phys., 2003, 119, 2801 CrossRef CAS PubMed.
- L. Zhang, G. Wu and J. Jiang, J. Phys. Chem. C, 2014, 118, 8788 CAS.
- F. Salles, H. Jobic, A. Ghoufi, P. L. Llewellyn, C. Serre, S. Bourrelly, G. Frey and G. Maurin, Angew. Chem., Int. Ed., 2009, 48, 8335 CrossRef CAS PubMed.
- J. A. C. Silva, K. Schumann and A. E. Rodrigues, Microporous Mesoporous Mater., 2012, 158, 219 CrossRef CAS PubMed.
- K. A. Forrest, T. Pham, A. Hogan, K. McLaughlin, B. Tudor, P. Nugent, S. D. Burd, A. Mullen, C. R. Cioce, L. Wojtas, M. J. Zaworotko and B. Space, J. Phys. Chem. C, 2013, 117, 17687 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta04241k |
| This journal is © The Royal Society of Chemistry 2015 |