
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(phenylene oxide) functionalized with quaternary ammonium groups via flexible alkyl spacers for high-performance anion exchange membranes†
Hai-Son
Dang
,
Eva Annika
Weiber
and
Patric
Jannasch
*
Polymer & Materials Chemistry, Department of Chemistry, Lund University, P.O. Box 124, SE-221 00 Lund, Sweden. E-mail: patric.jannasch@chem.lu.se
First published on 2nd February 2015
Abstract
We have attached quaternary ammonium (QA) groups to poly(phenylene oxide) via flexible and stable heptyl side chains by using a straightforward synthetic route involving bromoalkylation and quaternization. Membranes based on these polymers showed efficient phase separation, significantly enhanced hydroxide ion conductivity and far superior alkaline stability in relation to corresponding polymers with QA groups placed in benzylic positions directly on the backbone.
There is currently a growing interest in robust anion exchange membranes (AEMs) for use in various electrochemical energy storage and conversion systems such as alkaline membrane fuel cells (AMFCs) and electrolysers, redox flow batteries and reverse electrodialysis.1,2 The alkaline operating conditions bring distinct and attractive advantages to AMFCs in comparison to for example proton-exchange membrane fuel cells. These include faster oxygen reduction kinetics, efficient water management, greater flexibility in the choice of fuels, and the use of non-precious metal electrocatalysts.1,2 In AMFCs, oxygen reduction gives OH− ions that are transported through a solid alkaline membrane to the anode. Here, they react with the fuel to produce water and electrons. Today the greatest challenge for this technology is the development of new polymeric membrane materials that combine high OH− conductivity with sufficient long-term stability under the very highly basic and aggressive conditions. A wide range of polymers including polyethers, polysulfones, polyketones, and polystyrenes have been functionalized with various cationic groups such as QAs, imidazoliums, phosphoniums and guanidinium.1–7 The vast majority of the studies up until now have however been performed with polymers functionalized with benzyltrimethylammonium groups positioned directly on the backbone. These materials are readily accessed via chloromethylation or benzylic bromination of suitable aromatic polymers, followed by quaternization with trimethylamine. Although these polymers are currently regarded as benchmark materials in the field, several recent reports show that alkyl-substituted cations possess a significantly higher stability than benzyl-substituted cations in the presence of aqueous OH−.1,3,8 Moreover, the position of the benzylic cations directly on the backbone may induce polymer backbone scission.9 Finally, the placement of cations close to the polymer backbone will restrict their mobility and prevent the efficient phase separation necessary for high anion conductivity. To facilitate the phase separation in AEMs, ions have been highly concentrated to single units in statistical copolymers,10,11 stiff side groups12 or to segments in block copolymers.13 However, this approach may cause limitations in ion-dissociation, and thus conductivity, because of the close proximity of the ions in the polymer structure.10,14
It has very recently been pointed out that there is a current need for new synthetic approaches to functionalize aromatic polymers with cationic groups via alkyl spacers, which calls for non-conventional routes.1,3 In one of only a few examples,15,16 Hibbs functionalized a polyphenylene with hexyltrimethylammonium side chains by using 6-bromo-1-hexanoyl chloride in a Friedel–Crafts acylation, followed by reduction of the ketone link to a methylene link, and quaternization with trimethylamine.15 He found that the stability increased significantly in comparison with that of a corresponding benzyltrimethylammonium functionalized polymer. However, the poor accessibility of polyphenylenes combined with their very high chain rigidity and limited solubility, often leading to brittle materials, bring clear disadvantages. In the present paper we report on a straightforward method to attach QA groups to poly(2,6-dimethyl-1,4-phenylene oxide) (PPO) via long flexible heptyl spacers devoid of any hydrolysable group (Scheme 1). Full details of the synthesis and characterization of the polymers and corresponding membranes are available as ESI.† We envisage a number of distinct advantages from this approach. The presence of a spacer will facilitate phase separation to concentrate the QA groups in the AEM. This will in turn favour the formation of a well-hydrated and percolating phase resulting in high ionic conductivity. Moreover, the avoidance of the benzylic attachment and efficient hydration of the QA groups can be expected to increase the stability under alkaline conditions. PPO is readily accessible, has good solubility, outstanding mechanical properties, and high chemical and thermal stability. In addition, it can be modified in a variety of ways in either the aromatic or benzylic positions. Specifically, PPO has previously been shown to have a good stability under alkaline conditions, and can be expected to be an improvement over, e.g., poly(arylene ether sulfone)s were the ether bridges are activated for nucleophilic attack by OH− causing chain-scission.17 The first step in our modification of PPO involved benzylic lithiation. PPO (Mn = 20 kg mol−1, MwMn−1 = 2.3) was dissolved in tetrahydrofuran (THF), carefully degassed, and lithiated using n-butyllithium (n-BuLi) under a blanket of argon at −78 °C. Under these conditions, aryl and benzyl positions on PPO are lithiated at a molar ratio of approximately 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.18 However, only the benzyl lithiated sites are sufficiently nucleophilic to, e.g., react with bromoalkyl compounds and initiate anionic polymerisation of vinyl monomers.19 After 3 h, 1,6-dibromohexane was quickly added to quench all the benzyl lithiated sites, leading to the grafting of 6-bromohexyl chains to the corresponding benzylic positions of the PPO via displacement of bromine (Scheme 1). An excess of the dibromoalkane was used to suppress potential crosslinking reactions arising from the difunctionality of the dibromohexane.
:1.18 However, only the benzyl lithiated sites are sufficiently nucleophilic to, e.g., react with bromoalkyl compounds and initiate anionic polymerisation of vinyl monomers.19 After 3 h, 1,6-dibromohexane was quickly added to quench all the benzyl lithiated sites, leading to the grafting of 6-bromohexyl chains to the corresponding benzylic positions of the PPO via displacement of bromine (Scheme 1). An excess of the dibromoalkane was used to suppress potential crosslinking reactions arising from the difunctionality of the dibromohexane.
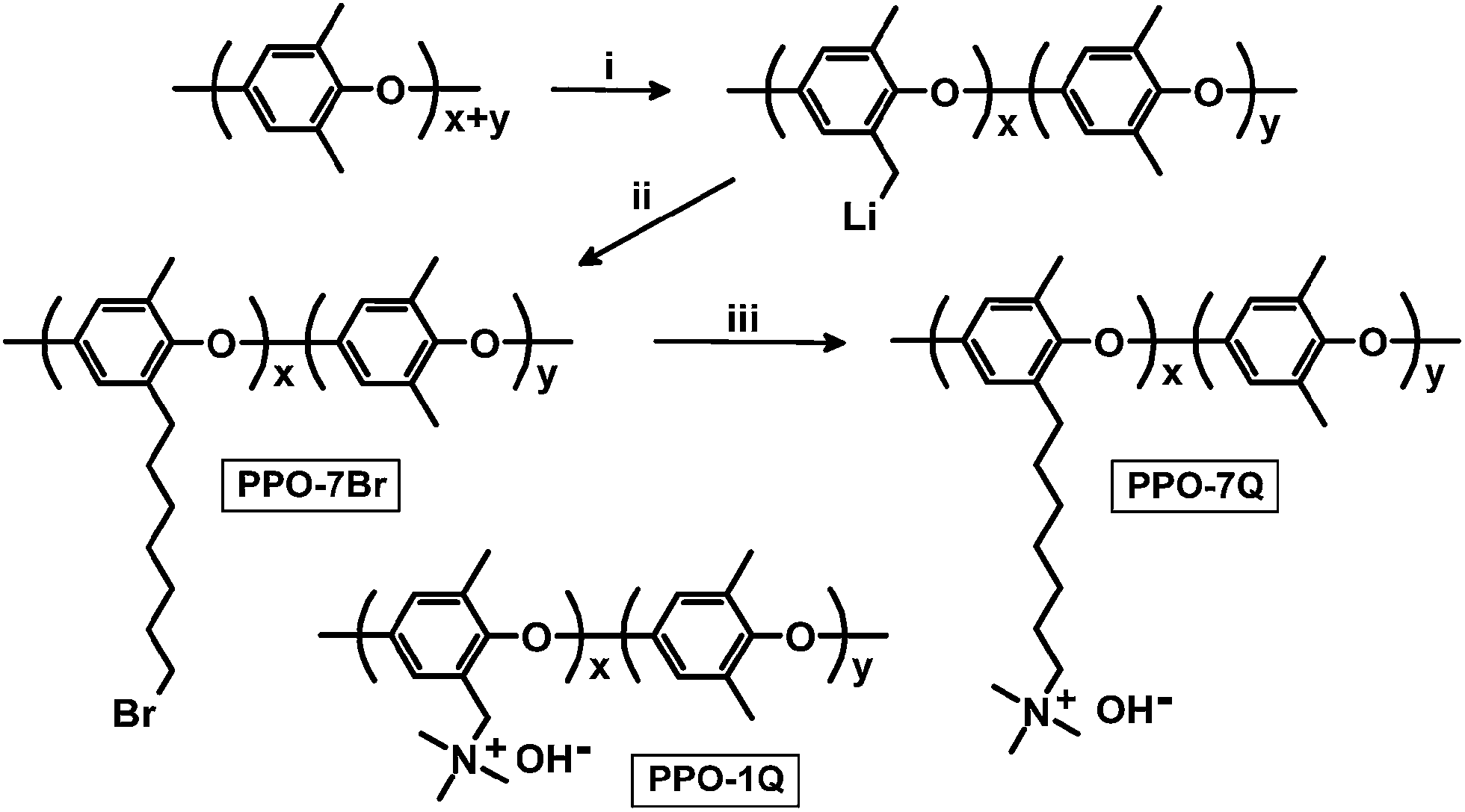 | ||
| Scheme 1 Synthetic pathway to PPO modified with heptyltrimethyl-ammonium side chains (PPO-7Q) via lithiation, bromoalkylation and quaternization. PPO modified with benzyltrimethylammonium groups (PPO-1Q) is used as a benchmark material [key: (i) n-BuLi, THF, −78 °C, (ii) 1,6-dibromohexane, THF, −78 °C, (iii) trimethylamine, NMP, 25 °C; NaOH]. | ||
Five samples were bromoalkylated with good precision to a degree of bromination (DB, percentage of bromoalkylated benzylic methyl groups of the PPO) between 11 and 29%, denoted PPO-7Br-DB, to obtain AEMs with a practical range of ionic content. The samples were precipitated, repeatedly washed in methanol, and dried before analysis. Fig. 1b shows the expected additional 1H NMR signals arising from the grafting in relation to the spectrum of native PPO in Fig. 1a. For example, a new signal (c) appeared at 3.4 ppm from the –CH2Br protons and a band of signals from additional –CH2– protons can be seen in the range 1.2–2.0 ppm. The DB values were calculated by taking into account the integrated signals (c and b), the latter centred at 6.4 ppm and originating from the aromatic PPO protons (Fig. 1b). Expectedly, the molecular weight and the polydispersity (MwMn−1) were found by size exclusion chromatography to increase after the bromoalkylation (Table S1†). No gel formation was detected in any of the modified polymers. As anticipated, the glass transition temperature (Tg) analyzed by differential scanning calorimetry (DSC, 10 °C min−1) decreased with increasing DB, from 216 °C for native PPO to 147 °C for PPO-7Br-29 because of internal plasticization caused by the flexible side chains (Fig. S1†). Thermogravimetric analysis (TGA, N2 at 10 °C min−1) showed that the thermal stability of the bromoalkylated PPOs decreased with DB showing an initial degradation between 340 and 360 °C (Table S1†). In comparison, the native PPO decomposed in a single step at 419 °C. The bromoalkylated samples were efficiently quaternized by the Menshutkin reaction to form the cationic heptyltrimethylammonium side chains. The reaction was performed under homogeneous conditions in 5 wt% solutions of the polymers in N-methyl-2-pyrrolidone (NMP) using an excess of trimethylamine over 4 days at 20 °C. After precipitation and repeated washing in diethylether, 1H NMR analysis showed signals from the methyl protons of the QA groups at 3.0 ppm (Fig. 1c) and Mohr titrations of the Br− content confirmed full quaternization in agreement with the DB value of the precursor PPO-7Br polymer. Flexible, transparent and mechanically tough AEMs with ion exchange capacities (IECs) between 0.8 and 1.8 meq. g−1 were cast from NMP solutions at 80 °C and designated as PPO-7Q-IEC where IEC is the ion exchange capacity (IEC) as evaluated by Mohr titrations (Table S2†). Similarly, two PPO samples were conventionally functionalized with benzyltrimethylammonium groups via radical-mediated benzylbromination using N-bromosuccinimide and azobisisobutyronitrile, followed by quaternization with trimethylamine to form benchmark AEMs with IECs of 0.8 and 1.5 meq. g−1 (designated as PPO-1Q-IEC, see Scheme 1). Dry membranes in the Br− form were analyzed by small angle X-ray scattering (SAXS) to study their morphology. While the PPO-1Q membranes showed very weak (if any) ionomer peaks (scattering maxima), all the PPO-7Q membranes showed distinct peaks at q ∼ 1.9 nm−1, corresponding to a characteristic distance of d ∼ 3.3 nm (Fig. 2, SAXS data of all AEMs available in Fig. S2†). The scattering profiles indicated ionic clusters organized in the short range, but without any long-range order. The scattering intensity of the PPO-7Q AEMs increased with the ionic content. These findings clearly demonstrated that the flexible alkyl spacers gave a significant mobility to the QA groups during membrane formation. This facilitated a much more efficient phase separation of the ionic groups than that found in the PPO-1Q AEMs.
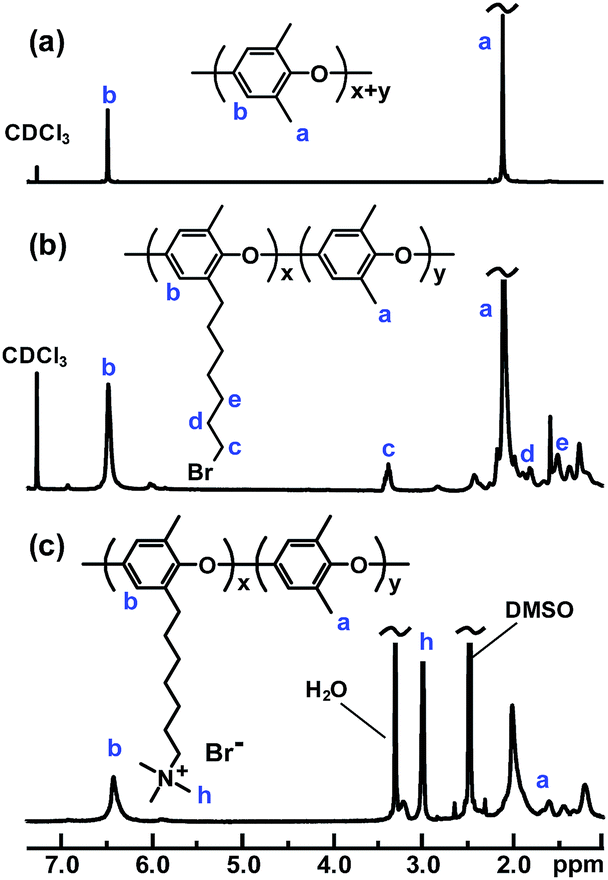 | ||
| Fig. 1 1H NMR spectra of unmodified PPO (a) and after bromoalkylation (b) and quaternization (c). | ||
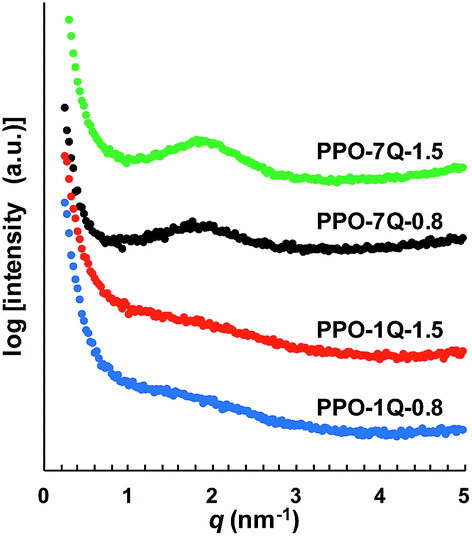 | ||
| Fig. 2 SAXS profiles of AEMs functionalized with heptyltrimethyl (PPO-7Q) and benzyltrimethyl (PPO-1Q) ammonium groups, respectively, at IEC 0.8 and 1.5 meq. g−1 (membranes in Br− form). | ||
Water is critical for the formation and transport of charge carriers by facilitating ion pair separation and promoting the development of a percolating conducting phase domain. However, too much water uptake will lead to high swelling and compromised mechanical properties of the membrane. Fig. 3a shows the temperature dependence of the water uptake of the AEMs in the OH− form fully hydrated between 20 and 80 °C. As expected the water uptake increased with IEC and temperature. PPO-7Q-0.8 showed remarkably low values in the studied temperature range with a nearly constant number of water molecules per QA group (λ) close to 6. The reason may be the presence of a low concentration of well segregated QA groups which results in isolated ionic clusters dispersed in an intact high-Tg glassy PPO matrix which counters the osmotic pressure that drives the water uptake. At higher IEC values, the water uptake was higher for the PPO-7Q series than for the PPO-1Q one, which may indicate that the well-clustered QA groups of the former polymers promoted the formation of distinct and localized water rich domains. Even at the highest IEC value of 1.8 meq. g−1, the water uptake remained restricted around 60 wt% at 80 °C (λ = 18). In contrast, the dispersed QA groups of the PPO-1Q membranes led to a morphology where the water was more evenly distributed (delocalised). The high but controlled water uptake of the PPO-7Q membranes may be an advantage for anion conductivity under reduced relative humidity which will be studied in the near future. At 20 °C, λ for the PPO-7Q membranes varied between 6 (PPO-7Q-0.8) and 13 (PPO-7Q-1.8). All the λ-values are given in Fig. S3.†
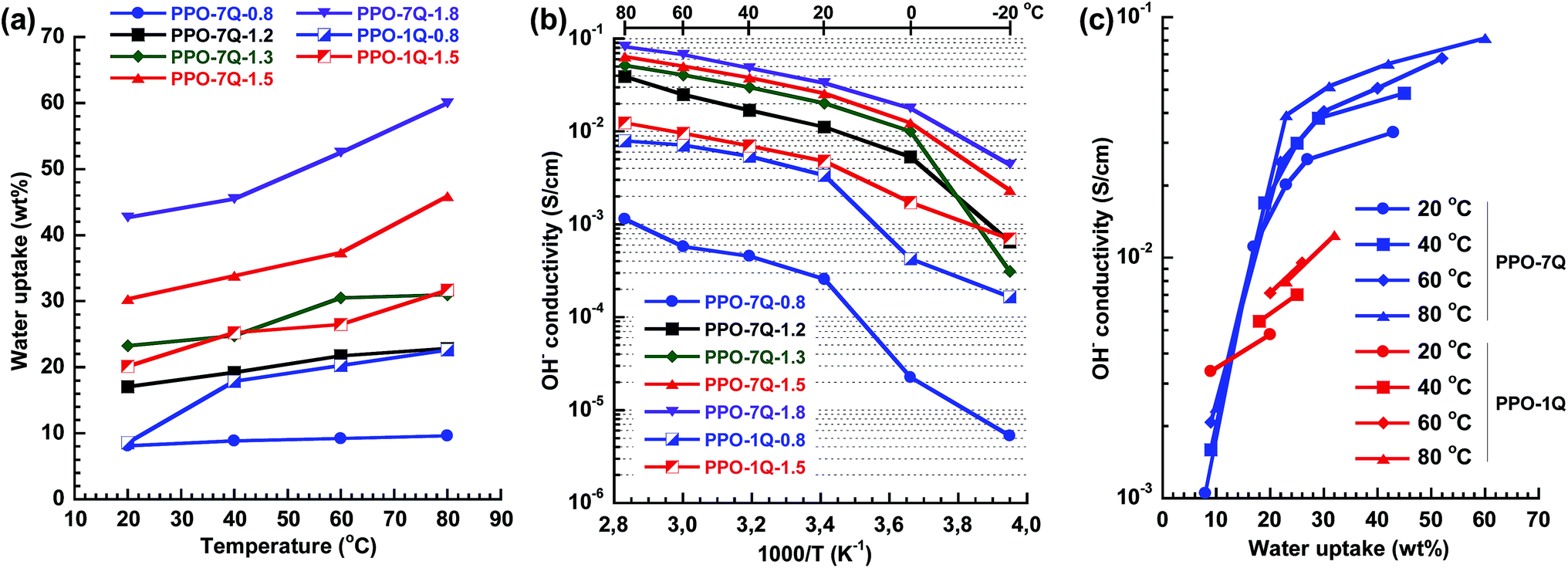 | ||
| Fig. 3 Water uptake of fully hydrated AEMs in the OH− form as a function of temperature (a), Arrhenius plot of the OH− conductivity under fully humidified conditions (b), and OH− conductivity under fully humidified conditions as a function of the water uptake for data measured between 20 and 80 °C (c). | ||
The temperature dependence of the OH− conductivity of the AEMs fully immersed in water was measured by electrochemical impedance spectroscopy using a two probe cell. As expected, the conductivity increased with increasing ion and water content (Fig. 3b). In the range between −20 and 20 °C the conductivity increased more markedly because of the melting of ice in the membranes. Because of its very low water uptake, PPO-7Q-0.8 displayed the lowest conductivity in the series. At higher IEC values the conductivity was found to be significantly higher for the PPO-7Q membranes than for the corresponding PPO-1Q ones. At an IEC of 1.5 meq. g−1, PPO-7Q-1.5 reached an ion conductivity of 26 and 64 mS cm−1 at 20 and 80 °C, respectively, while PPO-1Q-1.5 only reached 5 and 12 mS cm−1, respectively, at the same temperatures. By increasing the IEC value to 1.8 meq. g−1 (PPO-7Q-1.8), the conductivity increased to 33 and 85 mS cm−1 at 20 and 80 °C, respectively. At these high IECs, the conductivity showed Arrhenius behaviour above 20 °C with an estimated apparent activation energy of ∼15 kJ mol−1. This is on par with previously reported values and indicated decoupling from the slow polymer chain dynamics.20 Thus, the PPO-7Q AEMs clearly showed superior conductivity over the conventional PPO-1Q membranes in the interesting IEC range, at least partly because of a higher water content and a more localized water domain.
Fig. 3c shows the OH− conductivity plotted against the water uptake of the PPO-7Q and -1Q AEMs between 20 and 80 °C. The former AEMs clearly displayed two behavioural regimes. Between ∼10 and ∼20 wt% water, all the data points fell on the same straight line and the conductivity increased steeply with the IEC and the water content because of an increasing degree of percolation. Above, ∼25 wt% the percolation was fully developed and the conductivity increase with the water uptake was significantly lower. Here, the conductivity was primarily activated by temperature. The data in Fig. 3c also demonstrated that at a given water uptake above 15 wt%, the PPO-7Q AEMs showed a significantly higher conductivity than the PPO-1Q materials, i.e., the water in the former materials was used more efficiently to facilitate ion conductivity than that in the latter membranes.
The long-term stability of AEMs under alkaline conditions is critical because of the known degradation reactions including β-hydrogen Hoffmann elimination, direct nucleophilic substitution at an α-carbon and via ylide formation.1–7 An initial evaluation of the stability of the PPO-7Q-1.5 and PPO-1Q-1.5 AEMs was performed by immersing membrane samples in 1 M aq. NaOH at 80 °C. After 4 and 8 days, samples were removed and analyzed by 1H NMR spectroscopy to monitor the intensity ratio between the signal of the QA group at 3.0 ppm and the aromatic signal at 6.3–6.7 ppm (IQA:IAr, Fig. 1c). As seen in Fig. 4, this ratio did not change for PPO-7Q-1.5, to indicate a high stability. On the other hand, the intensity ratio fell quite dramatically for PPO-1Q-1.5, to 50% and then to only 16% of the original ratio after 96 and 192 h, respectively. The full NMR spectra are found in Fig. S4.† The reason for the superior stability of the PPO-7Q materials may be the higher stability of alkyltrimethylammonium in comparison to benzyltrimethylammonium, and also that the spacer units allowed a higher degree of hydration of the QA groups (well-solvated OH−). The findings are in agreement with previous results on low-molecular weight analogues.8 The thermal stability of the AEMs in the Br− form was investigated by thermogravimetric analysis (TGA, N2 at 10 °C min−1). All the AEMs were found to degrade in two steps, with the first occurring in the range between 230 to 270 °C by the decomposition of the QA groups (Fig. S5 and S6†). Thus, the thermal decomposition temperatures were well above the typical operating temperatures of most electrochemical energy systems.
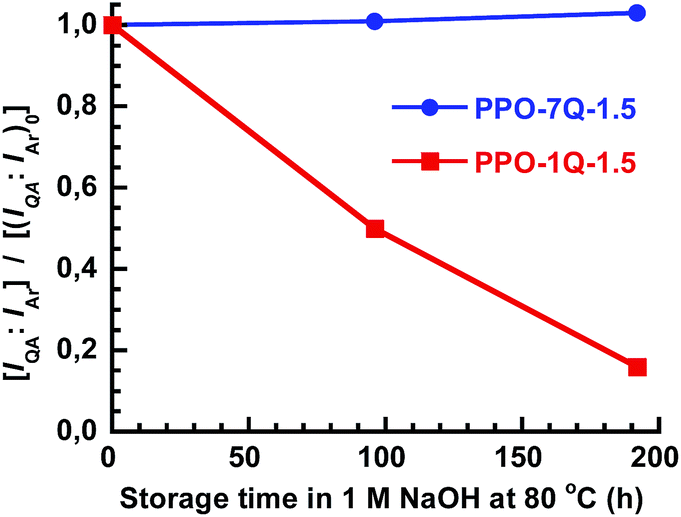 | ||
| Fig. 4 Evolution of the 1H NMR signal ratio of the QA protons and the aromatic protons [IQA:IAr] during immersion in 1 M aq. NaOH at 80 °C, in comparison with the original ratio [(IQA:IAr)0]. | ||
The properties exhibited by the present AEMs can be benchmarked against those reported by Zhuang et al. on membranes based on polysulfones carrying long alkyl chains separated from the QA groups along the backbone.21 The phase separated AEMs reached OH− conductivity of 35 mS cm−1 at 20 °C and >100 mS cm−1 at 80 °C. These are very high conductivity values, close to the range usually reported for the proton conductivity of Nafion®. Still, the membranes were found to degrade to some extent in 1 M aq. KOH at 60 °C. In addition, Zhuang and co-workers functionalized polysulfone with side chains containing one benzylic and one alkyl QA group.22 These membranes reached 80 mS cm−1 (fully hydrated at 80 °C, IEC = 2.1 meq. g−1), but degradation was notable in 3 M aq. KOH at 60 °C. Polyetherketones having alkyl chains with terminal QA groups were reported by Xu et al.23 The AEMs had an OH– conductivity of 91 mS cm−1 at 60 °C and no degradation was detected after aging in 6 M NaOH at 60 °C for 40 days. Bai et al. functionalized PPO with aromatic side chains carrying three benzylic QA groups.12 The OH− conductivity reached 72 mS cm−1 (fully hydrated at 60 °C, IEC = 1.5 meq. g−1), and the AEMs “maintained acceptable properties” after 12 days in 1 M NaOH at 80 °C. Finally, Binder et al. functionalized PPO with benzylic QA groups containing a long alkyl chains.24 These AEMs reached OH− conductivities up to 35 mS cm−1 (fully hydrated at room temperature, IEC = 1.9 meq. g−1). The membranes degraded by ∼20% during 500 h immersion in 1 M aq. NaOH at 80 °C and no high-temperature conductivity data was reported. In comparison, the present PPO-7Q materials combine a OH− conductivity of 85 mS cm−1 at 80 °C (fully hydrated, IEC = 1.8 meq. g−1) with no detectable degradation in 1 M NaOH at 80 °C during 196 h. This demonstrates their high-performance as AEMs.
In conclusion, we have successfully synthesized PPO functionalized with QA groups via flexible heptyl spacer units by using straightforward bromoalkylation and quaternization steps. This procedure gives mechanically stable AEMs with good control of the IEC. The spacer arrangement provides a high local mobility of the tethered cations which promotes ionic clustering and phase separation during membrane formation. In turn, this allows for the development of a localised and continuous water- and ion-rich phase upon hydration which results in an excellent level of conductivity above a critical IEC level. Initial results indicate a high stability of the AEMs under alkaline conditions, far superior to the benchmark materials functionalized with benzylic QA groups. The combined results demonstrate the excellent properties of the heptyltrimethylammonium substituted PPO membranes in relation to the benchmark materials functionalized with benzyltrimethylammonium groups. A more detailed study of the influence of the nature of the QA group and the alkyl spacer length on the stability and conductivity of AEMs is underway.
Acknowledgements
We thank the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (Formas) for financial support, and Marc Obiols-Rabasa for assistance with the SAXS measurements and data treatment.Notes and references
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. W. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135 CAS.
- N. W. Li and M. D. Guiver, Macromolecules, 2014, 47, 2175 CrossRef CAS.
- M. A. Hickner, A. M. Herring and E. B. Coughlin, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1727 CrossRef CAS.
- G. Couture, A. Alaaeddine, F. Boschet and B. Ameduri, Prog. Polym. Sci., 2011, 36, 1521 CrossRef CAS PubMed.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377, 1 CrossRef CAS PubMed.
- Y. J. Wang, J. L. Qiao, R. Baker and J. J. Zhang, Chem. Soc. Rev., 2013, 42, 5768 RSC.
- C. Vogel and J. Meier-Haack, Desalination, 2014, 342, 156 CrossRef CAS PubMed.
- A. D. Mohanty and C. Bae, J. Mater. Chem. A, 2014, 2, 17314 CAS.
- C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2490 CrossRef CAS PubMed.
- E. A. Weiber and P. Jannasch, ChemSusChem, 2014, 7, 2621 CrossRef CAS PubMed.
- D. Y. Chen and M. A. Hickner, Macromolecules, 2013, 46, 9270 CrossRef CAS.
- Q. Li, L. Liu, Q. Q. Miao, B. K. Jin and R. K. Bai, Chem. Commun., 2014, 50, 2791 RSC.
- (a) M. Tanaka, K. Fukasawa, E. Nishino, S. Yamaguchi, K. Yamada, H. Tanaka, B. Bae, K. Miyatake and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 10646 CrossRef CAS PubMed; (b) X. H. Li, Y. F. Yu, Q. F. Liu and Y. Z. Meng, J. Membr. Sci., 2013, 436, 202 CrossRef CAS PubMed; (c) A. Jasti and V. K. Shahi, J. Power Sources, 2014, 267, 714 CrossRef CAS PubMed; (d) Q. Li, L. Liu, Q. Q. Miao, B. K. Jin and R. K. Bai, Polym. Chem., 2014, 5, 2208 RSC; (e) D. Y. Park, P. A. Kohl and H. W. Beckham, J. Phys. Chem. C, 2013, 117, 15468 CrossRef CAS.
- M. G. Marino, J. P. Melchior, A. Wohlfarth and K. D. Kreuer, J. Membr. Sci., 2014, 464, 61 CrossRef CAS PubMed.
- M. R. Hibbs, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1736 CrossRef CAS.
- M. Tomoi, K. Yamaguchi, R. Ando, Y. Kantake, Y. Aosaki and H. Kubota, J. Appl. Polym. Sci., 1997, 64, 1161 CrossRef CAS.
- S. A. Nunez and M. A. Hickner, ACS Macro Lett., 2013, 2, 49 CrossRef CAS.
- (a) A. J. Chalk and T. J. Hoogeboom, J. Polym. Sci., Part A-1: Polym. Chem., 1969, 7, 1359 CrossRef CAS; (b) A. J. Chalk and A. S. Hay, J. Polym. Sci., Part A-1: Polym. Chem., 1969, 7, 691 CrossRef CAS.
- (a) M. Ingratta, M. Elomaa and P. Jannasch, Polym. Chem., 2010, 1, 739 RSC; (b) M. Ingratta, E. P. Jutemar and P. Jannasch, Macromolecules, 2011, 44, 2074 CrossRef CAS.
- J. Pan, S. F. Lu, Y. Li, A. B. Huang, L. Zhuang and J. T. Lu, Adv. Funct. Mater., 2010, 20, 312 CrossRef CAS.
- J. Pan, C. Chen, Y. Li, L. Wang, L. S. Tan, G. W. Li, X. Tang, L. Xiao, J. T. Lu and L. Zhuang, Energy Environ. Sci., 2014, 7, 354 CAS.
- J. Pan, Y. Li, J. J. Han, G. W. Li, L. S. Tan, C. Chen, J. T. Lu and L. Zhuang, Energy Environ. Sci., 2013, 6, 2912 CAS.
- Z. H. Zhang, L. Wu, J. Varcoe, C. R. Li, A. L. Ong, S. Poynton and T. W. Xu, J. Mater. Chem. A, 2013, 1, 2595 CAS.
- N. W. Li, T. Z. Yan, Z. Li, T. Thurn-Albrecht and W. H. Binder, Energy Environ. Sci., 2012, 5, 7888 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description of synthesis and characterization of polymers and AEMs; full thermal, water uptake, SAXS and NMR degradation data of the AEMs. See DOI: 10.1039/c5ta00350d |
| This journal is © The Royal Society of Chemistry 2015 |