
Visible light enhanced removal of a sulfur mustard gas surrogate from a vapor phase on novel hydrous ferric oxide/graphite oxide composites†
Javier A.
Arcibar-Orozco
and
Teresa J.
Bandosz
*
Department of Chemistry, The City College of New York, New York, NY 10031, USA. E-mail: tbandosz@ccny.cuny.edu; Fax: +1-212-650-6107; Tel: +1-212-650-6017
First published on 28th October 2014
Abstract
In this work, novel composites of iron hydroxide and graphite oxide (GO), initial or modified with urea, were synthesized and used as media for a 2-chloroethyl ethyl sulfide (CEES) removal/decontamination process. The results of surface characterization, using various physical and chemical methods, indicated that oxygen groups in GO act as nucleation centers for the hydrous ferric oxide formation/aggregation. Addition of the graphene phase increased the surface area and the amount of reactive adsorption centers. The iron hydroxide particles were highly dispersed between and on the graphene layers. Mesoporous 2 and 6-line ferrihydrites with a surface area higher than 200 m2 g−1 were identified as the main inorganic phase of the composites. An alteration in the optical band gap was found, depending on the chemical properties of the graphite oxide. The composites demonstrated a marked CEES adsorption capacity and outperformed the unmodified iron hydroxide. Visible light enhanced the removal of CEES owing to its photocatalytic properties. As a result of this, CEES degradation products migrate to very small pores of the composites, releasing the adsorption centers for further reactive adsorption of the CEES molecules.
Introduction
Bis(2-chloroethyl)sulfide, also known as mustard gas (HD), is a toxic compound that was used in the past as a chemical warfare agent.1 HD is a vesicant and bifunctional alkylating agent with a high degree of toxicity. It rapidly causes erythema, edema, and severe blistering after a short period of exposure.2 2-Chloroethyl ethyl sulfide (CEES) is a surrogate of a mustard gas that contains the same functional group (SCH2CH2Cl) as HD, which is responsible for alkylation of proteins, and therefore it simulates the toxicological effects of a mustard gas. Exposure to liquid HD or CEES causes severe skin damage; however, an exposure to its vapors might also cause a temporal or permanent incapacitation.1 Even though one of the mechanisms by which CEES can be dispersed as a chemical warfare agent (CWA) is by volatilization and generation of vapors, studies on vapor adsorption of CEES are less frequent than those addressing its behavior in aqueous or in organic solutions.3–5 Due to the similarity of CEES to HD and the possibility of using this compound as a chemical warfare agent, there is large interest in the research on new and efficient detoxification and decontamination technologies.Hydrolysis of the C–Cl bond is commonly reported as an important path for the detoxification of CEES, however, sometimes numerous intermediate compounds are formed on the surface of catalysts, deactivating them.5 Alternatively, two of the main products of HD and CEES oxidation are their corresponding sulfones and sulfoxides.1,6 The former ones are less toxic than HD and have a lower vapour pressure. Nevertheless, they still may cause lacrimation and sneezing. On the other hand, sulfoxide is not a vesicant and has less toxicological effects. Since either hydrolysis of the C–Cl bond or oxidation of the S atom reduces the toxicological effect of CEES, these two mechanisms have been investigated as means of decontamination in the presence of oxidant materials with photocatalytic activity.1
The surface of metal oxides is known for its oxidation potential and semiconducting properties.7 Iron(III) oxides and hydroxides collectively named hydrous ferric oxides (HFOs) have been reported to be excellent oxidizing agents for several compounds, including sulfur.8,9 The HFOs possess a band gap that ranges from very low (<0.1 eV) to medium high (2.2 eV) energy values, depending on the crystal structure, crystallinity level, and crystal size of the specific HFO.10 It has been demonstrated that hydrous ferric oxides can be photocatalytically active in the visible light range.11 Moreover, the HFOs have been proven to be excellent adsorbents, due to their porosity and a relatively high surface area.12,13 In addition, they can be fast and easily produced on laboratory and industrial scales, using a low cost synthesis process. All of these make them interesting materials for the destruction of toxic compounds.
In recent years a great amount of interest has emerged in the study of graphene-based materials.14–16 Owing to their unique properties they can provide beneficial features to their composites with metal oxides.17,18 Graphite oxide (GO) is formed when graphite is treated with strong oxidizing agents, resulting in a material with a large amount of surface oxygen-containing groups attached to the graphene layers.19 Recent literature reports have presented the benefits of graphite oxide addition to metal oxides for various applications of the resulting composites. Seredych and co-workers found an enhancement in the electrical conductivity of zinc hydr(oxide)s after GO addition,16 and an increase in the efficiency of H2S20 and NO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 reactive adsorption. This synergistic effect of GO has also been reported as increasing the uptake of H2S on CuO/GO composites,22 the uptake of NO2 on metal organic frameworks (MOFs),23 and as enhancing the degradation of methyl orange on TiO2.24
21 reactive adsorption. This synergistic effect of GO has also been reported as increasing the uptake of H2S on CuO/GO composites,22 the uptake of NO2 on metal organic frameworks (MOFs),23 and as enhancing the degradation of methyl orange on TiO2.24
Morishige and Hamada reported a successful pillaring of α-Fe2O3 and Fe3O4 between the thin graphene layers. Bashkova and Bandosz25 demonstrated the efficacy of this kind of composite for the removal of NO2 at room temperature, as a result of an increase in the composite surface area after the GO addition. It has also been reported that the electrical conductivity of iron hydroxides changes after the GO addition.26
The chemical composition of GO is an important factor that influences the formation and performance of the composites. Not only did the addition of aminated graphite oxide (GOU) to MOF significantly enhance the CO2 adsorption on the composites27 but that adsorption was much higher compared to that on the composite with unmodified GO.27 The superior performance was attributed to the incorporation of amino groups into the edges of graphene oxide that provided reaction sites for copper complexation and thus increased the structural and chemical heterogeneity of the composite products.
Based on the state-of-the-art research results on the GO-containing composites, we hypothesize that owing to the oxidizing and photocatalytic properties of GO and iron oxyhydroxides, both composites will have the capability to efficiently remove/destroy CEES via reactive adsorption. To the best of our knowledge, iron oxyhydroxide composites have not been studied for the detoxification of CEES. Therefore, the objective of this work is to evaluate the decontamination efficiency of a mustard gas surrogate on the graphite oxide/and aminated graphite oxide/iron oxyhydroxide composites (FeO–GO and FeO–GOU, respectively), as well as on the un-modified iron oxyhydroxide (FeO). An extensive surface characterization of iron/oxyhydroxides/graphite oxide and iron/oxyhydroxides/aminated graphite oxide composites was conducted, and the physicochemical properties of the materials were linked to their capacity for the reactive adsorption of CEES.
Results and discussion
To evaluate if the treatment with urea changed the surface chemistry of GO, the following analyses were carried out on the initial and modified samples: X-ray powder diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), and potentiometric titration. The results are summarized in Fig. 1. The XRD patterns show an increase in the intensity of the peak at 43° (2θ) associated with graphite (12128), which suggests the possibility of graphite oxide reduction after urea treatment. The d002 in the GO changes from 9.6 to 7.9 after the urea exposure, indicating a decrease in the interlayer distance due to the reactions of urea with GO functional groups29 (Fig. 1A). The FTIR spectra reveal the characteristic bands of the GO oxygen functional groups30 at 1726, 1621, 1047 and 985 cm−1 (Fig. 1B). The band at 1228 cm−1 can be attributed to S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O asymmetric stretching vibrations of sulfonic groups,31 due to the application of sulfuric acid for graphite oxidation.14 After urea treatment there is a shift in the wavenumbers of the bands corresponding to the carbonyl groups, and the band at 1621 cm−1 is detected at 1616 cm−1. These changes indicate a change in the chemical environment of the oxygen groups that interact with amine groups in urea. The band at 1616 cm−1 might also be due to N–H bending vibrations characteristic of secondary amines.32
O asymmetric stretching vibrations of sulfonic groups,31 due to the application of sulfuric acid for graphite oxidation.14 After urea treatment there is a shift in the wavenumbers of the bands corresponding to the carbonyl groups, and the band at 1621 cm−1 is detected at 1616 cm−1. These changes indicate a change in the chemical environment of the oxygen groups that interact with amine groups in urea. The band at 1616 cm−1 might also be due to N–H bending vibrations characteristic of secondary amines.32
 | ||
| Fig. 1 (A) X-ray diffraction patterns, (B) FTIR spectra, (C) proton binding curves and (D) pKa distributions of the graphite oxide (GO) and aminated graphite oxide (GOU). | ||
Further evidence of the graphite oxide amination is provided by the titration experiments. The proton binding curves (Fig. 1C) show an increase in the alkalinity of the surface of GOU and a slight but repeatable and consistent change in the surface pH (from 2.08 to 2.24). The increase in the basic properties of the graphite oxide is also demonstrated by the shift in the pKa positions (Fig. 1D) of some oxygen functionalities (marked with arrows), especially the ones at high acidity. We link it to their reactions with amine groups.29 In both samples, groups having pKa values between 10 and 11 predominate. We assign them to the OH/phenolic groups of GO and GOU.33 In the latter sample some basic nitrogen surface compounds can also contribute to these species.
The parameters of the porous structure of the materials studied are shown in Table 1. The surface area (SBET) of FeO is considered as being high and it is in the range of those of other HFOs reported in the literature (e.g. 230 m2 g−1 (ref. 34)). Those high surface areas are typical of highly amorphous materials, mainly 2-line ferrihydrite. The total pore volume (VT) is greater than 0.25 cm3 g−1, and mainly mesopores contribute to this value. The addition of GO and GOU increases the porosity. In the former case the surface area increases to 243 m2 g−1 (21%), which is linked to increases in the total volume of pores (15%) and in the volume micropores (Vmic) (19%). For the composite with GOU, there is an even greater increase in the surface area (88 m2 g−1; 44%). The micropore and total pore volumes increase by 17% and 40%, respectively. Even though the ratios of the mesopore to micropore volumes in all composites remain higher than 2, the smallest value obtained for FeO–GOU suggests its least mesoporous character.
| Sample | S BET [m2 g−1] | V T [cm3 g−1] | V mic [cm3 g−1] | V meso [cm3 g−1] | V meso/Vmic |
|---|---|---|---|---|---|
| FeO | 200 | 0.265 | 0.073 | 0.192 | 2.6 |
| FeO–GO | 243 | 0.304 | 0.087 | 0.217 | 2.5 |
| FeO–GOU | 288 | 0.312 | 0.102 | 0.210 | 2.1 |
Pore size distributions calculated from the nitrogen adsorption isotherms are compared in Fig. 2A. The distribution of FeO shows that the predominant pore size is 4–5 nm, demonstrating the mesoporous nature of the pure hydrous ferric oxide. The PSDs of the composites are similar to that of FeO in shape, although the pore volume increased. Another important feature is a shift in the predominant pore size to smaller values for the composite with GOU, and the appearance of pores with sizes of about 2.5 nm. Kaiser and co-workers35 reported that the uptake of CEES from a solution of hydrofluoroethers on commercial activated carbon fabrics was the most efficient on those with a mean pore size of 2.9 nm. Depending on the mechanism of decontamination, the mesopores of these sizes could also play an important role in the reactive adsorption of CEES on our materials.
 | ||
| Fig. 2 (A) Pore size distributions of ferric hydrous oxide and the composites with graphite oxide. (B) X-ray diffraction patterns of the hydrous ferric oxide and the composites with graphite oxide, ● 6-line ferrihydrite, ☆ akaganeite. | ||
The high surface area of the composites is also affected by their specific crystallographic structure. X-ray diffraction patterns are shown in Fig. 2B. The XRD pattern of FeO shows broad peaks at 35, 40, 53, 59 and 63° (2θ) that correspond to 6-line ferrihydrite with a small crystal size (110, 112, 114, 115 and 300 Miller indices respectively36), or a metaphase between 6-line and 2-line ferrihydrite.37 The size of the crystallites calculated using the Scherrer equation is about 2.9 nm (see details in the ESI†), which is an indication of the microcrystalline nature of this material.38 Besides the peaks associated with ferrihydrite, the diffraction pattern of FeO also reveals low intensity peaks corresponding to akaganeite at 21 (110), 27 (311), 40 (211) and 46° (301). The existence of this crystallographic phase might be linked to a minor transformation of 2-line ferrihydrite caused by the precursor salt and partial dehydration taking place during the synthesis process (temperature effect).37
After the GO addition there is a marked change in the crystallographic pattern, and only the broad peaks at 35° and 63° (2θ) are visible indicating the presence of 2-line ferrihydrite. The peaks corresponding to akaganeite decreased in their intensity, suggesting that GO inhibits the minor transformation of the 2-line ferrihydrite. Applying the Scherrer equation gives a crystallite size of about 2.1 nm. This implies that GO induces a decrease in the particle size of the inorganic iron hydroxide phase. After GOU addition the XRD pattern is similar to that of FeO with the main crystallite size of 4.0 nm. Thus the addition of GOU does not considerably change the crystal structure of the composite. It only alters its porosity.
The Scanning Electron Microscopy (SEM) images of the composites are shown in Fig. 3. FeO shows a rugous surface with large pores formed between small clusters of round-shaped nanoparticles ranging between 60 and 110 nm (as shown in the rectangle). The shape of the particles is a characteristic of 6-line ferrihydrite.39 The images suggest that the presence of GO in the FeO–GO composite increased the apparent level of amorphicity, since the round-shaped nanocrystals detected are in the range of 20–70 nm, 50% smaller than those in FeO.
 | ||
| Fig. 3 SEM images of the surfaces of the samples studied. The enlarged area in the image of FeO–GOU corresponds to a band-pass filter image obtained by applying Fast Fourier Transform (FFT) to the area marked in the square. A mask removal at high and low frequencies was performed, and the inverse FFT yielded the band-passed image (see ESI† for details of the image processing). | ||
The morphology of FeO–GOU differs from those of FeO and FeO–GO. In the former material the presence of the graphene layers of the aminated graphite oxide is evident. Small particles are visible in the vicinity of the layers. A filtered image of the selected area in the SEM image of FeO–GOU shows the presence of the graphene layers in the composite and the hydrous iron oxide particles deposited on their surface.
The differences in the morphology of the composites are also evident from HRTEM images presented in Fig. 4. FeO exhibits a marked, amorphous nature, showing the typical structure of 2-line ferrihydrite (Fig. 4A). The image also reveals the presence of an ordered structure around the amorphous phase (Fig. 4B), which we link to the graphene based phase embedded within the amorphous structure of the 2-line ferrihydrite. In the case of FeO–GOU clusters of particles of about 100–200 nm with some crystallinity level are seen, as observed in the electron diffraction pattern (Fig. 4C and D). As mentioned above, in this sample the 6-L ferrihydrite is present and therefore the crystallinity can be attributed to this phase. The crystal sizes of about 3–5 nm (Fig. 4E) are in agreement with the XRD results. Lattice spacing is of about 0.23 nm, (Fig. 4F, inset) and it corresponds to the (110) d-space of 6-line ferrihydrite.10
 | ||
| Fig. 4 (A) TEM image of FeO–G and (B) a highlighted zone showing the graphene phase in FeO–GO and a small crystal of the iron hydroxide phase. (C) TEM image of FeO–GOU. (D) Electron diffraction pattern revealing some degree of crystallinity in the samples. (E) Image of the FeO–GOU nanocrystals. (F) Image of the nanocrystals; the inset represents the inverse Fourier transform of the selected area, the fridge distance is 0.25 nm. | ||
The particle size found based on the TEM images of FeO–GOU is slightly larger than that calculated from the Scherrer equation. This is due to other factors (besides particle size) that influence the peak broadening, such as dislocations and lattice imperfections. However, both methods give the sizes of similar magnitudes and both of them confirm the nanoscale range of our materials.
Not only the morphology, but also the surface chemistry of the iron oxyhydroxides are affected by the addition of GO and GOU. The proton binding curves (Q) and the pKa distributions of the samples studied show the marked differences in surface chemistry (Fig. 5). The proton binding curve of FeO shows a negatively charged surface with a point of zero charge (pHPZC) of 7.5, which is in the range of hydrous ferric oxides, especially ferrihydrites.10,40 The addition of GO or GOU acidifies the surface of the composites and causes a shift in the pHPZC to 6.8. This can be caused by the contribution of acidic groups from the graphite oxide or changes in the chemical environment of iron containing acidic groups at the interface between the GO and hydroxide phases. These changes are less pronounced for the composite with GOU than for that with GO.
 | ||
| Fig. 5 Potentiometric titration results: proton binding curves (A) and pKa distributions (B). | ||
The pKa distribution in Fig. 5B provides more details on the surface chemical changes caused by the GO and GOU additions. For FeO three species at pKas 7.1, 8.6 and 10 are revealed. The ones at 7.1 and 8.6 might correspond to the acidity constants of the protolysis reactions of the OH surface groups of ferrihydrite;10 and the one at 10 can be attributed to the presence of polynuclear Fe(III) (hydroxyl) species.41 On the surface of FeO–GO new groups at a pKa of 6.6 appear, and the species represented by pKa between 7 and 8, and between 8 and 9 in the case of FeO have developed a more basic character. Also, the contribution of the most basic group at pKa > 10 representing terminal hydroxyl groups clearly increases. This increase is likely caused by an increase in the amorphicity level of the inorganic phase and thus more terminal hydroxyl groups present on the surface.37 The OH groups of GO can also contribute to these values. In the case of FeO–GOU, the peak at pKa 6.6 is still detected; however, instead of the peak at pKa 9.6 attributed to the presence of the OH groups from ferrihydrite, there are two new peaks at 8.1 and 9.1. The population of species at pKa 10.5 increases compared to those on FeO but their contribution seems to be smaller than that of FeO–GO. The total number of groups in the composites with GO and GOU increased 250% and 220%, respectively, when compared to the numbers of acidic groups detected on FeO (Table S1 in the ESI†).
The FTIR-ATR spectra (Fig. 6) show subtle but interesting differences between the composites. Two bands are observed for all samples; the one at a wavenumber of 3410 cm−1 corresponds mainly to the stretch of OH bulk groups in the ferrihydrite structure,42 and the band at 1650 cm−1 corresponds to adsorbed water.43 Low intensity bands at 1469 and 1353 cm−1 can be attributed to Fe–O and Fe–OH, respectively.42 Finally the band at 694 cm−1 corresponds to the deformation of bulk hydroxyl groups coming from the hydrated ferric oxides.37 For FeO–GO a band at 1598 cm−1 suggests a possible link to the remaining carbonyl groups of GO. A band at 1336 cm−1 represents the shift of the Fe–OH band due to a change in the chemical environment caused by the GO addition. The same assignment applies to the band at 684 cm−1 that corresponds to iron hydroxyl group deformation.
 | ||
| Fig. 6 FTIR-ATR spectra of the materials studied. | ||
In the composite synthesized with GOU there is a decrease in the intensities of the bands representing Fe–O bonds, and the band corresponding to GO’s carbonyl groups disappears. As for the composite with GO, the Fe–OH band shifts to 1332 cm−1 and that associated with the hydroxyl groups shifted to 670 cm−1. Also there is an appearance of a small shoulder at a wavenumber of 910 cm−1. This band might correspond to the N–H waging of primary and secondary amines in GOU.44
The FTIR results suggest the interaction of iron with oxygen groups of GO. This is in agreement with other studies that have reported reactions of GO’s oxygen groups (mainly epoxy) with metal oxide surfaces.22–24
The changes in the chemistry of our materials were also studied by Raman spectroscopy (Fig. 7). The Raman spectrum of FeO reveals weak bands at 500 cm−1 and 1230 cm−1, and a strong band at 715 cm−1, which correspond to those reported for 2-line ferrihydrite.45 In the spectra of the composites with GO and GOU, the bands assigned to the carbon phase appear; one at 1320 cm−1 that is related to the disorder in the graphene layers (D band), and one at 1590 cm−1 that is attributed to the sp2 domains in the graphene lattice (G band).46 It is important to mention that for the composite formed with GOU, the D and G bands are less intense and wider than those of FeO–GO. The ID/IG ratio of the composite with GO is 0.97 and of that with GOU is 0.83. It is well known that a higher disorder in graphite leads to broader G bands and a larger ID/IG ratio.47 Thus a higher ID/IG ratio and a broader G band for the composite with GO than for that with GOU indicate its higher level of structural defects. These results are in agreement with the SEM images in which the GOU surface shows a layered structure that has fewer visible defects than that of GO. The structural defects in the graphene phase of the composites affects their electronic properties.48 Even though the GO addition is expected to enhance the electron transport in the composite, the structural defects might change the electron trajectories.49 On the other hand, the amination of graphite oxide led to a layered composite with less structural defects, and therefore the better electron transport. These features might be important for potential redox reactions taking place on the surface of these materials. Owing to these differences in the morphology, it is expected that the composites with GO and GOU will perform in a different way as CEES adsorption/degradation media.
 | ||
| Fig. 7 Raman spectra of FeO and its composites with GO or GOU. | ||
The Ultraviolet-Visible-Near Infrared Spectroscopy (UV-Vis-NIR) spectra of the composites are shown in Fig. 8A. The shoulder at 760 nm originates from the electronic transitions of the Fe3+ ligand field, and magnetically coupled Fe3+ cations and also from ligand-to-metal charge transfer (LMCT).50 The difference in the maximum and minimum absorption branches after the GO and GOU additions is a result of the change in the chemical environment caused by the incorporation of graphene layers to the composite.29 Since any change in the material's physics and chemistry has an effect on the band gap width, the band gap energy (Eg) was estimated from the UV-Vis-NIR spectrum.51 Details of the Eg calculation are provided in the ESI.† The extrapolation of the linear fit of the plot of [F(R∞)hv]2versus the photon energy (hv) yields the value of Eg. The plots are shown in Fig. 8B.
 | ||
| Fig. 8 (A) UV-Vis-NIR spectra of the synthesized composites and (B) [F(R∞)hv]2versus photon energy. The lines show the cut-off employed to calculate the band gap energy. | ||
The calculated band gap energy of the FeO is 1.74 eV, which is in the range of poorly crystallized hydrous iron oxides.37 On the other hand, the samples with GO and GOU exhibit energy gaps of 1.63 and 1.79 eV, respectively. Therefore, the addition of GO decreases the extent of Eg, and in the case of the aminated graphite oxide, a slight increase is caused by the addition of GOU. These different effects on the optoelectronic properties of the composites, depending on the chemical nature of the graphite oxide, are associated with the specific interactions of hydrous ferric oxides and graphite oxide.
Since FeO–GO absorbs a broader range of visible light than FeO, graphite oxide photosensitizes the composite. This effect has been observed for wide band semiconductors in which the band gap is not able to be photoexcited, and no holes are formed. In those cases the graphene phase acts like a macromolecular photosensitizer.52 For Fe–GO it is possible that GO is photoexcited to such a state that it injects electrons into the conduction band of the hydrous ferric oxide. This process promotes the activation of oxygen and the formation of oxide radicals.52 On the other hand, the FeO–GOU material shows an absorption at lower wavelengths close to the NIR range, and also a higher absorption at wavelengths lower than 550 nm. It has been reported that this effect occurs due to the abundance of delocalized electrons in the graphite network, which might enhance the charge transport, and promote the formation of holes and electrons in the composite materials.20,53 It is possible that in this composite the carbon phase may act as a trap for the formation of holes in the irradiated composites.54 It has also been reported that nitrogen doping into Cu2O films increases Eg, due to the structural changes that generate numerous oxygen vacancies.55 Also, the incorporation of nitrogen moieties into the graphene matrix shifts the Fermi level and contributes to breaks in the symmetry of the graphite lattices, therefore causing an increase in Eg.56 It is possible that the combination of these effects takes place in the FeO–GOU composite. Both effects, photosensitization and narrowing of the band gap, can be beneficial for the process of the CEES oxidation/degradation. While wide band gap semiconductors have a good carrier mobility,57 narrow band gap materials are more active in the visible spectrum. It is important to mention that the small differences between the energies might be statistically insignificant. Nevertheless, the values of the band gap energies of our samples suggest their photoactivity in visible light.
Based on the results obtained, it is suggested that in the FeO–GO composite, HFOs were incorporated into the composite via interactions with oxygen groups located in the graphite oxide network structure. The incorporation mechanism can be explained as follows: in the first stage, interactions of the iron hexaaquo complexes in a solution with the oxygen groups of GO to form oxygen bridges take place (Fig. 9A); when the pH increases, in the second stage, the oxygen groups in the graphite oxide act as nucleation centers for the propagation reactions of the iron complexes to form HFOs (Fig. 9B). The propagation stage occurs when the increase in the pH causes the condensates to aggregate58 (Fig. 9C), thus promoting the formation of particles around the graphite oxide layers (Fig. 9D). In the current experimental conditions (concentration of the precipitation agent and addition rate), the particles are formed as an apparently amorphous phase consisting of microcrystals with a high surface area and mesoporosity.
 | ||
| Fig. 9 Interactions of HFO with the GO surface, (A) Nucleation of iron hexaaquo complexes, (B) oxygen bridge between oxygen groups in the GO and iron complexes, (C) condensation of the hydrous ferric oxide particles, and (D) evolution of porosity in the HFO aggregates. | ||
A 24 hour equilibrium time was established as an acceptable time to reach total CEES evaporation which led to a constant concentration in our systems (see Fig. S2 in the ESI†). After a 24 h duration of the adsorption process, an increase in the weight of the composites was recorded (Qads). It represents the capacity of the system to adsorb CEES and/or its reaction products. Besides the experiments in visible light, the experiments in the dark were also carried out to determine if the redox reactions stimulated by visible light might affect the adsorption capacity of the composites, or promote reactions that might result in a change in the adsorption capacity of our materials.
Table 2 shows the increases in the weight of the materials after CEES exposure. As mentioned above, these amounts include the adsorption of both CEES and all non-volatile compounds produced during reactive adsorption that might be deposited in the pore system. The uptakes on the composites are visibly higher than those on the pure hydrous oxides, either in the light or dark experiments. Exposure to visible light for 24 hours increases the amount adsorbed by 43% and 100% on the composite with GO and GOU, respectively, in comparison with the amount adsorbed on FeO. Interestingly, that increase in the dark experiments after 24 hours was 40% and 48%, respectively. This indicates the strong effect of light, especially in the case of FeO–GOU.
The increase in the exposure from 24 h to 7 days further increases the amount of CEES/its decomposition products adsorbed (Table 2). It is possible that this can be due to surface mediated slow transformation of molecules from the vapor phase followed by their reactive adsorption. The largest increase was recorded for FeO–GOU, whose high surface area and pore volume promotes the deposition of the reaction products on the surface. The graphene phase has a positive effect on the enhancement of the CEES reactive adsorption under visible light exposure. Thus for FeO the increase in the weight after 24 hours was 18%, and for the composites with GO and GOU it was 20% and 61%, respectively, in comparison with the corresponding results obtained after adsorption in the dark.
To understand the role of the surface features of our materials in the reactive adsorption process, the relationship between the adsorption capacities and the micro- (Vmic) and total pore volume (VT) of the composites was analyzed (Fig. 10). The adsorption capacities measured in visible light show an almost linear dependence on the micropore volumes (R2 = 0.99). Apparently the total pore volume is less important for this process (R2 = 0.81). On the other hand, the results obtained in the dark experiments show a perfect correlation with the total pore volume (R2 = 0.99) and much less dependence on the volume of micropores (R2 = 0.86).
 | ||
| Fig. 10 Dependence of the amount adsorbed (after 24 h of equilibrium) on the volume of pores in the materials. (A) Under light exposure; (B) experiments in the dark. | ||
Initially adsorbed CEES, besides being retained on the surface via dispersive forces, can also be transformed either into small gaseous molecules non-adsorbed on the material surfaces, or can undergo surface reactions with their products adsorbed in the pore system. Regardless of its fate, the analysis of the concentration of CEES in the headspace of the reactors (Ceq.) should indicate the total disappearance of this adsorbate. To better evaluate its fate, the quantities listed below were introduced:
The capacity of CEES removal (Qrem) was calculated according to the formula:
 | (1) |
The total CEES removal capacity (Mrem) can be calculated as a ratio between the initial and final CEES concentrations.
 | (2) |
Finally, the percentage of adsorbed molecules (Mad) is calculated as follows:
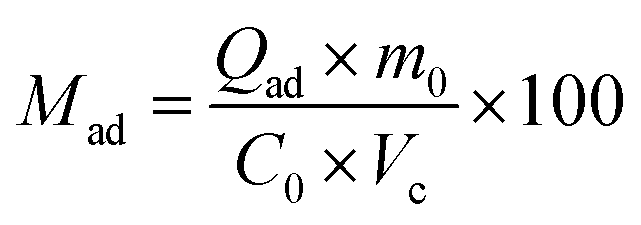 | (3) |
The weight percentage of volatile non-adsorbed molecules (Volatile) can be calculated as the difference between the percentage of CEES removed (Mrem) (which was either adsorbed or decomposed) and the percentage of the adsorbed molecules (Mad). The percentage of the molecules transformed into volatile compounds was calculated according to the formula:
| Volatile = Mrem − Mad | (4) |
Even though it is judged based on the unchanged concentration of CEES that its adsorption reaches an apparent equilibrium after 24 h, it is possible that slow surface reactions between compounds formed from CEES are still taking place on the surface of the adsorbents as hypothesized above. Following this assumption, we ran the experiments for seven days to promote more complete transformation of the molecules. After seven days, the concentration of CEES in the headspace and the increase in its weight were evaluated. The results are shown in Table 2.
The materials studied show a high capacity for the elimination of CEES. While on FeO 32% CEES from the reactor volume was adsorbed, the addition of GO increased this amount to 52%. The beneficial effect is reflected also by an increase in the percentage of CEES converted to the volatile, non-adsorbed surface reaction products.
The formation of new compounds, as a result of photoactivity, might explain the trends found in the dependence of the Qad and the volume of specific pores discussed above (Fig. 2A). If light promotes the hydrolysis of C–Cl bonds and therefore the transformation of CEES into smaller molecules, then it is plausible to assume that those small molecules would migrate to narrow pores (micropores) that are less accessible to the larger molecules, such as CEES. In the gaseous phase the CEES molecule has a critical diameter of 0.58 nm,35 small enough to migrate to the material micropores. However, in the presence of atmospheric water, the solvation of the molecule results in a molecular diameter of 1.88 nm.35 Therefore it is expected that mesopores play a relevant role as reactive adsorption centers for CEES, while micropores are more important for smaller molecules which could be the products of surface reactions/CEES decomposition.
In the absence of light, the extent of CEES transformation is smaller, due to the lack of energy that might promote redox reactions; therefore mesopore sizes are favorable for the accommodation of adsorbed molecules on the surface.35 Thus a better correlation of the amount adsorbed with the total pore volume was found.
The results suggest that several reactions between CEES and the surface of the materials take place simultaneously. However, the specific identification of the products of the CEES reactive adsorption, as well as the detailed mechanism of decontamination, is beyond the scope of this paper and it is the focus of our ongoing study. Regardless of the removal mechanism, it is worth mentioning that our composites exhibit a marked CEES removal capacity, which makes them promising materials for the decontamination of CWA.
Experimental
Materials
Commercial graphite (from Aldrich) was oxidized using the Hummers method.59 Briefly, commercial graphite (20 g) was mixed with concentrated sulfuric acid (230 mL) at 0 °C, next potassium permanganate (30 g) was slowly added by keeping the reaction temperature below 20 °C in a cooling bath. After this, deionized water (230 mL) was slowly added to prevent the temperature from exceeding 98 °C. The colloid was stirred for 15 min, followed by a dilution to 1.4 L, and finally H2O2 (100 mL, 30%) was added. The mixture was left to settle overnight and then rinsed until no remaining SO4−2 ions were detected. The brown precipitate was freeze-dried for 2 weeks and then stored at −4 °C before use. This material is referred to as GO. The aminated graphite was obtained by mixing GO (1 g) with a urea solution (100 mL, 0.3 M). The mixture was maintained under continuous stirring for 24 h, and then rinsed with deionized water until a constant pH. Finally, the resulting material was air dried at room temperature; the sample is referred to as GOU. The hydrous ferric oxide (FeO) particles were obtained by a precipitation method. FeCl3·6H2O (350 mL, 0.026 M) and NaOH (660 mL, 0.05 M) were used. NaOH was added using a Titronic Universal (SCHOTT) titrator at a rate of 40 mL min−1. After the red-brown precipitate was formed, it was rapidly decanted, and then rinsed several times with deionized water until no AgCl precipitate was present in the rinsing solution after AgNO3 addition. Finally, the FeO sample was dried for 24 h at 100 °C. To prepare the composites, GO or GOU was added to the composites to reach a mass content of 10% of the final materials. Either GO or GOU was mixed with FeCl3·6H2O (350 mL, 0.026 M) solution. The suspensions were sonicated for 1 h to promote graphite oxide dispersion, and then stirred for an extra hour to increase homogeneity. After that, NaOH (660 mL, 0.05 M) was added at a rate of 40 mL min−1. The precipitate was collected and rinsed until no chloride presence was detected, and finally dried at 100 °C for 24 h. Samples are referred to as FeO–GO and FeO–GOU, depending on the type of GO used.Determination of porosity
The surface area and porosity of the materials studied were calculated from N2 adsorption isotherms measured using ASAP 2020 (Micromeritics). The BET surface areas were calculated from the isotherms. The total pore volume was obtained from the amount adsorbed at a relative pressure of 0.99 and the micropore volume was calculated from the Dubinin–Astakhov equation.60 The mesopore volume was calculated by the difference between VT and Vmic and the Barret–Joyner–Halenda (BJH) method was used to calculate pore size distributions, PSDs.61X-ray diffraction
The X-ray powder diffraction patterns were collected from 10 to 90° (2θ) at absolute scan using a Philips X'Pert X-ray diffractometer with CuKα radiation at 40 mA and 40 kV.SEM
The SEM images of the surface of the materials were obtained using a Zeiss Supra 55 VP instrument with an acceleration voltage of 5 keV.TEM
The TEM images were obtained using a JEOL JEM-2100 Transmission Electron Microscope operating at 200 kV. Before analysis, the sample was ground, suspended in high purity isopropanol and sonicated for 20 min. After this, sample drops were applied to a grid holder.Potentiometric titration
Potentiometric titration measurements were carried out with a DMS Titrando 888 automatic titrator (Metrohm). The experimental procedure was as follows: the sample (100 mg) was mixed with a NaNO3 solution (50 mL, 0.01 M) and equilibrated for 12 h. The solution was purged with N2 and when a constant pH was reached, a titration with NaOH (0.1 M) was carried out. During the titration the suspension was continuously purged with N2 to eliminate the interference of atmospheric CO2. The experimental titration curves were transformed into proton binding curves (Q) using a proton balance with a theoretical blank reference.62Q represents the total amount of protonated sites on the surface of the materials. The intercept of the x-axis represents the point of zero charge of the materials (pHPZC). Q values that are below the zero line indicate proton release related to surface acidity. Q is related to the pKa distribution of all dissociated groups in the solid by the following equation:63 | (5) |
Application of this equation yields the pKa distribution of the species present on the surface. The integral equation was solved using SAEIUS software.64
Surface pH
The surface pH was measured by mixing 0.1 g of the material with 5 mL of distilled water and stirring overnight. After this, the pH of the solution was recorded.FTIR
The analyses were carried out using a Nicolet Magna-IR 380 spectrometer, by using the Smart MIRacle accessory that measures the attenuated total reflectance. The spectrum was collected 64 times and corrected for background noise. Experiments were done without KBr addition.Raman spectroscopy
Raman spectra studies were carried out with a MonoVista Confocal Raman microscope spectrometer using a 633 nm helium/neon laser at a 10× working distance. The analyses were carried out on the powder deposited on a glass holder.Ultraviolet-visible-near infrared spectroscopy
The spectra were obtained with a Cary500 Scan spectrometer (Varian) by using the Cary 500 diffuse reflectance accessory (integrated sphere). Before the analyses, the samples were compressed to form 0.65 mm thick pellets. The samples were mounted on a black tape and fitted into an integration sphere analysis port. The integration sphere was operated to collect diffuse reflection.CEES adsorption
The adsorption of CEES was studied in batch experiments. A glass vial containing 150 mg of the sample was introduced into a 160 mL reaction vessel closed with a septum. After hermetically sealing the vessel, CEES (300 μL) was injected through a septum into a 5 mL beaker in the reaction vessel. The containers were kept under visible light (xenon lamp, Solar light Co., INC, XPS-150™) and/or in the dark at room temperature for 1 or 7 days, depending on the target experiment. The latter was done to ensure the equilibrium of any surface reaction and allow the complete evaporation of CEES in the container. After equilibrium was reached, vapor phases from the headspace of the containers were sampled with a syringe and injected into a GC-MS. Once the vapor sample was taken, the containers were opened and the adsorbent samples were equilibrated in air for 1 h at atmospheric pressure, in the absence of moisture. Finally, the containers with the adsorbents were weighed, and the mass gain as a result of adsorption was recorded. The total CEES removal capacity of the materials was calculated after the 7 day experiments as the ratio between the CEES concentration in the containers and the starting concentration. The percentage of non-bonded volatile compounds was estimated as the difference between the percentages of total removal capacity and the increase in the material mass after CEES exposure. Details on the calculations are provided in the Results and discussion section.Gas chromatography-mass spectrometry (GC-MS)
The analysis of the relative amounts of CEES and any reaction product present in the vapor phase was carried out using a GCMS-QP5050A (Shimadzu). The separation of the compounds was performed in a XTI-5 column (5% diphenyl–95% dimethyl polysiloxane) of 30 m length, 0.25 mm internal diameter, and 0.25 μm of liquid film thickness. The GC operation program was as follows: an increase from 50 °C to 100 °C at a rate of 5 deg min−1, then the rate was changed to 40 deg min−1 up to 280 °C. Helium was used as the carrier gas. The injection volume, total flow, and the split ratio were 40 μL, 17.8 mL and 8, respectively. CEES was detected at the elusion time of 6.4 min. The mass spectrometer detector was used in an electron impact ionization mode. A calibration curve was prepared by adding 100, 200, 300 and 400 μL of CEES into empty containers. After a complete evaporation of each volume of CEES, the area of the peak at 6.4 min was correlated with the concentration of CEES.Conclusions
The results presented in this paper demonstrate the benefits of the incorporation of graphite oxide and aminated graphite oxide into hydrous ferric oxides for the elimination of CEES from air. The graphene-based phase significantly increased the dispersion of ferric particles, enhancing the adsorption capacity. It is suggested that oxygen and/or nitrogen groups in the graphite oxide act as nucleation centers for iron aquo-complexes; when the pH increases, the particles grow, around the graphite oxide flakes. The resulting iron/graphite oxide composites have higher surface areas and mesopore volumes than the parent hydrous ferric oxide. They are composed mainly of 2 and 6-line ferrihydrites, with a minor amount of akaganeite. The FT-IR and UV-Vis-NIR results showed the interactions between the hydrous ferric hydroxide and the oxygen groups in the graphite oxide surface. The composites exhibit a high efficiency for removing CEES vapors from air under both light and dark experimental conditions. Apparently the addition of GO and GOU causes a shift in the band gap energy, causing a clear enhancement in the elimination capacity under visible light radiation. This is associated with the transformation of CEES to simpler/smaller molecules and their migration to smaller pores of higher adsorption energy. Under light exposure, a linear correlation of the amount adsorbed with the micropore volume was found. On the other hand, when the experiments were run in the dark, the total pore volume of composites was found as the most important factor for the adsorption process, owing to the size of the hydrated CEES molecules. The results highlighted the hydrous ferric oxide/graphite oxide composites as interesting materials for the detoxification of CEES vapors. Even though we showed the applicability of these materials for CWA detoxification, their catalytic activity might find potential application for other industrially important processes.Acknowledgements
This study was supported by the ARO (Army Research Office) grant W911NF-13-1-0225 and NSF collaborative SBET Grant no. 1133112. The assistance of Drs Jorge Morales and Alexei Bykov during microscopy studies and UV-Vis measurements, respectively, is appreciated.References
- T. T. Marrs, R. L. Maynard and F. Sidell, Chemical Warfare Agents: Toxicology and Treatment, Wiley, West Sussex, England, 2007 Search PubMed.
- N. A. Monteiro-Riviere, A. O. Inman, M. C. Babin and R. P. Casillas, J. Appl. Toxicol., 1999, 19, 313–328 CrossRef CAS.
- A. Roy, A. K. Srivastava, B. Singh, T. H. Mahato, D. Shah and A. K. Halve, Microporous Mesoporous Mater., 2012, 162, 207–212 CrossRef CAS PubMed.
- M. Fox, Y.-S. Kim, A. A. Abdel-Wahab and M. Dulay, Catal. Lett., 1990, 5, 369–376 CrossRef CAS.
- A. V. Vorontsov, C. Lion, E. N. Savinov and P. G. Smirniotis, J. Catal., 2003, 220, 414–423 CrossRef CAS.
- L. Bromberg, N. Pomerantz, H. Schreuder-Gibson and T. A. Hatton, Ind. Eng. Chem. Res., 2014 DOI:10.1021/ie501055g.
- J. P. Jolivet, M. Henry and J. Livage, Metal Oxide Chemistry and Synthesis: from Solution to Solid State, Wiley, West Sussex, England, 2000 Search PubMed.
- J. A. Arcibar-Orozco, J. R. Rangel-Mendez and T. J. Bandosz, J. Hazard. Mater., 2013, 246–247, 300–309 CrossRef CAS PubMed.
- B. C. Faust, M. R. Hoffmann and D. W. Bahnemann, J. Phys. Chem., 1989, 93, 6371–6381 CrossRef CAS.
- R. M. Cornell and U. Schwetmann, The Iron Oxides Structure, Propeties, Reactions, Ocurrences and Uses, Winheim, 2003 Search PubMed.
- X. Zhou, H. Yang, C. Wang, X. Mao, Y. Wang, Y. Yang and G. Liu, J. Phys. Chem. C, 2010, 114, 17051–17061 CAS.
- R. R. Gadde and H. A. Laitinen, Anal. Chem., 1974, 46, 2022–2026 CrossRef CAS.
- J. A. Arcibar-Orozco, M. Avalos-Borja and J. R. Rangel-Mendez, Environ. Sci. Technol., 2012, 46, 9577–9583 CrossRef CAS PubMed.
- C. Petit, M. Seredych and T. J. Bandosz, J. Mater. Chem., 2009, 19, 9176–9185 RSC.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- M. Seredych, O. Mabayoje, M. M. Kolesnik, V. Krstic and T. J. Bandosz, J. Mater. Chem., 2012, 22, 7970–7978 RSC.
- Y. H. Ng, A. Iwase, N. J. Bell, A. Kudo and R. Amal, Catal. Today, 2011, 164, 353–357 CrossRef CAS PubMed.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2009, 4, 380–386 CrossRef PubMed.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- M. Seredych, O. Mabayoje and T. J. Bandosz, Langmuir, 2011, 28, 1337–1346 CrossRef PubMed.
- M. Seredych, O. Mabayoje and T. J. Bandosz, J. Phys. Chem. C, 2012, 116, 2527–2535 CAS.
- O. Mabayoje, M. Seredych and T. J. Bandosz, ACS Appl. Mater. Interfaces, 2012, 4, 3316–3324 CAS.
- B. Levasseur, C. Petit and T. J. Bandosz, ACS Appl. Mater. Interfaces, 2010, 2, 3606–3613 CAS.
- T. Jiang, Z. Tao, M. Ji, Q. Zhao, X. Fu and H. Yin, Catal. Commun., 2012, 28, 47–51 CrossRef CAS PubMed.
- S. Bashkova and T. J. Bandosz, Ind. Eng. Chem. Res., 2009, 48, 10884–10891 CrossRef CAS.
- V. K. Singh, M. K. Patra, M. Manoth, G. S. Gowd, S. R. Vadera and N. Kumar, New Carbon Mater., 2009, 24, 147–152 CrossRef CAS.
- Y. Zhao, M. Seredych, Q. Zhong and T. J. Bandosz, ACS Appl. Mater. Interfaces, 2013, 5, 4951–4959 CAS.
- J. Fayos, J. Solid State Chem., 1999, 148, 278–285 CrossRef CAS.
- Y. Zhao, M. Seredych, Q. Zhong and T. J. Bandosz, RSC Adv., 2013, 3, 9932–9941 RSC.
- T. Szabó, O. Berkesi and I. Dékány, Carbon, 2005, 43, 3186–3189 CrossRef PubMed.
- U. Bohner and G. Zundel, J. Phys. Chem., 1985, 89, 1408–1413 CrossRef.
- B. H. Stuart, Infrared Spectroscopy: Fundamentals and Applications, Wiley, West Sussex, England, 2004 Search PubMed.
- B. Konkena and S. Vasudevan, J. Phys. Chem. Lett., 2012, 3, 867–872 CrossRef CAS.
- D. Masih, Y. Seida and Y. Izumi, Water, Air, Soil Pollut.: Focus, 2009, 9, 203–211 CrossRef CAS.
- R. Kaiser, A. Kulczyk, D. Rich, R. J. Willey, J. Minicucci and B. MacIver, Ind. Eng. Chem. Res., 2007, 46, 6126–6132 CrossRef CAS.
- I. Carabante, M. Grahn, A. Holmgren, J. Kumpiene and J. Hedlund, Colloids Surf., A, 2009, 346, 106–113 CrossRef CAS PubMed.
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, Wiley, Weinheim, Germany, 2006 Search PubMed.
- R. L. Penn, Science, 2007, 316, 1704–1705 CrossRef CAS PubMed.
- D. E. Janney, J. M. Cowley and P. R. Buseck, Clays Clay Miner., 2000, 48, 111–119 CAS.
- L. Charlet and A. A. Manceau, J. Colloid Interface Sci., 1992, 148, 443–458 CrossRef CAS.
- T. J. Bandosz, Pol. J. Chem., 1998, 72, 1202–1214 CAS.
- K. Rout, M. Mohapatra and S. Anand, Dalton Trans., 2012, 41, 3302–3312 RSC.
- J. D. Russell, Clay Miner., 1979, 14, 109–114 CAS.
- B. C. Smith, Infrared Spectral Interpretation: a Systematic Approach, Taylor & Francis, Boca Raton, 1998 Search PubMed.
- L. Mazzetti and P. J. Thistlethwaite, J. Raman Spectrosc., 2002, 33, 104–111 CrossRef CAS.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2007, 8, 36–41 CrossRef PubMed.
- A. Kaniyoor, T. T. Baby and S. Ramaprabhu, J. Mater. Chem., 2010, 20, 8467–8469 RSC.
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2010, 5, 26–41 CrossRef PubMed.
- A. Cortijo and M. A. H. Vozmediano, Nucl. Phys. B, 2007, 763, 293–308 CrossRef PubMed.
- J. Torrent and V. Barrón, Diffuse reflectance spectroscopy of iron oxides, Boca Raton, 2002 Search PubMed.
- A. E. Morales, E. S. Mora and U. Pal, Rev. Mex. Fis. S, 2007, 53, 18–22 CAS.
- Y. Zhang, N. Zhang, Z.-R. Tang and Y.-J. Xu, ACS Nano, 2012, 6, 9777–9789 CrossRef CAS PubMed.
- O. Mabayoje, M. Seredych and T. J. Bandosz, Appl. Catal., B, 2013, 132–133, 321–331 CrossRef CAS PubMed.
- S. Y. Treschev, P.-W. Chou, Y.-H. Tseng, J.-B. Wang, E. V. Perevedentseva and C.-L. Cheng, Appl. Catal., B, 2008, 79, 8–16 CrossRef CAS PubMed.
- Y. Nakano, S. Saeki and T. Morikawa, Appl. Phys. Lett., 2009, 94, 022111-1–022111-3 CrossRef PubMed.
- P. Rani and V. K. Jindal, RSC Adv., 2013, 3, 802–812 RSC.
- T. P. Chow and R. Tyagi, Electron Devices, IEEE Transactions on, 1994, 41, 1481–1483 CrossRef CAS.
- J.-P. Jolivet, C. Chaneac and E. Tronc, Chem. Commun., 2004, 481–483 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- M. M. Dubinin, in Progress in Surface and Membrane Science, ed. D. A. Cadenhead, Academic Press, New York, US, 1975, pp. 1–70 Search PubMed.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- C. Contescu, J. Jagiello and J. A. Schwarz, Langmuir, 1993, 9, 1754–1765 CrossRef CAS.
- T. J. Bandosz, J. Jagiello, C. Contescu and J. A. Schwarz, Carbon, 1993, 31, 1193–1202 CrossRef CAS.
- J. Jagiello, Langmuir, 1994, 10, 2778–2785 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Peak positions on the pKa distributions, calculations of the crystallite size, details of the energy band gap calculations, image processing of Fig. 3, and CEES chromatograms. See DOI: 10.1039/c4ta04159c |
| This journal is © The Royal Society of Chemistry 2015 |