
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co-adsorption of peptide amphiphile V6K and conventional surfactants SDS and C12TAB at the solid/water interface†
Dharana
Jayawardane‡
a,
Fang
Pan‡
b,
Jian R.
Lu
*b and
Xiubo
Zhao
*a
aDepartment of Chemical and Biological Engineering, University of Sheffield, Sheffield, S1 3JD, UK. E-mail: Xiubo.zhao@sheffield.ac.uk; Tel: +44-114-2228256
bBiological Physics Group, Schuster Building, Oxford Road, University of Manchester, Manchester, M13 9PL, UK. E-mail: J.lu@manchester.ac.uk; Tel: +44-161-3063926
First published on 19th August 2015
Abstract
Recent research has reported many attractive benefits from short peptide amphiphiles. A practical route for them to enter the real world of applications is through formulation with conventional surfactants. This study reports the co-adsorption of the surfactant-like peptide, V6K, with conventional anionic and cationic surfactants at the solid/water interface. The time-dependant adsorption behaviour was examined using spectroscopic ellipsometry whilst adsorbed layer composition and structural distribution of the components were investigated by neutron reflection with the use of hydrogen/deuterium labelling of the surfactant molecules. Both binary (surfactant/peptide mixtures) and sequential (peptide followed by surfactant) adsorption have been studied. It was found that at the hydrophilic SiO2/water interface, the peptide was able to form a stable, flat, defected bilayer structure however both the structure and adsorbed amount were highly dependent on the initial peptide concentration. This consequently affected surfactant adsorption. In the presence of a pre-adsorbed peptide layer anionic sodium dodecyl sulfate (SDS) could readily co-adsorb at the interface; however, cationic dodecyl trimethyl ammonium bromide (C12TAB) could not co-adsorb due to the same charge character. However on a trimethoxy octyl silane (C8) coated hydrophobic surface, V6K formed a monolayer, and subsequent exposure to cationic and anionic surfactants both led to some co-adsorption at the interface. In binary surfactant/peptide mixtures, it was found that adsorption was dependent on the molar ratio of the surfactant and peptide. For SDS mixtures below molar unity and concentrations below CMC for C12TAB, V6K was able to dominate adsorption at the interface. Above molar unity, no adsorption was detected for SDS/V6K mixtures. In contrast, C12TAB gradually replaced the peptide and became dominant at the interface. These results thus elucidate the adsorption behaviour of V6K, which was found to dominate interfacial adsorption but its exact adsorbed amount and distribution were affected by interfacial hydrophobicity and interactions with conventional surfactants.
1. Introduction
The great versatility of short designed peptide materials lies in the ability to either alter amino acid sequences of known peptide structures found in nature or create completely novel sequences. De novo designed short peptides are biocompatible, biodegradable and have found applications in a wide range of areas including tissue engineering, drug delivery, membrane protein stabilisation, cosmetics and skin care.1,2 To further explore their potential applications, it is important to reveal not only their interfacial behaviour alone but also investigate their interactions with other molecules, in particular, conventional surfactants.One of the important classes of designed peptides are peptide amphiphiles which are designed to mimic the basic structural features of common surfactants. They generally bear distinct hydrophilic and hydrophobic moieties and thus share many common functions to conventional surfactants such as sodium dodecyl sulfate (SDS) and dodecyl trimethyl ammonium bromide (C12TAB).3 However the amino acid sequence which builds up a peptide surfactant renders them far more complex than simple alkyl chain surfactants. The resulting increased functionality of these peptides is appreciable in their antimicrobial and antitumor activity.4–6 In addition their surfactant-like amphiphilicity is attractive to a wide range of biotechnological applications including regenerative medicine and drug delivery systems.7–10 Thus peptide amphiphiles are promising alternatives to synthetic surfactants derived from petrochemical sources as a more environmentally responsible option showing strong sustainability, degradability and biocompatibility.11,12
We have previously reported the self-assembling properties and surface adsorption dynamics of a class of short designed peptides (VmKn, where m = 3–6 and n = 1–3) at the solid/liquid interface.13–15 It was found that changes in salt, pH and peptide concentration all have significant effects on their interfacial adsorption.15 At the silicon oxide/water interface V6K was found to be the most surface active peptide with the highest adsorption amount. It reached the steady-state adsorption faster than V6K2 and V3K. Both V6K and V6K2 were capable of self-assembling into nanostructures at the hydrophilic silica/water interface. Over high peptide concentrations, it was found that V6K formed a peptide bilayer that also incorporated some defects and peptide stacks or vesicles. The bilayer arrangement is also common to many other surfactants such as C12E6.16,17 Over lower peptide concentrations, V6K was shown to adsorb onto the silicon oxide interface and formed distinct flat cylindrical micellar structures.13 Once adsorbed, the V6K layers could not be removed easily by rinsing, showing great stability across a wide range of pH values and peptide concentrations.14 Short peptides that demonstrate good adsorption dynamics are attractive to the personal care industry as they can be formulated in skin care products.18–20 Similarly they are also attractive in the pharmaceutical industry to help drug solubilisation.21 To realize the potential of such peptides in formulations, it is important to rationalise their basic interfacial behaviour in the presence of other conventional surfactants such as SDS and C12TAB which have been extensively studied as they are relevant to numerous applications including biotechnology, oil recovery, detergency, and personal care.22
Adsorption at the solid/liquid interface is generally a result of a complex combination of forces including hydrophobic interactions, electrostatic interactions and hydrogen bonding.22–24 Extensive research has been carried out to study adsorption from surfactant mixtures and surfactant/polymer mixtures.25–29 Recent research has also focused on adsorption involving biosurfactants such as rhamnolipids and proteins such as hydrophobin and their interactions with conventional surfactants.11,30–32 Due to the lack of understanding of interfacial behaviour the commercial use of short peptides or other biosurfactants is still at an early stage. Thus detailed studies are required to explore their basic adsorption behaviour when mixed with conventional surfactants in different manners.
This paper aims to investigate how a surfactant-like peptide, V6K, interacts with conventional surfactants, SDS and C12TAB, at the solid/water interface by performing selective spectroscopic ellipsometry and neutron reflection measurements and elucidate the main interactions taking place in order to maximize understanding for potential applications.
2. Materials and method
Materials
The peptide was synthesized using the Solid Phase Peptide Synthesis (SPPS) procedure with C-terminal amides attached and N-terminal acetylated. The peptide was then purified by Gel Permeation Chromatography (GPC) twice, giving the final purity >95%. The peptide solution was made freshly by dissolving 5 mg peptide in 5.0 ml ultrahigh quality (UHQ) water (Purelab UHQ, Vivendi Water Systems Ltd) and subsequent serial dilution to the desired concentration. The peptide solutions were adjusted to pH 7 using minimal amounts of either HCl or NaOH. The critical aggregation concentration (CAC) for V6K was determined to be 0.15 mM, from conductance measurements in pure water and by fluorescence probe measurements.13,14 SDS and C12TAB were purchased respectively from Lancaster and Sigma, UK. Both of them were purified by recrystallization more than 3 times, in ethanol + water for SDS and in acetone + absolute ethanol for C12TAB, till the surface tension around their CMC showed no minimum.33Substrates
Silicon wafers were purchased from Compart Technology Ltd, UK. The silicon surface has a native oxide layer and bears silanol (Si–OH) groups and when in contact with water or moisture, it becomes very hydrophilic. Prior to each experiment the silicon wafers were cleaned by Piranha treatment (95% H2SO4/30% H2O2 = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 90 °C for 1 min) before wash by 5% Decon90 solution (from Decon Laboratory, UK), and followed by copious rinsing with UHQ water. To mimic the hydrophobic nature of different surfaces, silica surfaces were modified with C8 hydrocarbon (trimethoxy octyl silane) as described in our previous work.34,35 It is the model hydrophobic surface that has been most widely used for adsorption. It is useful at this stage to establish their basic interfacial behaviour using model substrate surface so that results can be compared with adsorption at more complex interfaces.
:1 at 90 °C for 1 min) before wash by 5% Decon90 solution (from Decon Laboratory, UK), and followed by copious rinsing with UHQ water. To mimic the hydrophobic nature of different surfaces, silica surfaces were modified with C8 hydrocarbon (trimethoxy octyl silane) as described in our previous work.34,35 It is the model hydrophobic surface that has been most widely used for adsorption. It is useful at this stage to establish their basic interfacial behaviour using model substrate surface so that results can be compared with adsorption at more complex interfaces.
Spectroscopic ellipsometry
Measurements were determined using a Jobin-Yvon UVISEL spectroscopic ellipsometer. The SE measurements were performed over a wavelength range between 300 and 600 nm. A liquid cell with fused quartz windows was used to enable the SE measurement at the solid/liquid interface with the incident light beam at 70°. The experimental data were analysed using a software called DeltaPsi II developed by Jobin-Yvon. The ellipsometer measured the change in the polarization state of light reflected from the surface of the sample. By studying the changes in the state of polarization, information about layer thickness and refractive index was revealed through the simultaneous analysis of two ellipsometric angles ψ and Δ. The changes in amplitude and phase of polarization of the light after reflection were determined in two components, the plane of reflection (p-plane), and that perpendicular to it (s-plane). The sample ellipticity, F, is defined as the ratio of the Fresnel coefficients of the p and s planes (Rp and Rs) and is expressed as36 | (1) |
 | (2) |
Neutron reflection (NR)
Measurements were carried out on SURF at RAL, Oxford, UK, using a neutron beam of wavelength 0.5 to 6.5 Å. The silicon 〈111〉 blocks used were polished by Crystran Ltd, UK and treated with Piranha solution at 90 °C for 1 min. Solution samples (2 ml) were filled into the lumen cell made by clamping a Perspex trough against the polished face of a silicon block with dimensions of 6 × 5 × 1.2 cm3. The sample cell was mounted on a goniometer stage controlled by the computer terminals. The neutron beam entered the small face of the silicon block, was reflected from the solid/solution interface and exited from the opposite end of the small face. The neutron beam was collimated by two sets of horizontal and vertical slits placed before the sample cell, creating a typical beam illuminated area around 4 × 3 cm2. Each reflectivity experiment was carried out at three incidence angles of 0.35, 0.8 and 1.8° and the resulting reflectivity profiles combined to cover a wave vector (κ) between 0.012 and 0.5 Å−1. Reflectivity profiles below the critical angle were theoretically equal to unity and all the data measured were scaled accordingly. Constant background was subtracted using the average reflectivity between 0.3 and 0.5 Å−1. The background was found to be typically around 2 × 10−6 in D2O.Model fitting of neutron reflection data has been extensively used to quantitatively analyse information regarding thickness and composition of adsorbed layers and has been found to be ideally suited to investigate multi component mixed layers adsorbed at an interface.37–40 The Motofit package was used for the data fitting.41 When fitting the data a structural model was assumed and the interface was divided into a suitable number of uniform sublayers. A model with minimum number of sublayers is preferred as it reduces the complexity of the system and overfitting of the data. Reflectivity from the model layers is calculated using the optical matrix formulism this is then compared with the measured reflectivity and the structural parameters (mainly thickness (τ) and scattering length density (ρ)) are modified in a least-square iteration until a good fit is obtained. For a two component adsorption, such as surfactant and peptide in water, the volume fraction and scattering length density of a layer can be expressed using eqn (3) and (4)
| ρ = ρpφp + ρsφs + ρwφw | (3) |
| φp + φs + φw = 1 | (4) |
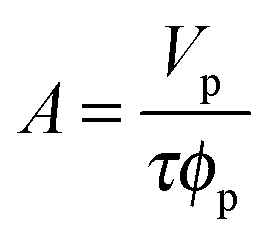 | (5) |
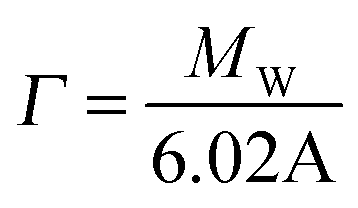 | (6) |
3. Results and discussion
(A). SDS and V6K system
:1 (SDS/V6K), the adsorption reached the same plateau as V6K alone but the process was slower, and as the ratio was further increased the adsorption dynamics slowed down further. It took over 100 min to reach a lower plateau, at a ratio of 0.78:1, with a final surface adsorbed amount of 2.8 mg m−2. When the ratio increased to unity and above, only a negligible amount (less than 0.2 mg m−2) of adsorption was detected. The SE results indicated that peptide adsorption was strongly inhibited by the increasing molar ratio of SDS in the solution. As the molar ratio of SDS/V6K is also equal to the charge ratio, the adsorption dynamics in Fig. 1 is explained by the charge neutralization of cationic V6K molecules by SDS in the solution. At and above unity, the amount of free peptide was minimal, hence adsorption was negligible. Below molar unity, SDS was not enough to neutralize all the peptide molecules in the solution, thus excess peptides drove the surface adsorption. The adsorbed amount and dynamic process both reduced and slowed down as the ratio was increased, similar to the SE adsorption curves at concentrations below 100 μg ml−1 reported in our previous studies.14
 | ||
| Fig. 1 The adsorption of V6K (×) peptide and co-adsorption of SDS and V6K at the silica/water interface with the molar ratio of SDS:V6K at 0.5:1 (□), 0.78:1 (Δ), 1:1 (◇), 3.9:1 (*), and 7.8:1 (○). V6K was fixed at 100 μg ml−1, pH 7. | ||
SE results alone cannot reveal how much SDS contributed to the total adsorbed amount and whether the presence of SDS in the mixture altered the self-assembling properties of V6K at the solid/liquid interface. Therefore, NR was carried out for the adsorption of SDS/V6K (0.78/1) at pH 7, the reflectivity profiles are shown in Fig. 2A for both hydrogenated SDS (h-SDS) and deuterated SDS (d-SDS). The fitted data for the curves along with the calculated volume fraction and adsorbed amounts for each layer are shown in Table 1.
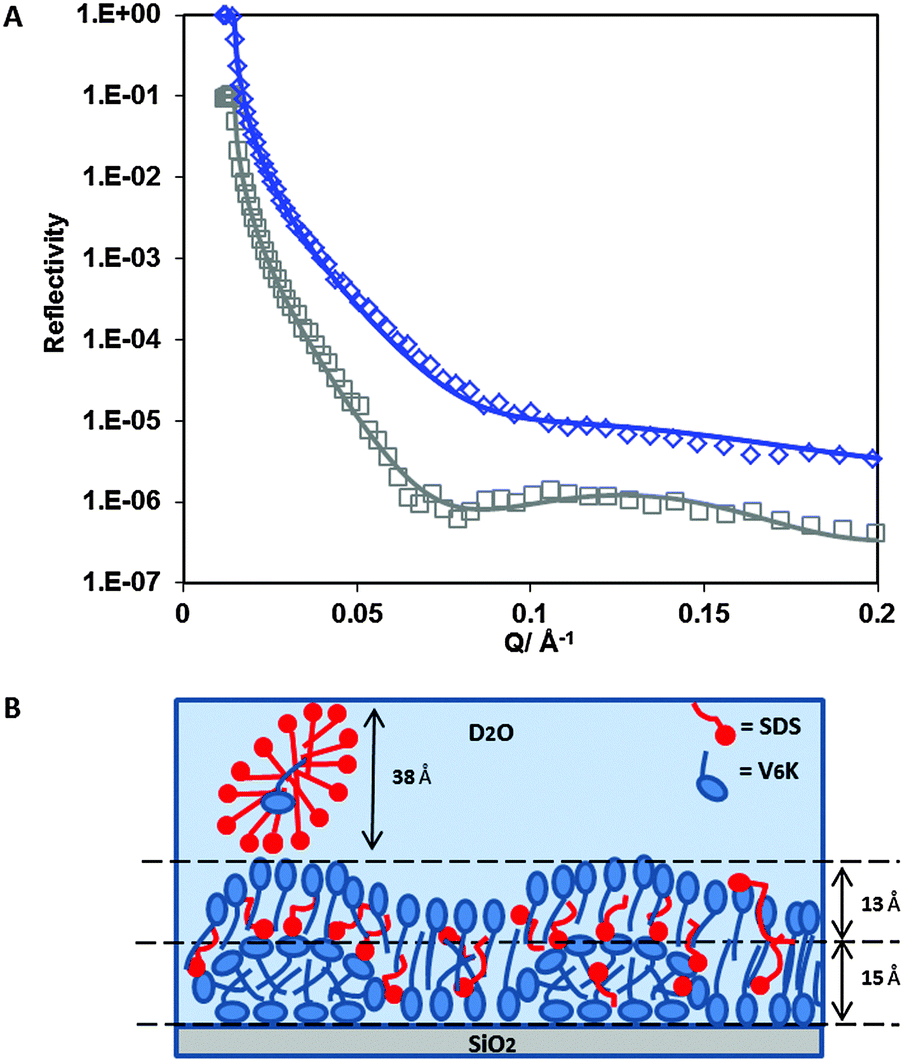 | ||
| Fig. 2 (A) Reflectivity profiles for h-SDS/V6K (0.78/1) mixture (□) and d-SDS/V6K (0.78/1) (◇) at the SiO2 interface in D2O pH 7. Solid lines through the data points correspond to the model fits for the corresponding measured reflectivity data points. (B) Schematic diagram showing the arrangement of V6K molecules (blue), SDS (red), at SDS/V6K = 0.78/1. | ||
| Sample/contrast | Fitted thickness ±2 Å | Fitted SLD ±0.1 × 10−6/Å−2 | Sample SLD (V6K/SDS) ±0.01 × 10−6/Å−2 | Volume fraction (V6K/SDS) ± 0.005 | Γ (V6K/SDS) ± 0.1 mg m−2 |
|---|---|---|---|---|---|
| h-SDS/V6K (0.78/1)/D2O | 15 | 2.1 | 1.71/0.37 | 0.815/0.080 | 1.4/0.1 |
| 13 | 4.6 | 1.71/0.37 | 0.255/0.095 | 0.4/0.2 | |
| 38 | 5.6 | 1.71/0.37 | 0/0.115 | 0/0.5 | |
| Γ Total 2.6 | |||||
| d-SDS/V6K (0.78/1)/D2O | 15 | 2.6 | 1.71/6.72 | 0.815/0.080 | 1.4/0.1 |
| 13 | 5.2 | 1.71/6.72 | 0.255/0.095 | 0.4/0.2 | |
| Γ Total 2.1 | |||||
Due to the electrostatic repulsion SDS does not adsorb directly onto the hydrophilic SiO2 surface. However, the presence of cationic V6K peptides at the SiO2 interface can aid the subsequent adsorption of SDS through increased hydrophobic interaction and charge reversal of the surface. Careful evaluation and analysis of the NR data revealed that three distinct layers were found at the interface (Fig. 2B). The innermost layer was 15 Å thick and densely packed, above it a more diffuse 13 Å layer followed by an even more diffuse SDS layer, as shown by the volume fraction values in Table 1. The peptide structures in the innermost layer were reminiscent of the V6K structures adsorbed at 20 μg ml−1 (discussed later). The thickness of the outermost SDS layer (38 Å) suggested the formation of surface micelles or lamellar structures which have frequently been reported to occur on positively charged hydrophilic surfaces.27,42–44 Since the outermost layer was predominantly occupied by SDS and D2O, the layer was essentially invisible and cannot be detected by the d-SDS run as both d-SDS and D2O have similar SLDs. Co-adsorption of SDS was likely aided by a combination of hydrophobic/electrostatic interactions with excess V6K peptides which had readily adsorbed at the interface. The adsorbed SDS reduced the electrostatic repulsion between peptides and allowed for a tight packing of V6K in the inner leaflet of the peptide bilayer. Overall the total adsorbed amount measured by NR, 2.6 mg m−2, closely matched ellipsometry results, but V6K was revealed to have contributed only 1.8 mg m−2 (2.3 × 10−3 mM m−2) to the total amount, significantly less peptide compared to adsorption at the same peptide concentration without surfactant. On the other hand, adsorption from the binary mixture significantly aided SDS adsorption and resulted in 0.8 mg m−2 (2.9 × 10−3 mM m−2) at the interface.
Analysis of the reflectivity curves (Fig. 3A and Table 2) from the pre-adsorbed peptide layer, at 20 μg ml−1, supports the formation of flattened V6K cylindrical micelles. The adsorbed peptide layer was very reproducible and had a surface excess of 1.4 mg m−2 after 20 minutes adsorption at pH 7, matching adsorption results from ellipsometry measurements reported by Pan et al.14 The thickness of the peptide layer was found to be 19 Å with a volume fraction of 0.66.
 | ||
| Fig. 3 (A) Reflectivity profiles for 20 μg ml−1 V6K pH 7 (□), 20 μg ml−1 V6K pH 7 + 4 mM d-SDS (◇) and 20 μg ml−1 V6K pH 7 + 4 mM h-SDS (Δ). Solid lines through the data points correspond to the best fits for the corresponding reflectivity data points. (B) Schematic diagram showing the arrangement of pre-adsorbed V6K peptides at 20 μg ml−1 (blue) and adsorption of SDS (red) at the SiO2/D2O interface at pH 7. | ||
| Sample/contrast | Fitted thickness ±2 Å | Fitted SLD ±0.1 × 10−6/Å−2 | Sample SLD (V6K/SDS) ±0.01 × 10−6/Å−2 | Volume fraction (V6K/SDS) ±0.005 | Γ (V6K/SDS) ±0.1 mg m−2 |
|---|---|---|---|---|---|
| 20 μg ml−1 V6K/D2O | 19 | 3.3 | 1.71/— | 0.660/— | 1.4/— |
| Γ Total 1.4 | |||||
| 20 μg ml−1 V6K + 4 mM d-SDS/D2O | 19 | 3.3 | 1.71/6.72 | 0.660/0.095 | 1.4/0.2 |
| Γ Total 1.6 | |||||
| 20 μg ml−1 V6K + 4 mM h-SDS/D2O | 19 | 2.7 | 1.71/0.37 | 0.660/0.095 | 1.4/0.2 |
| 16 | 5.0 | —/0.37 | 0/0.225 | —/0.4 | |
| Γ Total 2.0 | |||||
Following the initial adsorption, the peptide solution was carefully rinsed out with D2O (peptide desorption was negligible) and 4 mM h-SDS solution was added. As expected, the addition of h-SDS resulted in an increased layer thickness at the interface.
To investigate whether SDS adsorption caused any changes to the initial structural arrangement of the pre-adsorbed peptide layer and quantify adsorption, the experiment was repeated using d-SDS (Table 2). Addition of 4 mM d-SDS revealed no significant changes in the pre-adsorbed V6K layer, suggesting that SDS adsorption was limited to the surface of the peptide structures. If SDS had strongly interfered with the pre-adsorbed peptide structures (e.g. disruption), the d-SDS run would reveal a significant change to the layer SLD and or its thickness. However, in this case, both the subsequent addition of d-SDS and peptide only runs could be fitted with the same model, indicating that SDS displaced D2O and adsorbed onto the peptide layer. On the other hand the changes in SLD between the h-SDS and d-SDS run (Table 2) were used to determine the adsorbed amounts at the interface. Volume fraction values show less than 10% penetration of SDS into the pre-adsorbed peptide layer with a surface excess amounting to 0.20 mg m−2 (0.7 × 10−3 mM m−2). A higher proportion of the adsorbed SDS, 0.4 mg m−2 (1.5 × 10−3 mM m−2) formed a distinct 16 Å thick layer on top of the peptide structures. Fig. 3B shows a cross-sectional diagram of the proposed arrangement of pre-adsorbed peptide, highlighting a compact flattened micellar structure with gaps between individual structures and SDS adsorbed around and on top.
Similar to ionic surfactant adsorption onto oppositely charged hydrophilic surfaces; individual surfactant molecules are likely to have initiated adsorption onto the peptide structures with their negatively charged heads. Subsequent adsorption of SDS molecules would be through hydrophobic interactions with the alkyl tails of SDS molecules already adsorbed or hydrophobic patches on the peptide structures. SDS molecules extending from the peptide structures in such a manner would indeed stretch to around 16 Å.44
At 100 μg ml−1, the resulting adsorbed V6K layer could withstand UHQ water rinse and a series of rinses with SDS concentrations ranging from 0.1 mM to 4 mM (Fig. S1 in the ESI†). Initially, during the UHQ water rinse, minimal amounts (less than 0.2 mg m−2) of loosely adsorbed peptide were removed leaving a very stable and firmly absorbed peptide layer. Subsequent SDS rinses with increasing SDS concentration, up to 4 mM, resulted in increasing surface adsorbed amounts measured by SE at the solid interface.
NR data (Fig. 4A) confirmed the presence of a bilayer structure of V6K at the SiO2/water interface. The pre-adsorbed peptide formed three distinctive layers (Table 3) detectable due to the ordering of the lysine heads and valine tails, the latter having a lower SLD than lysine. Due to irregularities in the bilayer structure, the three layers could not be modelled to have perfectly defined K–V–K (lysine head–valine core–lysine head) layers, instead, the peptide layer was fitted by incorporating at least one V in the head region, hence KV–V–KV was used. Similar structure arrangement was found in our previous study of the interfacial structure of V6K2 peptide at the SiO2/water interface.15
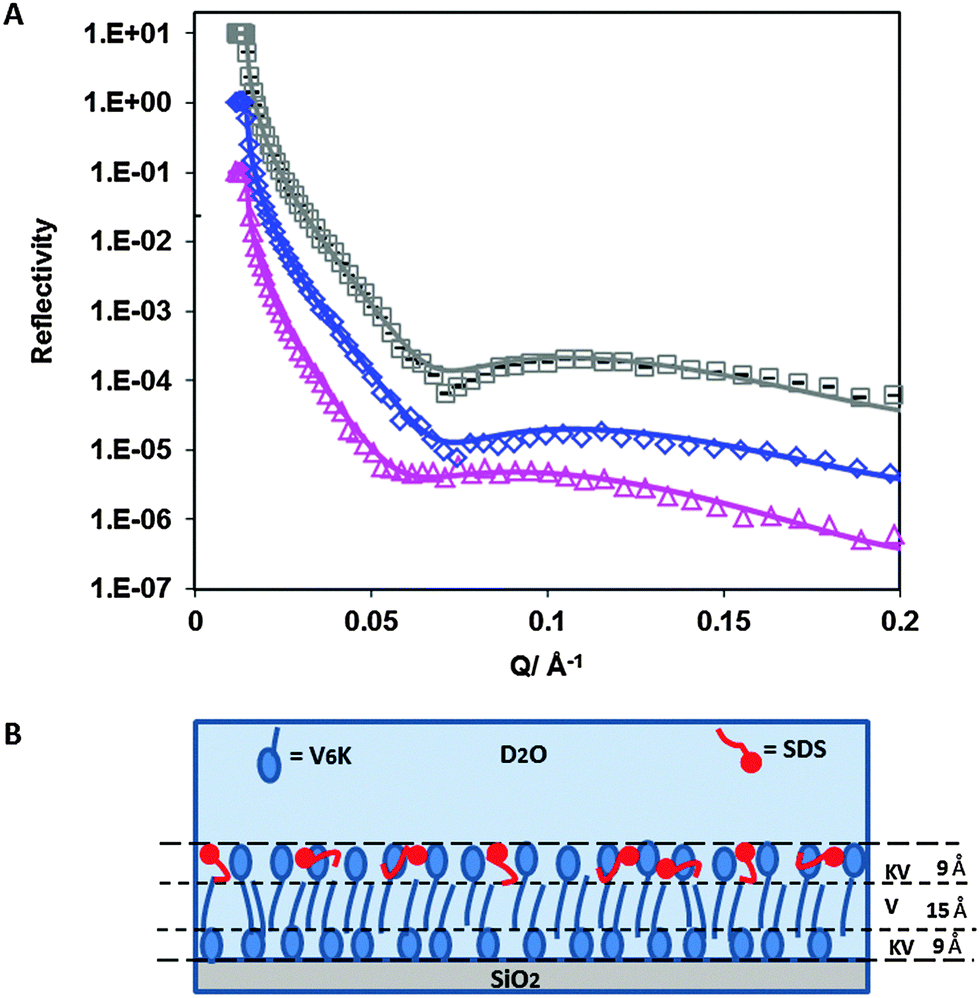 | ||
| Fig. 4 (A) Reflectivity profiles for: 100 μg ml−1 V6K pH 7 (□), 100 μg ml−1 V6K pH 7 + 4 mM d-SDS (◇) and 100 μg ml−1 V6K pH 7 + 4 mM h-SDS (Δ). Solid lines through the data points correspond to the model fits for the corresponding reflectivity data points. (B) Schematic diagram showing the arrangement of pre-adsorbed 100 μg ml−1 V6K peptides (blue) and the effect of SDS (red) on its arrangement at the SiO2/D2O interface at pH 7. | ||
| Sample/contrast | Fitted thickness ±2 Å | Fitted SLD ±0.1 × 10−6/Å−2 | Sample SLD (V6K/SDS) ±0.01 × 10−6/Å−2 | Volume fraction (V6K/SDS) ±0.005 | Γ (V6K/SDS) ±0.1 mg m−2 | |
|---|---|---|---|---|---|---|
| 100 μg ml−1 V6K/D2O | KV | 9 | 4.7 | 2.13/— | 0.390/— | 0.5/— |
| V | 15 | 2.7 | 1.54/— | 0.760/— | 1.3/— | |
| KV | 9 | 4.7 | 2.13/— | 0.390/— | 0.5/— | |
| Γ Total 2.3 | ||||||
| 100 μg ml−1 V6K + 4 mM d-SDS/D2O | KV | 9 | 4.7 | 2.13/6.72 | 0.390/0 | 0.5/0 |
| V | 15 | 2.7 | 1.54/6.72 | 0.760/0 | 1.3/0 | |
| KV | 9 | 4.9 | 2.13/6.72 | 0.390/0.420 | 0.5/0.5 | |
| Γ Total 2.8 | ||||||
| 100 μg ml−1 V6K + 4 mM h-SDS/D2O | KV | 9 | 4.7 | 2.13/0.37 | 0.390/0 | 0.5/0 |
| V | 15 | 2.7 | 1.54/0.37 | 0.760/0 | 1.3/0 | |
| KV | 9 | 2.2 | 2.13/0.37 | 0.390/0.420 | 0.5/0.5 | |
| Γ Total 2.8 | ||||||
The valine core had a thickness of 15 Å and SLD of 2.7 × 10−6 Å−2 whilst the inner and outer layer were both 9 Å thick with an SLD of 4.7 × 10−6 Å−2. This corresponds to the peptide molecules being tightly packed with a highly interdigitated valine tail core. The total thickness of the pre-adsorbed bilayer was 33 Å, which is in good agreement with the extended length of a V6K molecule around 2.5–3 nm. The total surface excess value calculated from the VK–V–VK layer was slightly lower than ellipsometry results (3.1 mg m−2) shown in Fig. S1 in ESI† but is in agreement with the values found in the literature ranging between 2.5 and 3.0 mg m−2.13
Addition of 4 mM h-SDS solution after careful rinsing of the peptide solution caused a significant change in the reflectivity curves (Fig. 4A) and fitted SLD (Table 3) confirming that SDS strongly interacted with the pre-adsorbed peptide layer. Measurements using d-SDS also showed a change in the reflectivity curve indicating a change had occurred to the pre-adsorbed peptide layer upon SDS addition. Careful evaluation of the reflectivity curves revealed that SDS had penetrated into the outer peptide layer (schematic diagram shown in Fig. 4B). The surfactant was able to insert itself in the spaces between adjacent lysine heads by displacing D2O. However, SDS was not able to penetrate further into the bilayer valine core to a significantly appreciable amount. There was no additional SDS adsorbed on top of the peptide bilayer, suggesting that the adsorbed SDS was enough to cause an overall charge neutralization of the peptide surface and limit any further SDS adsorption. The total SDS adsorbed amount was 0.5 mg m−2 (1.6 × 10−3 mM m−2) and is comparable to the surfactant adsorbed amount found on the top layer of the 20 μg ml−1 V6K run.
Subsequent addition of SDS solution (up to 4 mM) onto the peptide monolayer resulted in an additional adsorbed amount of 1.2 mg m−2 (Fig. S2 in the ESI†). This is significantly higher than the SDS adsorption onto the peptide bilayer found at the hydrophilic SiO2 interface of only 0.5 mg m−2 under the same conditions with the same peptide concentration. The increased adsorption of SDS is consistent with the peptide being arranged flat on the hydrophobic surface. In this arrangement, the hydrophobic valine tails of the peptide are more exposed allowing strong hydrophobic interactions with the SDS alkyl chains.
(B). C12TAB and V6K system
:1 was similar to the pure peptide but had a slightly lower surface excess. Increasing the ratio to 20:1 caused the final plateau to reduce further with a noticeable slowing of the adsorption process. A further increase in the ratio to 94:1 resulted in a significant drop of the surface excess. At 94:1, C12TAB had reached its CMC and was highly in excess. Indeed, both the surface excess and the adsorption dynamics closely resembled the adsorption of pure C12TAB at 12 mM (CMC).
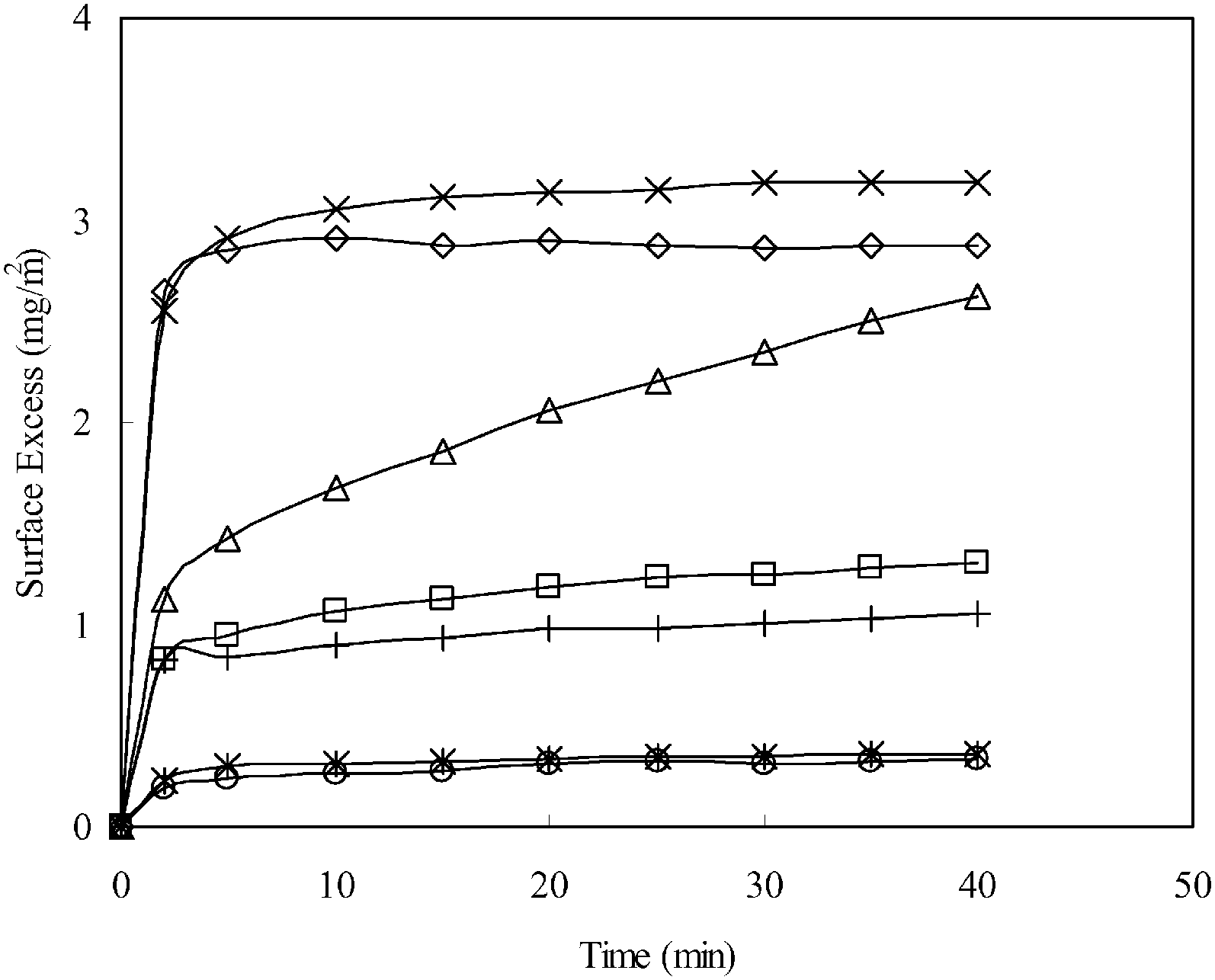 | ||
| Fig. 5 The adsorption isotherm of V6K at 100 μg ml−1 (×) and C12TAB at 0.128 mM (○); 2.63 mM (*); 12 mM (□). The co-adsorption was at the molar ratio of C12TAB and V6K of 1:1 (◇), 20:1 (Δ) and 94:1 (+). V6K was fixed at 100 μg ml−1 (0.128 mM), all at pH 7. | ||
The SE results show both the surfactant and the peptide competing for adsorption at the interface. At the highest ratio (94:1) the adsorption was dominated by C12TAB, limiting total adsorption to under 1.3 mg m−2. However, it is not clear from the SE results alone how the two components coexisted and arranged themselves at the interface.
NR was used to probe the arrangement of the two molecules at a molar ratio of 20:1. Fig. 6A shows neutron reflection data and model fits for the adsorption of C12TAB and d-C12TAB with V6K in D2O pH 7. Analysis of the data revealed a bilayer structure retained by the V6K peptide but had significant amounts of C12TAB incorporated within it (Table 4). The inner layer was dominated by V6K and only 0.1 mg m−2 (0.3 × 10−3 mM m−2) of C12TAB present. The bilayer core was densely packed and housed the largest amount of C12TAB, 0.3 mg m−2 (1.0 × 10−3 mM m−2), and the outer layer had no surfactant present as shown in the schematic diagram in Fig. 6B. Peptide molecules adsorbed faster and in higher amount than C12TAB, allowing the peptide to form a bilayer and in the process trapping C12TAB within its core. Following the formation of the bilayer, electrostatic repulsion stopped additional adsorption onto the co-adsorbed layer. This supports the SE results which suggest competitive adsorption of C12TAB at the interface with increasing amounts of surfactant at increasing molar ratio.
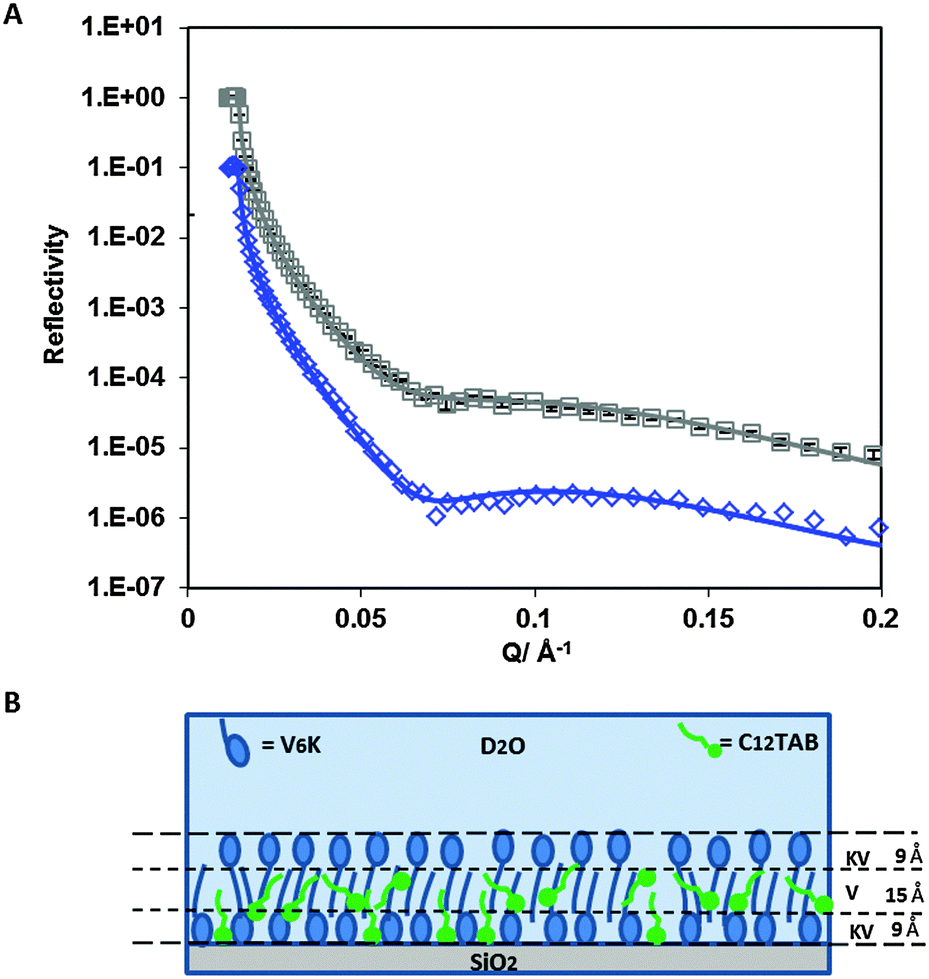 | ||
| Fig. 6 (A) Reflectivity profiles for: C12TAB/V6K (20/1) mixture (□) and d-C12TAB/V6K (20/1) (◇). Solid lines through the data points correspond to the model fits for the corresponding reflectivity data points. (B) Schematic diagram showing the arrangement of V6K molecules (blue), C12TAB (green) at C12TAB/V6K = 20/1, at the SiO2 interface in D2O pH 7. | ||
| Sample/contrast | Fitted thickness ±2 Å | Fitted SLD ±0.1 × 10−6/Å−2 | Sample SLD (V6K/C12TAB) ±0.01 × 10−6/Å−2 | Volume fraction (V6K/C12TAB) ±0.005 | Γ (V6K/C12TAB) ±0.1 mg m−2 | |
|---|---|---|---|---|---|---|
| C12TAB/V6K (20/1)/D2O | KV | 9 | 4.4 | 2.13/−0.24 | 0.375/0.055 | 0.5/0.1 |
| V | 15 | 1.4 | 1.54/−0.24 | 0.750/0.205 | 1.3/0.3 | |
| KV | 9 | 4.6 | 2.13/−0.24 | 0.385/0.020 | 0.5/0.0 | |
| Γ Total 2.7 | ||||||
| d-C12TAB/V6K (20/1)/D2O | KV | 9 | 4.7 | 2.13/5.13 | 0.375/0.055 | 0.5/0.1 |
| V | 15 | 2.5 | 1.54/5.13 | 0.750/0.205 | 1.3/0.3 | |
| KV | 9 | 4.7 | 2.13/5.13 | 0.385/0.020 | 0.5/0.0 | |
| Γ Total 2.7 | ||||||
4. Conclusions
In this study, spectroscopic ellipsometry and neutron reflection were used to evaluate the adsorption behaviour of the V6K peptide and its interaction with conventional surfactants SDS and C12TAB. When the peptide was pre-adsorbed it was able to form a stable bilayer with different coverage and stability. At the highest concentration of 100 μg ml−1, the bilayer formed was very stable against rinsing by UHQ water. The positively charged lysine heads exposed on pre-adsorbed peptide layers facilitated SDS adsorption but hampered C12TAB adsorption. When SDS and V6K are mixed together in solution, adsorption showed a strong dependence on the molar ratio of the two components. SDS has a neutralizing effect on adsorption and when the molar unity was reached no adsorption was detected. Below molar unity, excess V6K aided SDS adsorption onto the surface. This behaviour is analogous to several polymer and polyelectrolyte systems which adsorb and cause charge reversal of the SiO2 surface and increase hydrophobic interactions thus aiding SDS adsorption.26,27,30,42 On the other hand, when C12TAB is combined with V6K in solution, the adsorption became competitive. Above surfactant CMC, V6K was stopped from adsorbing at the interface but at lower molar ratios V6K was able to adsorb at the interface faster than C12TAB and could coexist with C12TAB at the interface. These results are also consistent with other studies involving cationic surfactant mixtures.30,42 Overall, the results indicate that V6K was able to adsorb faster at the solid/liquid interface with the exact amount depending on the ratio and other factors. The fast adsorption and stability of V6K in the presence of surfactants shows the potential for the peptide to be used as a biosurfactant at the solid/liquid interface.Acknowledgements
The authors would like to thank Royal Society (RG120061), EPSRC (EP/N007174/1) and University of Sheffield for support and ISIS Neutron Facility for beam time. Dharana Jayawardane also thanks EPSRC for a research studentship.References
- X. Zhao, F. Pan, H. Xu, M. Yaseen, H. Shan, C. A. Hauser, S. Zhang and J. R. Lu, Chem. Soc. Rev., 2010, 39, 3480–3498 RSC.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- M. R. Infante, L. Pérez, A. Pinazo, P. Clapés, M. C. Morán, M. Angelet, M. T. García and M. P. Vinardell, Cron. Chim., 2004, 7, 583–592 CrossRef CAS PubMed.
- C. Chen, J. Hu, P. Zeng, F. Pan, M. Yaseen, H. Xu and J. R. Lu, Biomaterials, 2014, 35, 1552–1561 CrossRef CAS PubMed.
- A. Dehsorkhi, V. Castelletto, I. W. Hamley, J. Seitsonen and J. Ruokolainen, Langmuir, 2013, 29, 14246–14253 CrossRef CAS PubMed.
- J. Hu, C. Chen, S. Zhang, X. Zhao, H. Xu and J. R. Lu, Biomacromolecules, 2011, 12, 3839–3843 CrossRef CAS PubMed.
- S. G. Zhang, D. M. Marini, W. Hwang and S. Santoso, Curr. Opin. Chem. Biol., 2002, 6, 865–871 CrossRef CAS.
- X. Zhao and S. Zhang, Macromol. Biosci., 2007, 7, 13–22 CrossRef CAS PubMed.
- S. Vauthey, S. Santoso, H. Gong, N. Watson and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5355–5360 CrossRef CAS PubMed.
- G. v. Maltzahn, S. Vauthey, S. Santoso and S. Zhang, Langmuir, 2003, 19, 4332–4337 CrossRef.
- J. Penfold, R. K. Thomas and H. H. Shen, Soft Matter, 2012, 8, 578–591 RSC.
- A. F. Dexter and A. P. J. Middelberg, Ind. Eng. Chem. Res., 2008, 47, 6391–6398 CrossRef CAS.
- S. Y. Han, W. W. Xu, M. W. Cao, J. Q. Wang, D. H. Xia, H. Xu, X. B. Zhao and J. R. Lu, Soft Matter, 2012, 8, 645–652 RSC.
- F. Pan, X. Zhao, S. Perumal, T. A. Waigh, J. R. Lu and J. R. Webster, Langmuir, 2010, 26, 5690–5696 CrossRef CAS PubMed.
- X. B. Zhao, F. Pan, S. Perumal, H. Xu, J. R. Lu and J. R. P. Webster, Soft Matter, 2009, 5, 1630–1638 RSC.
- E. M. Lee, R. K. Thomas, P. G. Cummins, E. J. Staples, J. Penfold and A. R. Rennie, Chem. Phys. Lett., 1989, 162, 196–202 CrossRef CAS.
- R. Atkin, V. S. J. Craig, E. J. Wanless and S. Biggs, Adv. Colloid Interface Sci., 2003, 103, 219–304 CrossRef CAS.
- T. J. Deming, Soft Matter, 2005, 1, 28–35 RSC.
- X. Huang, W. Hong and W. Ying, US Pat., US 07790147, 2010 Search PubMed.
- H. Heerklotz and J. Seelig, Biophys. J., 2001, 81, 1547–1554 CrossRef CAS.
- V. P. Torchilin, J. Controlled Release, 2001, 73, 137–172 CrossRef CAS.
- R. Zhang and P. Somasundaran, Adv. Colloid Interface Sci., 2006, 123–126, 213–229 CrossRef CAS PubMed.
- S. Paria and K. C. Khilar, Adv. Colloid Interface Sci., 2004, 110, 75–95 CrossRef CAS PubMed.
- F. Tiberg, J. Brinck and L. Grant, Curr. Opin. Colloid Interface Sci., 1999, 4, 411–419 CrossRef CAS.
- I. M. Tucker, J. T. Petkov, J. Penfold and R. K. Thomas, Langmuir, 2012, 28, 10223–10229 CrossRef CAS PubMed.
- A. Mohr, T. Nylander, L. Piculell, B. Lindman, V. Boyko, F. W. Bartels, Y. Liu and V. Kurkal-Siebert, ACS Appl. Mater. Interfaces, 2012, 4, 1500–1511 CAS.
- X. Zhang, D. Taylor, R. Thomas, J. Penfold and I. Tucker, Langmuir, 2011, 27, 3569–3577 CrossRef CAS PubMed.
- C. D. Bain, P. M. Claesson, D. Langevin, R. Meszaros, T. Nylander, C. Stubenrauch, S. Titmuss and R. von Klitzing, Adv. Colloid Interface Sci., 2010, 155, 32–49 CrossRef CAS PubMed.
- T. Nylander, Y. Samoshina and B. Lindman, Adv. Colloid Interface Sci., 2006, 123–126, 105–123 CrossRef CAS PubMed.
- X. L. Zhang, J. Penfold, R. K. Thomas, I. M. Tucker, J. T. Petkov, J. Bent and A. Cox, Langmuir, 2011, 27, 10464–10474 CrossRef CAS PubMed.
- J. Penfold, R. K. Thomas, P. Li, J. T. Petkov, I. Tucker, A. R. Cox, N. Hedges, J. R. Webster and M. W. Skoda, Langmuir, 2014, 30, 9741–9751 CrossRef CAS PubMed.
- X. L. Zhang, J. Penfold, R. K. Thomas, I. M. Tucker, J. T. Petkov, J. Bent, A. Cox and R. A. Campbell, Langmuir, 2011, 27, 11316–11323 CrossRef CAS PubMed.
- E. A. Simister, R. K. Thomas, J. Penfold, R. Aveyard, B. P. Binks, P. Cooper, P. D. I. Fletcher, J. R. Lu and A. Sokolowski, J. Phys. Chem., 1992, 96, 1383–1388 CrossRef CAS.
- X. Zhao, F. Pan, P. Coffey and J. R. Lu, Langmuir, 2008, 24, 13556–13564 CrossRef CAS PubMed.
- X. Zhao, F. Pan, L. Garcia-Gancedo, A. J. Flewitt, G. M. Ashley, J. Luo and J. R. Lu, J. R. Soc., Interface, 2012, 9, 2457–2467 CrossRef CAS PubMed.
- Y. Tang, J. R. Lu, A. L. Lewis, T. A. Vick and P. W. Stratford, Macromolecules, 2002, 35, 3955–3964 CrossRef CAS.
- R. J. Green, T. J. Su, J. R. Lu and J. Penfold, J. Phys. Chem. B, 2001, 105, 1594–1602 CrossRef CAS.
- J. R. Lu, T. J. Su and R. K. Thomas, J. Phys. Chem. B, 1998, 102, 10307–10315 CrossRef CAS.
- R. J. Green, T. J. Su, J. R. Lu and J. R. P. Webster, J. Phys. Chem. B, 2001, 105, 9331–9338 CrossRef CAS.
- J. R. Lu, T. J. Su, R. K. Thomas and J. Penfold, Langmuir, 1998, 14, 6261–6268 CrossRef CAS.
- A. Nelson, J. Appl. Crystallogr., 2006, 39, 273–276 CrossRef CAS.
- J. Penfold, I. Tucker, E. Staples and R. K. Thomas, Langmuir, 2004, 20, 7177–7182 CrossRef CAS PubMed.
- J. Penfold, I. Tucker and R. K. Thomas, Langmuir, 2005, 21, 11757–11764 CrossRef CAS PubMed.
- C. Tanford, J. Phys. Chem., 1972, 76, 3020–3024 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sm01670c |
| ‡ The authors equally contributed to this paper. |
| This journal is © The Royal Society of Chemistry 2015 |