
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembly of a dual functional bioactive peptide amphiphile incorporating both matrix metalloprotease substrate and cell adhesion motifs†
Ashkan
Dehsorkhi
a,
Ricardo M.
Gouveia
b,
Andrew M.
Smith‡
a,
Ian W.
Hamley
*a,
Valeria
Castelletto
a,
Che J.
Connon
b,
Mehedi
Reza
c and
Janne
Ruokolainen
c
aSchool of Chemistry, Pharmacy and Food Biosciences, University of Reading, Whiteknights, Reading, RG6 6AD, UK. E-mail: I.W.Hamley@reading.ac.uk
bInstitute of Genetic Medicine, Newcastle University, International Centre for Life, Central Parkway, Newcastle upon Tyne, NE1 3BZ, UK
cDepartment of Applied Physics, Aalto University School of Science, P.O. Box 15100, FI-00076 Aalto, Finland
First published on 5th March 2015
Abstract
We describe a bioactive lipopeptide that combines the capacity to promote the adhesion and subsequent self-detachment of live cells, using template-cell-environment feedback interactions. This self-assembling peptide amphiphile comprises a diene-containing hexadecyl lipid chain (C16e) linked to a matrix metalloprotease-cleavable sequence, Thr-Pro-Gly-Pro-Gln-Gly-Ile-Ala-Gly-Gln, and contiguous with a cell-attachment and signalling motif, Arg-Gly-Asp-Ser. Biophysical characterisation revealed that the PA self-assembles into 3 nm diameter spherical micelles above a critical aggregation concentration (cac). In addition, when used in solution at 5–150 nM (well below the cac), the PA is capable of forming film coatings that provide a stable surface for human corneal fibroblasts to attach and grow. Furthermore, these coatings were demonstrated to be sensitive to metalloproteases expressed endogenously by the attached cells, and consequently to elicit the controlled detachment of cells without compromising their viability. As such, this material constitutes a novel class of multi-functional coating for both fundamental and clinical applications in tissue engineering.
Introduction
One of the main challenges in developing new biomaterials is the creation of a suitable environment in which cells can be seeded, grown, induced to differentiate towards the formation of complex tissues, and eventually detach from the surface leaving the synthetic component behind. Such an environment must be suitable for proper cell adhesion and survival, allowing nutrient and metabolite diffusion, and presenting adequate biochemical and mechanical cues. Presently, considerable efforts are being made towards the development of materials that incorporate bioactive moieties for use in tissue engineering and regenerative medicine.1–3 However, few of these materials have reached the market, possibly due to the difficulty to transfer and implement such technologies and/or methodologies between research groups with very distinct sets of expertise.In this context, the use of simple, synthetic biomimetic molecules has gained considerable popularity. For example, bio-inspired peptides now have important applications in many natural processes relevant to skin care and wound healing including inflammation, cell proliferation, angiogenesis (growth of new blood vessels), and melanogenesis (production of the skin pigment melanin).4 In these applications, presentation of the active peptide motif is important, as is delivery across the stratum corneum (outer layer of the epidermis containing keratin-rich cells) to the basement membrane. In order to enhance bioactivity, biocompatibility and delivery, lipid chains may be attached to peptides to create peptide amphiphiles (PAs).
One type of PA comprises a lipidic hydrophobic tail connected to a hydrophilic peptide segment. This configuration leads to self-assembled structures with a hydrophobic core surrounded by a peptide functionalized corona.5 Self-assembly most commonly leads to nanofibril or nanotape structures in which peptide epitopes are presented on the fibril surface.5–12 These nanostructures are being developed for diverse applications in bio-nanotechnology and regenerative medicine.7,8,13 Stupp and coworkers have undertaken seminal work to elucidate the principles of self-assembly of PAs,14–17 and explored several important applications in bioengineering, including the use of PA nanofibers as scaffolds for bio-mineralization to regenerate bone,18,19 in differentiation of progenitor20 or stem cells,21 or tissue scaffolding22 including cartilage regeneration.23 Other groups have examined the use of PAs for applications including cancer therapy,24–28 and drug release systems,29–31 among others. PAs enable the presentation of bioactive sequences such as cell adhesion motifs RGD18 or RGDS,32 protease substrates,33–35 or even cell growth factors such as TGF-β1 (ref. 23) at the surface of the nanostructures. In general, the peptide amphiphiles have biologically relevant lipid chain lengths, in particular palmitoyl (hexadecyl, C16).
We have recently been investigating the self-assembly of several series of bio-derived lipopeptides. For example, we showed that the PA C16-KTTKS,36 known as Matrixyl® (the trade name is registered to Sederma SA, Le Perray-en-Yvelines, France),37 is able to stimulate cells to produce collagen,38 consistent with its use in cosmeceutical applications. The self-assembly of this and two related PAs used in cosmeceuticals was investigated.39 We have also examined the self-assembly in aqueous solution of three lipopeptides derived from Bacillus subtilis which show antimicrobial activity.40 Ourselves and others have examined the self-assembly of PAs containing a saturated C16 lipid chain and an RGD or RGDS-containing headgroup (with tri-glycine, GGG spacers).41,42 We found that these PAs aggregate into extended nanotapes, and we also investigated co-assembly with C16-ETTES.42 We also prepared bioactive films from these PAs, which were shown to provide stable adhesive coatings on patterned hydrophobic surfaces composed of striated polytetrafluoroethylene (PTFE), significantly enhancing proliferation of aligned cells and increasing the complexity of the produced tissue.43 Very recently, we have developed PA coatings for cell and tissue therapies with a self-release action due to the incorporation within the peptide headgroup of matrix metalloprotease (MMP) substrate involved in remodelling the extracellular matrix as well as the RGDS cell adhesion motif. The peptide headgroup was attached to a simple (unsaturated) hexadecyl lipid chain.34
In the present paper, we describe the synthesis and self-assembly of a novel PA that incorporates the same bifunctional peptide headgroup. The PA also contains a diene-containing hexadecyl lipid chain in order to ultimately prepare polymerizable PA films with potential applications in wound healing, although this is not examined herein. The PA is denoted C16e-MMPRGDS, the term C16e indicating the diene-functionalized lipid chain. The peptide headgroup incorporates the N-terminal sequence Thr-Pro-Gly-Pro-Gln-Gly-Ile-Ala-Gly-Gln (TPGPQGIAGQ) was derived from the matrix metalloprotease (MMP) cleavage site of collagen type-I and functions as a specific proteolysis target for several MMPs, namely MMP1 and 2,44 with predicted cleavage between the Gly and Ile residues.45 The C-terminal sequence is the integrin-dependent cell-adhesion motif Arg-Gly-Asp-Ser (RGDS) found in several cell adhesion molecules and used extensively in biomaterial research to elicit cell attachment, promote survival, and control cell encapsulation and phenotype.2 As well as reporting on the synthesis and self-assembly of this PA, here we also show that the PA is cytocompatible, and is capable of performing as a stable but bio-degradable cell-adhesive coating template. Thus, the application of polymerizable PA films notwithstanding, the development of a contiguous MMP cleavage and cell adhesion peptide sequence facilitates the attachment of fibroblastic cells followed by their subsequent release (from the synthetic PA component) under endogenous enzyme control, combining to form a smart biomaterial for the generation of time-dependent cell release systems for future medical applications.
Experimental
Synthesis of hexadecadienoic acid
The basic method for synthesizing the diene-containing lipid chain was taken and modified from ref. 46, the reaction scheme is illustrated in Scheme 1. A mixture of 2.22 mL dodecanal, 4.88 mL triethyl 4-phosphonocrotonate, 0.46 g LiOH·H2O and 15 g 4 Å molecular sieves in 50 mL of tetrahydrofuran were refluxed under nitrogen gas for 12 hours. The resulting solution was crudely cleaned up through a short silica column using diethyl ether as the elution solution and the main yellow fraction was collected. This main fraction was concentrated on a rotary evaporator. Flash chromatography with a silica column and an elution solution of 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexane:ethylacetate was performed and fractions collected. Thin layer chromatography on silica plates was used to identify the contents of the fractions and those containing the product were pooled and concentrated on a rotary evaporator. To this concentrated solution 15 mL of a 1:1 solution of ethanol and 20% NaOH was added and heated to 70 °C and stirred for 3 hours. To this solution 15 mL of saturated cold NaCl was added and placed at 4 °C for 2 hours. The precipitated solution was then filtered and the resultant cake placed in a desiccator to finish drying. The resultant material was identified by mass spectrometry and was produced in 72% yield in relation to the starting material.
:1 hexane:ethylacetate was performed and fractions collected. Thin layer chromatography on silica plates was used to identify the contents of the fractions and those containing the product were pooled and concentrated on a rotary evaporator. To this concentrated solution 15 mL of a 1:1 solution of ethanol and 20% NaOH was added and heated to 70 °C and stirred for 3 hours. To this solution 15 mL of saturated cold NaCl was added and placed at 4 °C for 2 hours. The precipitated solution was then filtered and the resultant cake placed in a desiccator to finish drying. The resultant material was identified by mass spectrometry and was produced in 72% yield in relation to the starting material.
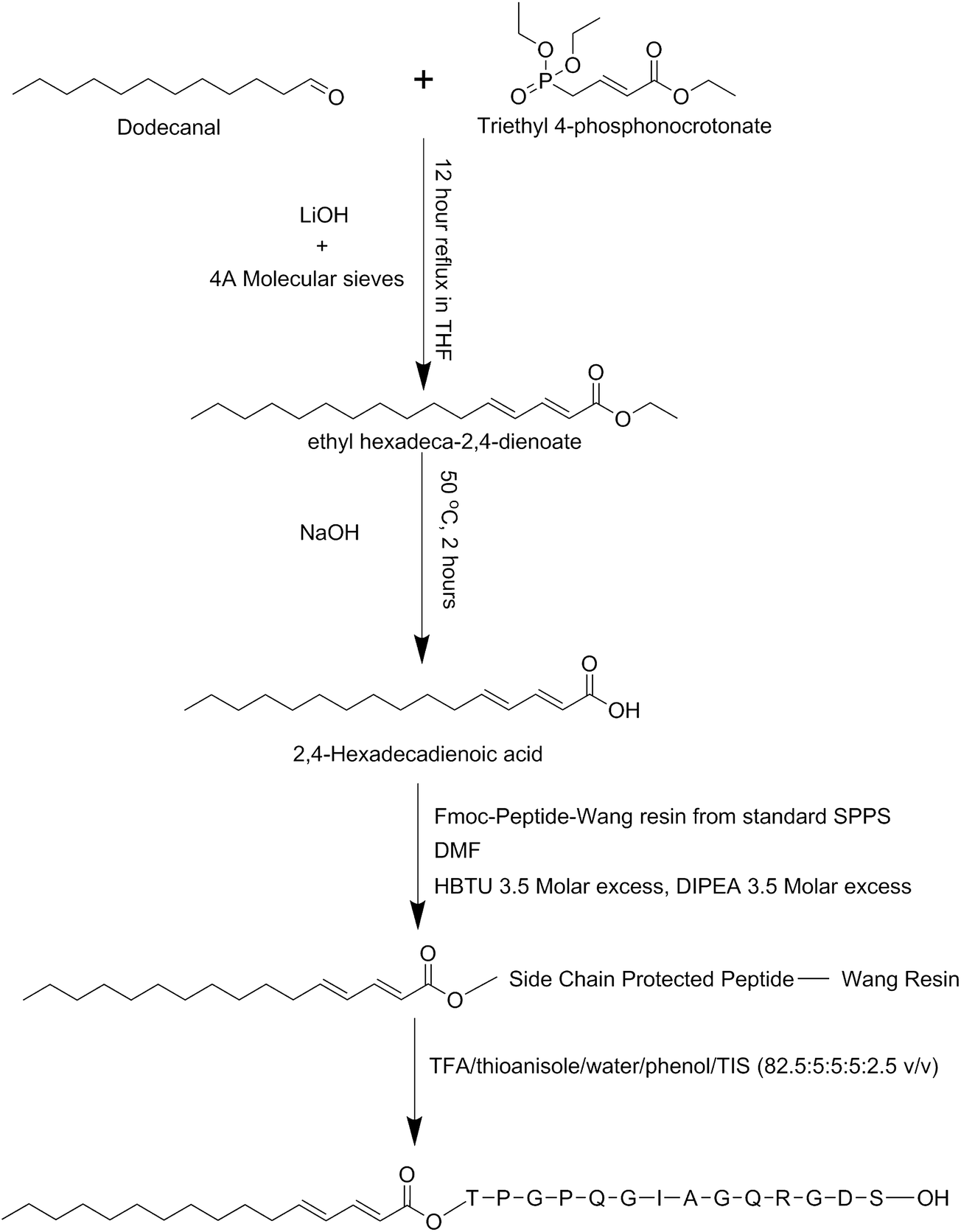 | ||
| Scheme 1 Synthesis pathway for C16e-MMPRGDS. | ||
Peptide synthesis
Peptides were synthesised using standard Merrifield solid phase peptide synthesis methods with 5 times molar excess of each Fmoc-amino acid at each couple step and 4.75 times excess of HBTU and DIPEA with a coupling time of 3 hours each. All synthesis was conducted on standard Wang resins with the last amino acid pre-coupled. Deprotection and cleavage of the final peptides from the resin was undertaken using the cleavage mixture of TFA/thioanisole/water/phenol/TIS (82.5:5:5:5:2.5 v/v). The crude peptides were purified using a Supelco Discovery Bio wide pore C18–10, 15 cm × 10 mm column with buffer A as acetonitrile, 0.01 M HCl and buffer B H2O, 0.01 M HCl. The following gradient was used in all purifications; 0–30% buffer A over 5 minutes, 30–54% buffer A over 9 minutes, 54–90% buffer A over 5 minutes, 90% buffer A for 3 minutes and 90–0% buffer A over 3 minutes. Elution of peptides from the column was detected using a UV detector at 214 nm, and all the main peptide peaks came off in the region of the second gradient. Multiple injections were pooled and freeze dried to give the pure peptide. Mass spectrometry was used to confirm the identity of the peaks in the chromatogram.
Cell culture
Films of C16e-MMPRGDS were prepared by dissolving the PA in ultrapure water to make 5, 25, 50, 75, and 150 nM solutions. A 600 μL aliquot of each concentration was deposited into each well of an ultra-low attachment 6 well plate (Costar, Corning, USA) which was then allowed to evaporate overnight to form a dry film on the bottom of the well. The film containing wells were then washed and rinsed with phosphate buffer saline (PBS) three times. Human corneal stromal fibroblasts (hCSFs) extracted from corneal rings were seeded at a density of 1.5 × 104 cells per cm2 onto each PA coated well to assess cell attachment, and cultured in 2 mL of serum-free medium (SFM, composed of DMEM/F12 with 1 mM ascorbic acid, 1% of ITS supplement (Sigma-Aldrich, USA), 1% penicillin/streptomycin), supplemented with 5 × 10−6 M of all-trans retinoic acid (RA) to minimize the expression of matrix metalloproteases from hCSFs.47 RA was prepared and dissolved in DMSO as a stock concentration of 5 × 10−2 M, sterile-filtered, and stored in a −80 °C freezer until use. Medium was replaced every other day. Cell proliferation assays were assessed over the course of seven days, and cell quantification was performed on day 1, 3, 5, and 7 to determine the number of cells growing on the PA films. The number of live cells was determined using the Alamar blue assay, by incubating the cells in 1:10 reasazurin:SFM solution for 4 hours, and measuring the fluorescence of 100 μL aliquots on a plate reader, in triplicate, using excitation/emission wavelengths of 530–560/590 nm, respectively. The signal from the triplicates was averaged and a standard curve was used to extrapolate fluorescence signals to cell numbers. Cell viability was assessed at the end of the culture period using the live/dead cell double staining kit (Merck Millipore, Germany), with live (calcein-positive) and dead (propidium iodide-positive) cells imaged in an Axiovert 200 microscope (Zeiss GMBH, DE). Cells were counted from ten fluorescence micrographs collected from random areas from each culture condition replicate. Data was analysed using two-way ANOVA followed by Bonferroni post-hoc tests, with differences deemed significant at p < 0.05. All experiments were performed in triplicate, and using cells isolated from three independent donors.
Coating stability and bio-degradability
C16e-MMPRGDS films were prepared from PA solutions at 50 nM as described above, imaged by phase-contrast microscopy, and then incubated in fresh serum-free medium (SFM) and cell-conditioned media at 37 °C for seven days. Conditioned media was produced by culturing hCSFs for five days with SFM alone or supplemented with RA, to either obtain matrix metalloprotease (MMP)-enriched (+MMP) or MMP-deprived (−MMP) conditioned medium, respectively.48 Changes in the shape and morphology of the films was evaluated after incubation by re-imaging coatings using a Nikon Eclipse inverted microscope (Nikon, JP) coupled with a Jenoptik CCD camera (Jenoptik AG, DE). All experiments were performed in triplicate.Circular dichroism (CD)
Spectra were recorded using a Chirascan spectropolarimeter (Applied Photophysics, UK). CD was performed on a solution of C16e-MMPRGDS in water which was then placed in a cover slip cuvette (0.01 mm thick). Spectra are presented with absorbance A < 2 at any measured point with a 0.5 nm step, 1 nm bandwidth and 1 second collection time per step at 20 °C, taking four averages.Fourier transform infra-red (FTIR) spectroscopy
Spectra were measured on a Nicolet Nexus spectrometer with DTGS detector. FTIR data was measured for a 1 wt% solution of C16e-MMPRGDS dissolved in D2O. Samples were sandwiched between two CaF2 plate windows (spacer 0.025 mm). Spectra were scanned 128 times over the range of 4000–900 cm−1. Data was corrected by baseline subtraction.X-ray diffraction (XRD)
X-ray diffraction was performed on stalks prepared by suspending drops of PA solution (1 wt% in water) between the ends of wax-coated capillaries, and allowing them to dry. The stalk was mounted vertically onto the four axis goniometer of a RAXIS IV++ X-ray diffractometer (Rigaku) equipped with a rotating anode generator. The XRD data was collected using a Saturn 992 CCD camera.Cryo-transmission electron microscopy (cryo-TEM)
Imaging was carried out using a field emission cryo-electron microscope (JEOL JEM-3200FSC), operating at 200 kV voltage. Images were taken in bright field mode and using zero loss energy filtering (omega type) with the slit with of 20 eV. Micrographs were recorded using a Gatan Ultrascan 4000 CCD camera. The specimen temperature was maintained at −187 °C during the imaging. Vitrified specimens were prepared using an automated FEI Vitrobot device using Quantifoil 3.5/1 holey carbon copper grids with the hole size of 3.5 μm. Just prior to use grids were plasma cleaned using a Gatan Solarus 9500 plasma cleaner and then transferred into an environmental chamber of a FEI Vitrobot having room temperature and 100% humidity. Thereafter 3 μl of sample solution (2 wt% concentration) was applied on the grid and it was blotted twice for 5 seconds and then vitrified in a 1/1 mixture of liquid ethane and propane at −180 °C. The most viscous gel (5 wt% concentration) was blotted 4 times for 5 s. The grid with vitrified sample solution was maintained at liquid nitrogen temperature and then cryo-transferred to the microscope.Fluorescence spectroscopy
The fluorescence of pyrene was excited at 335 nm at room temperature, and emission spectra were recorded from 350 to 450 nm, using a 10.0 mm × 5.00 mm quartz cell in a Varian Model Cary Eclipse spectrofluorimeter. Excitation and emission bandwidths of 2.5 nm were used throughout the experiments. The concentration of pyrene in water was 1.3 × 10−5 M. The same pyrene solution was used to dilute each peptide sample to avoid any dilution effect on pyrene fluorescence due to the addition of subsequent peptide amphiphile aliquots.Small-angle and X-ray scattering (SAXS)
Solution SAXS measurements was performed on the bioSAXS beamline BM29 at the ESRF, Grenoble, France. A solution of 1 wt% C16e-MMPRGDS was loaded in PCR tubes in an automated sample changer. This was injected into a fixed capillary tube for the SAXS measurements. SAXS data was collected using a Pilatus 1 M detector. The sample–detector distance was 2.84 m. The X-ray wavelength was 0.99 Å.SAXS data modelling
The cryo-TEM and SAXS data for 1 wt% C16-MMPRGDS at pH 2 indicate that it forms spherical micelles. The SAXS data was modelled using the software SASfit.49 A spherical shell model with a Gaussian polydispersity in outer radius was used. The model is based on scattering from a sphere with an outer radius R and an inner radius νR. The intensity is written as| I(q) = K(q,R,Δη) − K(q,νR,Δη(1 − μ)) |
With
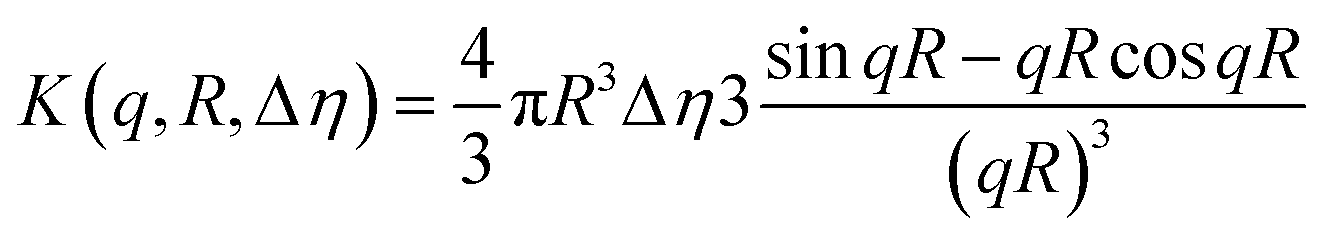
The Gaussian distribution function used to describe polydispersity in radius R is defined as follows:
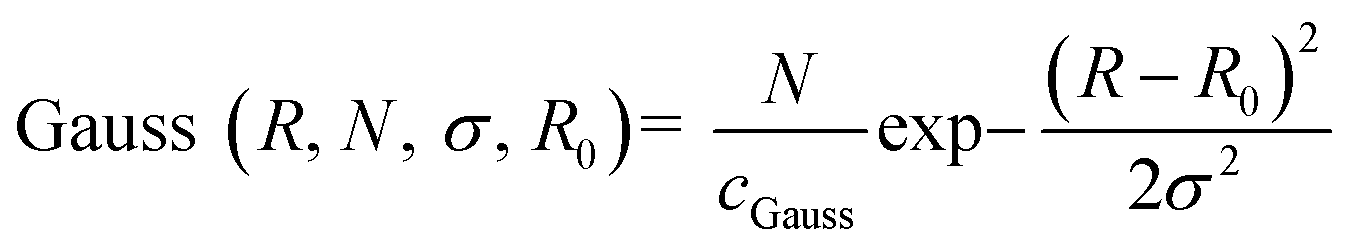
cGauss was chosen so that
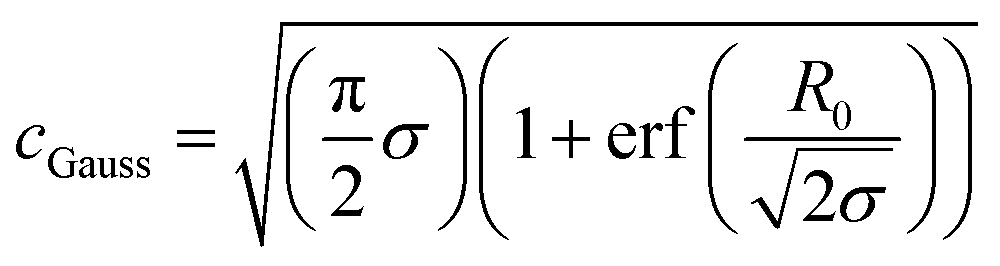
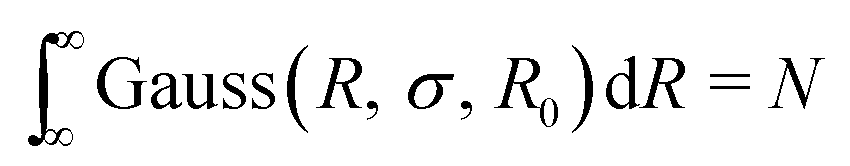 .
.
The parameters used to fit the data were; outer radius (R0), inner radius (vR0), scattering contrast relative to the core (μΔη), scattering contrast of shell (Δη), and Gaussian polydispersity parameters N and σ. A constant background was applied, BG.
Results
We investigated the self-assembly of C16e-MMPRGDS, a PA which can potentially act as a scaffold to support cell attachment, growth and subsequent release of cell-derived tissue from the initial scaffold. Cryo-TEM images were obtained for a 1 wt% solution of PA dissolved in water as illustrated by a representative example shown in Fig. 1. This image reveals that C16e-MMPRGDS self-assembles into very small spherical micelles with a diameter of approximately 3 nm. A similar size of lipopeptide micelles has been found in our previous work,40,50,51 and the radius is consistent with the length of the lipopeptide molecules. Interestingly, we observed that the micelles pack hexagonally in some areas with a lattice spacing of 6.7 nm (as shown in Fig. S1†). The hexagonal ordering is tentatively ascribed to enhanced packing of micelles within ultrathin regions of the cryo-TEM film. This is consistent with the fact that (bulk) SAXS measurements (to be discussed shortly) show no evidence of hexagonal ordering. | ||
| Fig. 1 Cryo-TEM image for a 1 wt% solution of C16e-MMPRGDS dissolved in water self-assembling into spherical micelles. | ||
Small-angle X-ray scattering (SAXS) was employed to further probe the C16e-MMPRGDS spherical micelle structures, complementing the cryo-TEM images and providing information on the internal structure of the micelles. A synchrotron SAXS profile for a 1 wt% solution of C16e-MMPRGDS is shown in Fig. 2a. The form factor profile was fitted to a spherical micelle model using the modelling software SASfit.49 The fitting parameters (listed in full in Table 1) obtained from the fit indicate an outer radius of 3.3 nm, which is in good agreement with the cryo-TEM images discussed above. The cryo-TEM (Fig. S1†) revealed that the spherical micelles adopt a hexagonal arrangement in some areas, but since hexagonal ordering is not obtained by SAXS this is believed, as mentioned above, to be an artefact arising from the sample preparation during vitrification. Temperature-dependent SAXS was performed for a fixed concentration (1 wt% solution) of the PA upon heating, from 5 °C to 55 °C (i.e. across the melting point for the lipid chain52,53) as shown in Fig. 2b. The temperature-dependent SAXS curves are similar to one another indicating that the spherical micelle structure is retained upon heating even across the lipid chain melting temperature (40–45 °C). The SAXS profiles for all three temperatures were fitted to the form factor for a core–shell spherical micelle (Fig. 2b). The parameters for the fits are listed in Table 1. The data reveals that at 5 °C the micelles have a radius of less than 3 nm and upon heating, an increase in radius of the micelles is observed with the largest increase from 5 °C to 30 °C. The core–shell contrast decreases with increasing temperature, possibly due to some solubilisation of water within the lipid interior of the micelle.
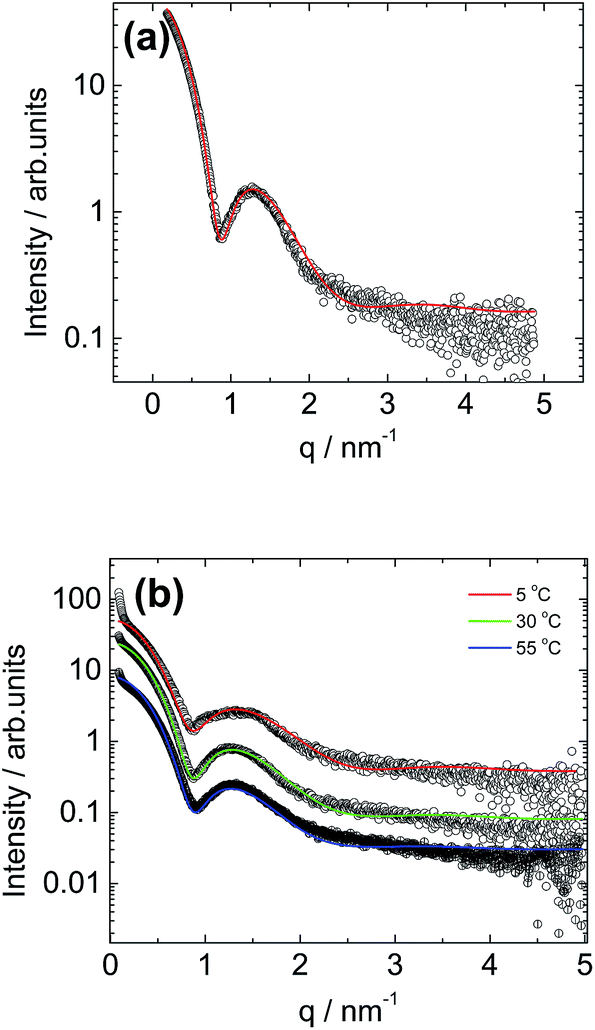 | ||
| Fig. 2 (a) SAXS profile for a solution of 1 wt% C16e-MMPRGDS fitted to a spherical shell form factor model (solid line) using SASfit. (b) Temperature dependent SAXS profiles for a solution containing 1 wt% C16e-MMPRGDS fitted to a spherical shell form factor models (solid lines). | ||
| 5 °C | 30 °C | 55 °C | |
|---|---|---|---|
| N | 1 | 1 | 1 |
| σ | 1.11 | 1.01 | 0.99 |
| R | 2.74 | 3.30 | 3.40 |
| vR | 1.62 | 1.65 | 1.70 |
| μΔη | −2.11 | −1.61 | −1.20 |
| Δη | 0.03 | 0.032 | 0.03 |
| BG | 0.19 | 0.16 | 0.18 |
Spectroscopic techniques were employed to elucidate the secondary structure of C16e-MMPRGDS. Circular dichroism (CD) spectroscopy measurements were performed to investigate secondary structure, the spectra being shown in Fig. 3a. A minimum at 230 nm was observed at room temperature, which suggests a lack of ordered structure.54,55 Temperature-dependent CD reveals no overall change in structure upon heating and cooling in the range of 20–90 °C. The FTIR spectrum obtained at room temperature in the amide I′ region shown in Fig. 3b indicates a disordered conformation, as a main peak at approximately 1650 cm−1 is observed.56–58 Thus, based on the data from both spectroscopic methods, this PA is characterised by a disordered secondary structure, with weak hydrogen bonding within the assembly which favours spherical micelle formation.8,53
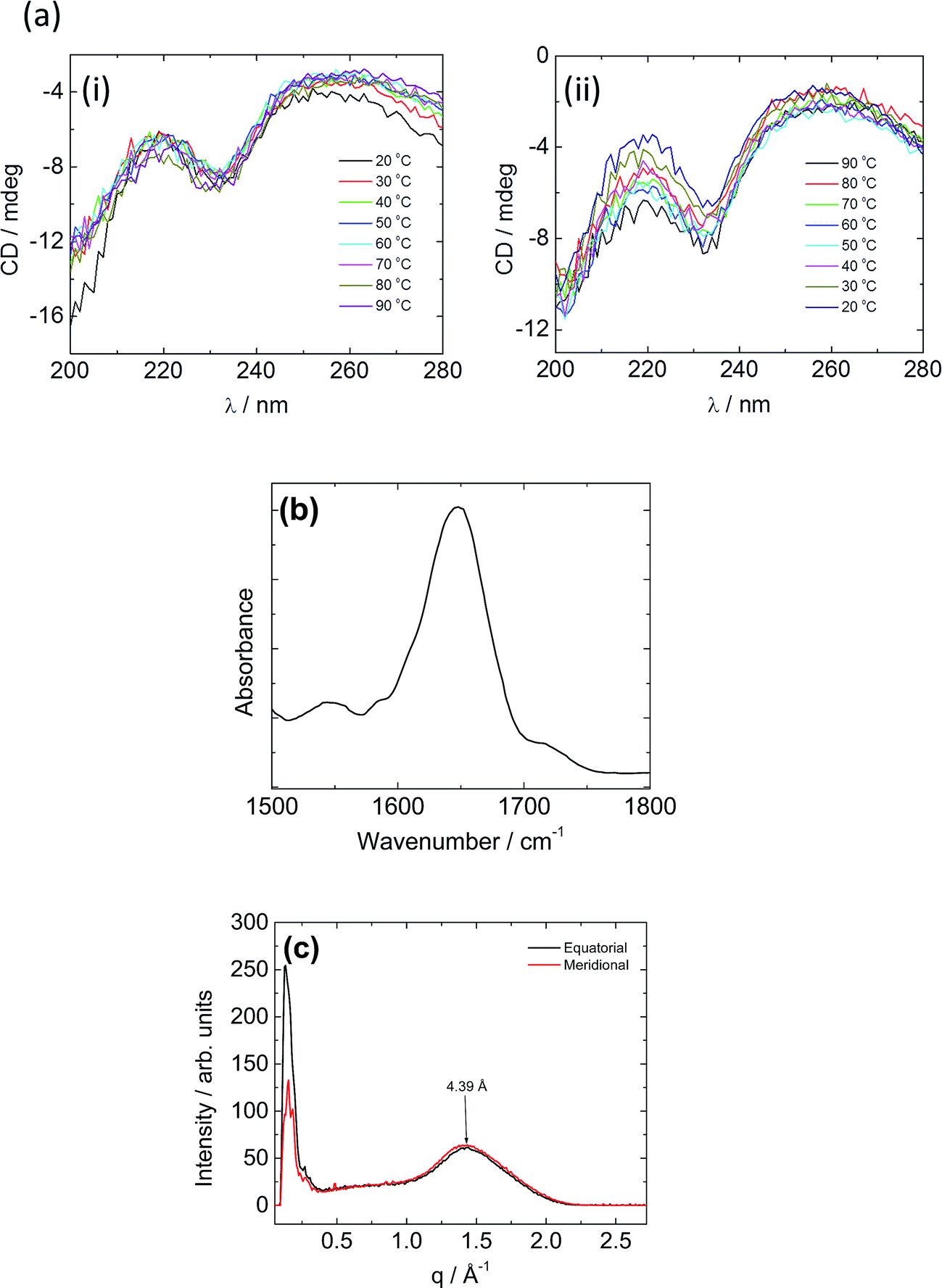 | ||
| Fig. 3 (a) Temperature dependent CD for C16e-MMPRGDS (i) heating and (ii) cooling. (b) FTIR spectrum for a 3 wt% concentration of C16e-MMPRGDS. (c) XRD performed on a dried stalk containing a 1 wt% concentration of C16e-MMPRGDS. | ||
To further probe the spherical micelle nanostructure, fibre XRD was employed using a stalk dried from a 1 wt% solution of C16e-MMPRGDS. The two-dimensional pattern was found to be isotropic, so it was integrated to one-dimensional profiles. The meridional and equatorial 1D intensity profiles are shown in Fig. 3c. A single peak is observed with a d spacing of 4.39 Å (calculated using d = 2π/q). A β-sheet structure is characterized by a β-strand spacing of 4.7 Å in the meridional direction along with a spacing of 10–13 Å in the equatorial direction, associated with the spacing of β-sheets.59–61 As no such peaks are present in the intensity profile a β-sheet structure can be ruled out and instead the observed peak is ascribed to the packing of the lipid chain, as it is broad and also unoriented. The XRD data is in good agreement with the spectroscopy data revealing a lack of intermolecular hydrogen bonded β-sheet structure and is consistent with the presence of spherical micelles which are unable to accommodate a β-sheet arrangement of PA molecules.
The critical aggregation concentration (CAC) of C16e-MMPRGDS was determined at room temperature using a fluorescence assays with pyrene. The fluorescence vibrational structure of pyrene is sensitive to its environmental hydrophobicity and has been successfully used previously to determine the CAC of amphiphilic molecules.62,63 The CAC of C16e-MMPRGDS in water was estimated by observing the fluorescence intensity at the I1 (λ = 383 nm) peak which corresponds to the first vibronic band of pyrene.62,64Fig. 4 shows that there is a break in the fluorescence intensity (I1) against concentration curve which denotes that C16e-MMPRGDS begins to self-assemble into spherical micelles above a concentration of 0.0065 wt% (4 mM). The self-assembly studies conducted in this research were well above this CAC, being performed at concentrations of 1 wt%.
 | ||
| Fig. 4 Pyrene fluorescence assay to determine the CAC of C16e-MMPRGDS. | ||
To understand the applicability of C16e-MMPRGDS as a bioactive, cell-adhesive template, the PA was drop-spotted in solution at 5–150 nM (i.e. well below the CAC) onto culture plates and allowed to dry overnight to form continuous film coatings. Data presented in Fig. 5 demonstrates that C16e-MMPRGDS coatings were stable in cell-culture compatible aqueous environment (in fresh serum-free medium for 7 days at 37 °C and 5% CO2). Moreover, the PA was susceptible to cleavage by MMPs, as shown by the degradation of the coating after incubation in cell-conditioned culture medium (Fig. 5, +MMPs panels). This degradation was specific, as conditioned medium deprived of cell-derived MMPs failed to alter the coating morphology and shape (Fig. 5, −MMPs panels).
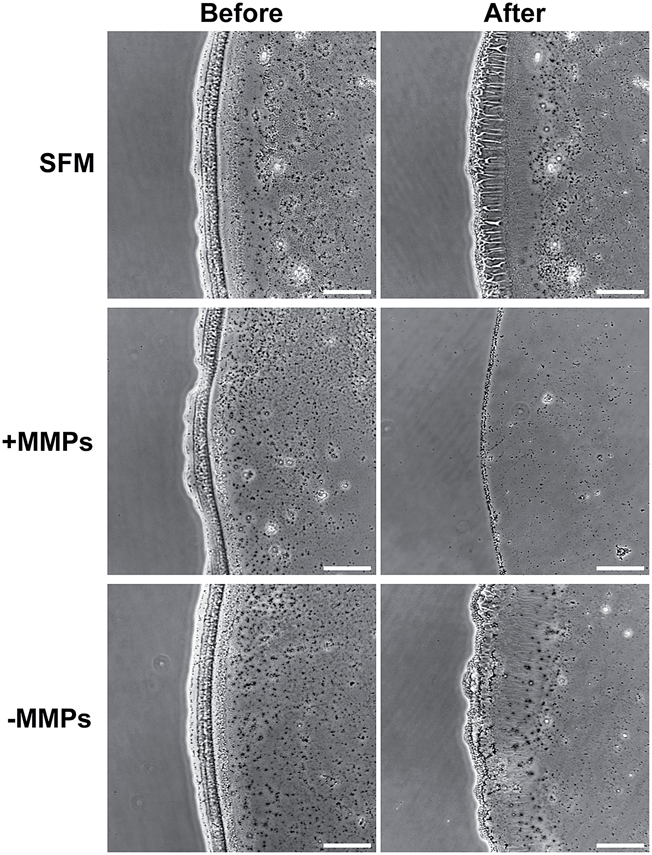 | ||
| Fig. 5 Stability and bio-degradability of C16e-MMPRGDS film coatings in aqueous environment. PA solutions at 50 nM were drop-spotted onto ultra-low attachment tissue culture plates, allowed to dry overnight, and imaged by phase-contrast microscopy before (left) and after 7 days incubation in different cell culture media (right): fresh serum-free medium (SFM) and cell-conditioned media with (+MMPs) or without matrix metalloproteases (−MMPs). Scale bars of representative micrographs = 100 μm. | ||
The potential of C16e-MMPRGDS as a material to create cytocompatible adhesive templates was further assessed by seeding human corneal stromal fibroblasts (hCSFs) onto the PA films. To restrict cell growth to the PA coatings and minimise unspecific cell adhesion, coatings were produced onto ultra-low attachment culture plates. As evident from Fig. 6a, PA coatings were stable on low-attachment plates, creating defined boundaries between the coated and non-coated surfaces. Upon seeding, hCSFs adhered to the bioactive PA films but not to the non-coated low-attachment plastic. This is especially evident at the edge of the films, where a clear delineation between the films and non-coated low-attachment surface was observed (Fig. 6b). In addition, hCSFs were able to proliferate within the area of the C16e-MMPRGDS coatings (Fig. 6c and 7), reaching semi-confluence while maintaining high viability levels (Fig. 6d). Quantification of cell numbers indicated that all tested concentrations of the PA were capable of supporting cell adhesion and growth at significantly higher levels compared to the non-coated control surfaces (Fig. 7). After seven days in culture, the number of hCSFs decreased. This effect was not caused by a reduction in cell viability (as demonstrated in Fig. 6d) but was due to the detachment of cells from the underlying adhesive surface, a consequence of the progressive cleavage of the C16e-MMPRGDS molecules by the endogenous proteolytic activity of hCSFs. Thus cells could be released from the scaffold under enzymatic control. This notion is also supported by the absence of such detachment from RGDS-containing PA coatings lacking an enzyme-cleavable peptide sequence.43 It is known that MMPs have a fundamental role in the proteolytic activity of cells, which can ultimately lead to the cleavage of ECM components and hence cell detachment.48 Due to supplementation of retinoic acid (RA) to the culture medium at 5 × 10−6 M, hCSFs retain the capability to secrete small amounts of matrix metalloproteases, namely MMP1.43 As the secreted MMP enzymes begin to accumulate in the culture supernatant, the C16e-MMPRGDS coatings start degrading, and the cells lose their anchoring support, detach from the surface, and float away. Together, these results indicate that C16e-MMPRGDS with RA enriched media can provide a coating suitable for initial cell adhesion, proliferation, and survival, but permit the subsequent self-detachment of cells though its specific sensitivity to cell-secreted, endogenous proteases over a number of days in culture.
 | ||
| Fig. 6 C16e-MMPRGDS film coatings allow specific attachment and proliferation of hCSFs. PA solutions at 5–150 nM were drop-spotted onto ultra-low attachment tissue culture plates, and allowed to dry overnight. (a) The PA formed a dense, continuous film with defined boundaries (traced line). (b) Seeded cells specifically attached to the coated, but not to the uncoated, surface. (c) Cells within the centre of the coated area were able to proliferate for up to 7 days (c), where they retained high-viability levels (d). Live and dead cells were correspondingly stained with calcein (green) and propidium iodide (red) fluorescent dyes. Scale bars of representative phase-contrast and fluorescence micrographs = 100 μm and 50 μm, respectively. | ||
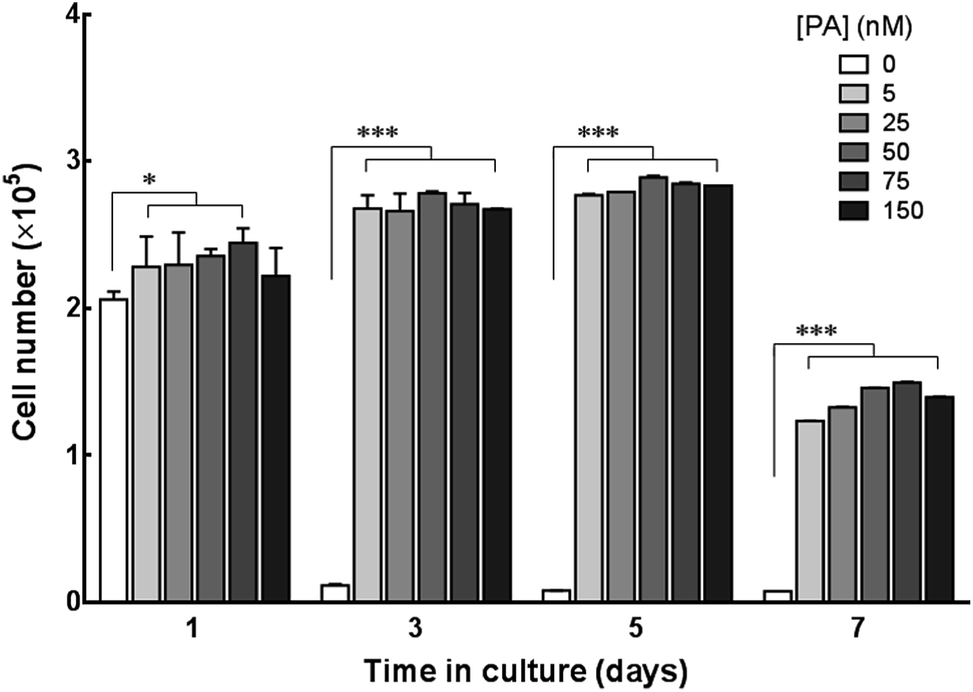 | ||
| Fig. 7 Quantification of viable hCSFs grown on C16e-MMPRGDS film coatings. Cells seeded onto uncoated (PA = 0 nM) and PA-coated (5–150 nM) ultra-low attachment culture plates were quantified upon seeding (day 1) and throughout the time in culture, up to 7 days, using the Alamar blue assay. All coating concentrations showed to promote cell attachment, whereas cells did not attach to uncoated surfaces (white bars). Statistical differences in cell numbers were indicated by two-way ANOVA followed by Tukey's post hoc tests (n = 3; * and *** correspond to p < 0.01 and 0.001, respectively). | ||
Summary and discussion
The PA C16e-MMPRGDS is shown to self-assemble into small spherical micelles above a critical aggregation concentration (0.0065 wt% in water). The peptide adopts a disordered conformation within the corona of the micelles, as probed by CD and FTIR spectroscopies, as well as X-ray diffraction. The secondary structure is not altered upon heating across the lipid chain melting temperature. The PA supports the growth of hCSFs for up to five days, whilst ensuring cell self-detachment at later time points.A remarkable feature of this study is the formation of spherical micelles by the PA with a disordered secondary structure. This is in complete contrast to the secondary structure and self-assembly properties of the homologue with saturated lipid chain C16-MMPRGDS.34 This latter PA adopts a β-sheet secondary structure and forms extended nanotape structures. In addition, this PA is not only capable of promoting the adhesion and growth of corneal or skin fibroblasts but can also stimulate them to deposit native extracellular matrix, thus forming layered tissues. These newly-shaped tissues governed their own release from the template via the action of endogenous proteases (MMPs), using the RA-supplementation system applied in the present study. However, the coatings formed by the saturated C16-MMPRGDS PA were less susceptible to the MMPs endogenously secreted by cells.34 A possible explanation for this discrepancy is that the diene functionality in C16e-MMPRGDS has a profound influence not only on the self-assembly but also on the bio-accessibility of proteolytic enzymes to the functional motifs within the nanostructure. Indeed, these two properties may be correlated, since it is possible that the formation of spherical micelles with peptides in a disordered conformation would allow the cleavable peptide sequence to be presented in a less compact configuration compared to that within β-sheet based nanotapes, and consequently enable less sterically-hindered access to the active site from bulky enzymes. In this context, investigating the relationship between self-assembly, molecule accessibility,65 and bioactivity will be of future interest in these and related PA systems. Moreover, these systems represent simple but highly versatile, highly-transferable multi-purpose materials with promising applications in both fundamental and clinical research.
Acknowledgements
This work was supported by BBSRC grant BB/J019836/1 to IWH and CJC and EPSRC grants EP/G067538/1 and EP/L020599/1 to IWH. We are grateful to Adam Round for assistance with SAXS experiments at the ESRF, Grenoble, France (beamtime ref. MX-1606).References
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- E. S. Place, N. D. Evans and M. M. Stevens, Nat. Mater., 2009, 8, 457–470 CrossRef CAS PubMed.
- E. S. Place, J. H. George, C. K. Williams and M. M. Stevens, Chem. Soc. Rev., 2009, 38, 1139–1151 RSC.
- L. Zhang and T. J. Falla, Clin. Dermatol., 2009, 27, 485–494 CrossRef PubMed.
- I. W. Hamley, Soft Matter, 2011, 7, 4122–4138 RSC.
- D. W. P. M. Löwik, E. H. P. Leunissen, M. van den Heuvel, M. B. Hansen and J. C. M. van Hest, Chem. Soc. Rev., 2010, 39, 3394–3412 RSC.
- D. W. P. M. Löwik and J. C. M. van Hest, Chem. Soc. Rev., 2004, 33, 234–245 RSC.
- H. G. Cui, M. J. Webber and S. I. Stupp, Pept. Sci., 2010, 94, 1–18 CrossRef CAS PubMed.
- J. B. Matson, R. H. Zha and S. I. Stupp, Curr. Opin. Solid State Mater. Sci., 2011, 15, 225–235 CrossRef CAS PubMed.
- J. Boekhoven and S. I. Stupp, Adv. Mater., 2014, 26, 1642–1659 CrossRef CAS PubMed.
- I. W. Hamley, Angew. Chem., 2014, 53, 6866–6881 CrossRef CAS PubMed.
- A. Dehsorkhi, V. Castelletto and I. W. Hamley, J. Pept. Sci., 2014, 20, 453–467 CrossRef CAS PubMed.
- S. Cavalli and A. Kros, Adv. Mater., 2008, 20, 627–631 CrossRef CAS.
- J. D. Tovar, R. C. Claussen and S. I. Stupp, J. Am. Chem. Soc., 2005, 127, 7337–7345 CrossRef CAS PubMed.
- H. A. Behanna, J. Donners, A. C. Gordon and S. I. Stupp, J. Am. Chem. Soc., 2005, 127, 1193–1200 CrossRef PubMed.
- H. Cui, T. Muraoka, A. G. Cheetham and S. I. Stupp, Nano Lett., 2009, 9, 945–951 CrossRef CAS PubMed.
- E. T. Pashuck, H. Cui and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 6041–6046 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- E. D. Spoerke, S. G. Anthony and S. I. Stupp, Adv. Mater., 2009, 21, 425–430 CrossRef CAS PubMed.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS PubMed.
- A. Mata, L. Hsu, R. Capito, C. Aparicio, K. Henrikson and S. I. Stupp, Soft Matter, 2009, 5, 1228–1236 RSC.
- M. A. Greenfield, J. R. Hoffman, M. O. de la Cruz and S. I. Stupp, Langmuir, 2010, 26, 3641–3647 CrossRef CAS PubMed.
- R. N. Shah, N. A. Shah, M. M. D. Lim, C. Hsieh, G. Nuber and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2009, 107, 3293–3298 CrossRef PubMed.
- R. H. Zha, S. Sur and S. I. Stupp, Adv. Mater., 2013, 2, 126–133 CAS.
- S. Soukasene, D. J. Toft, T. J. Moyer, H. Lu, H.-K. Lee, S. M. Standley, V. L. Cryns and S. I. Stupp, ACS Nano, 2011, 5, 9113–9121 CrossRef CAS PubMed.
- D. Missirlis, M. Farine, M. Kastantin, B. Ananthanarayanan, T. Neumann and M. Tirrell, Bioconjugate Chem., 2010, 21, 465–475 CrossRef CAS PubMed.
- D. Missirlis, H. Khant and M. Tirrell, Biochemistry, 2009, 48, 3304–3314 CrossRef CAS PubMed.
- A. G. Cheetham, P. C. Zhang, Y. A. Lin, L. L. Lock and H. G. Cui, J. Am. Chem. Soc., 2013, 135, 2907–2910 CrossRef CAS PubMed.
- J. B. Matson, C. J. Newcomb, R. Bitton and S. I. Stupp, Soft Matter, 2012, 8, 3586–3595 RSC.
- S. Bulut, T. S. Erkal, S. Toksoz, A. B. Tekinay, T. Tekinay and M. O. Guler, Biomacromolecules, 2011, 12, 3007–3014 CrossRef CAS PubMed.
- Y. A. Lin, A. G. Cheetham, P. C. Zhang, Y. C. Ou, Y. G. Li, G. S. Liu, D. Hermida-Merino, I. W. Hamley and H. G. Cui, ACS Nano, 2014, 8, 12690–12700 CrossRef CAS PubMed.
- M. O. Guler, L. Hsu, S. Soukasene, D. A. Harrington, J. F. Hulvat and S. I. Stupp, Biomacromolecules, 2006, 7, 1855–1863 CrossRef CAS PubMed.
- A. Dehsorkhi, I. W. Hamley, J. Seitsonen and J. Ruokolainen, Langmuir, 2013, 29, 6665–6672 CrossRef CAS PubMed.
- R. J. Gouveia, V. Castelletto, C. J. Connon and I. W. Hamley, Tissue Eng., Part A, 2015 DOI:10.1089/ten.TEA.2014.0671.
- M. Conda-Sheridan, S. S. Lee, A. T. Preslar and S. I. Stupp, Chem. Commun., 2014, 50, 13757–13760 RSC.
- V. Castelletto, I. W. Hamley, J. Perez, L. Abezgauz and D. Danino, Chem. Commun., 2010, 46, 9185–9187 RSC.
- K. Lintner, US Pat., US6620419 B1, 2003 Search PubMed.
- R. R. Jones, V. Castelletto, C. J. Connon and I. W. Hamley, Mol. Pharm., 2013, 10, 1063–1069 CrossRef CAS PubMed.
- V. Castelletto, I. W. Hamley, C. Whitehouse, P. Matts, R. Osborne and E. S. Baker, Langmuir, 2013, 29, 9149–9155 CrossRef CAS PubMed.
- I. W. Hamley, A. Dehsorkhi, P. Jauregi, J. Seitsonen, J. Ruokolainen, F. Coutte, G. Chataigné and P. Jacques, Soft Matter, 2013, 9, 9572–9578 RSC.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5133–5138 CrossRef CAS PubMed.
- V. Castelletto, R. J. Gouveia, C. J. Connon and I. W. Hamley, Faraday Discuss., 2013, 166, 381–397 RSC.
- R. J. Gouveia, V. Castelletto, S. G. Alcock, I. W. Hamley and C. J. Connon, J. Mater. Chem. B, 2013, 1, 6157–6169 RSC.
- J. Patterson and J. A. Hubbell, Biomaterials, 2010, 31, 7836–7845 CrossRef CAS PubMed.
- R. C. Billinghurst, L. Dahlberg, M. Ionescu, A. Reiner, R. Bourne, C. Rorabeck, P. Mitchell, J. Hambor, O. Diekmann, H. Tschesche, J. Chen, H. VanWart and A. R. Poole, J. Clin. Invest., 1997, 99, 1534–1545 CrossRef CAS PubMed.
- J. M. Takacs, M. R. Jaber, F. Clement and C. Walters, J. Org. Chem., 1998, 63, 6757–6760 CrossRef CAS.
- R. M. Gouveia and C. J. Connon, Invest. Ophthalmol. Visual Sci., 2013, 54, 7483–7491 CAS.
- P. Van Lint and C. Libert, J. Leukocyte Biol., 2007, 82, 1375–1381 CrossRef CAS PubMed.
- http://kur.web.psi.ch/sans1/SANSSoft/sasfit.html, in2015.
- A. Dehsorkhi, V. Castelletto, I. W. Hamley, J. Adamcik and R. Mezzenga, Soft Matter, 2013, 9, 6033–6036 RSC.
- I. W. Hamley, S. Kirkham, A. Dehsorkhi, V. Castelletto, M. Reza and J. Ruokolainen, Chem. Commun., 2014, 50, 15948–15951 RSC.
- V. Castelletto, I. W. Hamley, J. Perez, L. Abezgauz and D. Danino, Chem. Commun., 2010, 46, 9185–9187 RSC.
- J. F. Miravet, B. Escuder, M. D. Segarra-Maset, M. Tena-Solsona, I. W. Hamley, A. Dehsorkhi and V. Castelletto, Soft Matter, 2013, 9, 3558–3564 RSC.
- B. Nordén, A. Rodger and T. R. Dafforn, Linear Dichroism and Circular Dichroism: A Textbook on Polarized-Light Spectroscopy, RSC, Cambridge, 2010 Search PubMed.
- R. W. Woody, Methods Enzymol., 1995, 246, 34–71 CAS.
- Z. Ganim, H. S. Chung, A. W. Smith, L. P. Deflores, K. C. Jones and A. Tokmakoff, Acc. Chem. Res., 2007, 41, 432–441 CrossRef PubMed.
- B. Stuart, Biological Applications of Infrared Spectroscopy, Wiley, Chichester, 1997 Search PubMed.
- P. Haris and D. Chapman, Biopolymers, 1995, 37, 251–263 CrossRef CAS PubMed.
- I. W. Hamley, Angew. Chem., 2007, 46, 8128–8147 CrossRef CAS PubMed.
- L. C. Serpell, Biochim. Biophys. Acta, 2000, 1502, 16–30 CrossRef CAS.
- K. E. Marshall and L. C. Serpell, Open Biol. J., 2009, 2, 185–192 CrossRef CAS.
- P. Palladino, V. Castelletto, A. Dehsorkhi, D. Stetsenko and I. W. Hamley, Langmuir, 2012, 28, 12209–12215 CrossRef CAS PubMed.
- I. W. Hamley, A. Dehsorkhi and V. Castelletto, Langmuir, 2013, 29, 5050–5059 CrossRef CAS PubMed.
- J. Aguiar, P. Carpena, J. A. Molina-Bolívar and C. C. Ruiz, J. Colloid Interface Sci., 2003, 258, 116–122 CrossRef CAS.
- J. H. Ortony, C. J. Newcomb, J. B. Matson, L. C. Palmer, P. E. Doan, B. M. Hoffman and S. I. Stupp, Nat. Mater., 2014, 13, 812–816 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sm00459d |
| ‡ Currently at: Manchester Institute of Biotechnology and School of Materials, The University of Manchester, Oxford Road, Manchester, M13 9 PL, U.K. |
| This journal is © The Royal Society of Chemistry 2015 |