
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural basis of head to head polyketide fusion by CorB†
Georg
Zocher‡
a,
Joachim
Vilstrup‡
b,
Daniel
Heine
c,
Asis
Hallab
d,
Emilie
Goralski
e,
Christian
Hertweck
cf,
Mark
Stahl
g,
Till F.
Schäberle
*e and
Thilo
Stehle
*ah
aInterfaculty Institute of Biochemistry, University Tübingen, Hoppe-Seyler-Str. 4, 72076 Tübingen, Germany. E-mail: thilo.stehle@uni-tuebingen.de
bDepartment of Molecular Biology and Genetics, Aarhus University, Gustav Wieds Vej 10C, DK 8000 Aarhus C, Denmark
cLeibniz Institute for Natural Product Research and Infection Biology (HKI), 07745 Jena, Germany
dMax Planck Institute for Plant Breeding Research, Carl-von-Linné-Weg 10, 50829 Köln, Germany
eInstitute for Pharmaceutical Biology, University of Bonn, Nussallee 6, 53115 Bonn, Germany. E-mail: till.schaeberle@uni-bonn.de
fChair for Natural Product Chemistry, Friedrich Schiller University, 07743 Jena, Germany
gCenter for Plant Molecular Biology, University Tübingen, Auf der Morgenstelle 32, 72076 Tübingen, Germany
hDepartment of Pediatrics, Vanderbilt University School of Medicine, Nashville, TN 37232, USA
First published on 6th August 2015
Abstract
Corallopyronin A is a polyketide derived from the myxobacterium Corallococcus coralloides with potent antibiotic features. The gene cluster responsible for the biosynthesis of corallopyronin A has been described recently, and it was proposed that CorB acts as a ketosynthase to interconnect two polyketide chains in a rare head-to-head condensation reaction. We determined the structure of CorB, the interconnecting polyketide synthase, to high resolution and found that CorB displays a thiolase fold. Site-directed mutagenesis showed that the catalytic triad consisting of a cysteine, a histidine and an asparagine is crucial for catalysis, and that this triad shares similarities with the triad found in HMG-CoA synthases. We synthesized a substrate mimic to derivatize purified CorB and confirmed substrate attachment by ESI-MS. Structural analysis of the complex yielded an electron density-based model for the polyketide chain and showed that the unusually wide, T-shaped active site is able to accommodate two polyketides simultaneously. Our structural analysis provides a platform for understanding the unusual head-to-head polyketide-interconnecting reaction catalyzed by CorB.
Introduction
Controlling infectious diseases is often compromised by the development of resistance to a drug initially used to treat the infection. The development of antibiotic resistance in the infecting organism is a constant challenge, and the discovery of novel antibiotics is therefore urgently required.1 Secondary metabolites such as polyketides substantially contribute to the chemical diversity, providing a natural and valuable source for antibiotics, antiparasitic, immunosuppressive, and antitumoral agents.2,3Corallopyronin A (Cor) is a polyketide isolated from the myxobacterium Corallococcus coralloides.4,5 Cor contains an α-pyrone entity and shows promising antibiotical activity in vivo by targeting the so-called hinge region of the bacterial DNA-dependent RNA polymerase (RNAP), thereby preventing the initiation of transcription.6 Cor is thought to interact with conserved RNAP residues, thus exhibiting broad-spectrum antibacterial activity while leaving the eukaryotic transcription machinery intact. In addition, Cor interacts with RNAP residues that are remote from the binding sites of other characterized RNAP inhibitors, e.g. rifamycin, thus remaining active towards rifamycin-resistant bacteria.6,7 Cor is produced by Corallococcus coralloides strains, which are gliding, heterotrophic myxobacteria originally isolated from soil samples.8 As Cor shows substantial activity against various pathogenic bacteria including methicillin-resistant staphylococci (MRSA), it represents a promising lead compound for antibacterial drug development.9 While the activity of Cor against mycobacteria is only moderate, this is regarded as an advantage for the treatment of diseases such as lymphatic filariasis and onchocerciasis without concern for cross-resistance development in tuberculosis.9 These diseases affect over 150 million people in over 80 developing countries of the tropics and subtropics. They are caused by filarial nematodes, and it was recognized that depleting the obligate endosymbionts, i.e. Wolbachia, from the worms results in cure of the patients. Recently, Cor was shown to possess superior in vivo antimicrobial activity against Wolbachia.5 Thus, the compound is currently in the focus of a translational project that aims to establish Cor as a new antibiotic drug.9
The gene cluster responsible for the biosynthesis of Cor was described recently,4 and the authors showed by stable-isotope labeling experiments that two polyketide chains are synthesized, interconnected, and subsequently modified resulting in the formation of Cor (Fig. 1).4 Based on sequence analysis, CorB was assigned as a β-ketoacyl-ACP-synthase I. It was proposed that CorB performs an unusual head-to-head condensation reaction to interconnect two polyketide chains. Polyketide synthases (PKS) catalyzing head-to-head connections are rare but were described for plant type III PKSs.10,11 Ketosynthases usually catalyze the Claisen condensation of acylthioesters with malonylthioesters in a head-to-tail reaction, yielding an elongated carbon chain. Different classes of ketosynthases have been described according to the architecture and function of the system: (i) in modular type I PKSs, ketosynthases are part of large multifunctional enzymes in which a nascent chain is passed on from module to module, whereas (ii) in type II PKSs, the units are loosely associated, iteratively working, stand-alone enzymes, and (iii) in type III PKSs the enzymes are multifunctional and act iteratively.12 In addition to polyketide systems, ketosynthases are also crucial in fatty acid biosynthesis.13 In contrast to the head-to-tail fusion catalyzed by ketosynthases, hydroxymethylglutaryl-CoA synthases (HMG-CoA synthases) catalyze the condensation of acetoacetyl-CoA with a β-keto thioester such as acetoacetyl-CoA. In this way, 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) is formed in mevalonate-dependent isoprenoid pathways.14 Although the reaction is comparable to the biosynthesis of polyketides, the enzymatic reaction of HMG-CoA synthases lacks the decarboxylation process that is a common feature of the condensing enzymes in the thiolase superfamily.15 Irrespective of the different types and variants of ketosynthases, all of them share the thiolase fold.16 The reaction mechanism varies among enzymes of this superfamily. However, with a few exceptions,17,18 most superfamily members catalyze the formation of a carbon–carbon bond between a polyketide chain bound as a thioester to an activated cysteine and an α-carboxylated extender unit bound as a thioester to either an acyl carrier protein (ACP) or coenzyme A (CoA).15
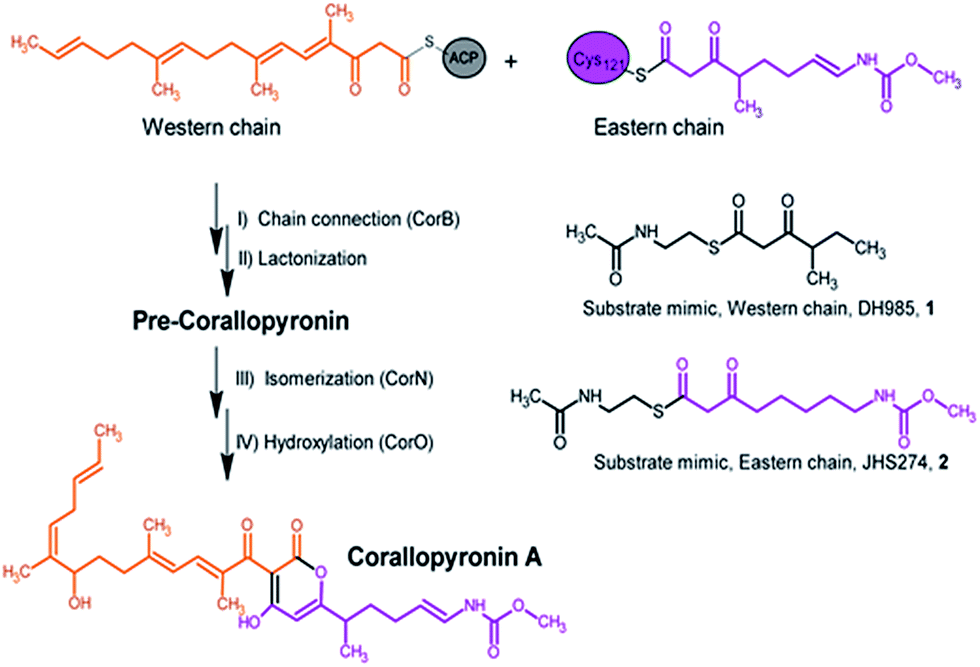 | ||
| Fig. 1 Section of the biosynthesis of corallopyronin A and the catalyzed reaction of CorB including the substrate mimics used in the in vitro enzyme assay. | ||
Here we report a structural and biochemical analysis of CorB, a member of a novel class of head-to-head condensing ketosynthases involved in the biosynthesis of α-pyrone antibiotics such as Cor. The enzyme is encoded by a stand-alone gene within the biosynthetic gene cluster. With an in vitro assay, we show that CorB catalyzes the interconnection of the two precursor chains (Fig. 1) with a Claisen-like condensation reaction. The subsequent lactonization eventually yields the central pyrone ring system of Cor.
Results and discussion
Substrate analogue synthesis and CorB activity assay
CorB was heterologously produced in E. coli. Due to the lack of commercially available substrate analogues, we synthesized mimics of the natural substrates. According to our hypothesis for pyrone ring formation4 the substrates should carry a β-keto residue. Moreover, natural substrates are tethered to a phosphopantetheine (PPant) arm, which represents the essential prosthetic group of carrier proteins within PKS/NRPS systems. We therefore synthesized substrate analogue 1 as a N-acetylcysteamine (SNAC) thioester derivative (Fig. 1, S2 and S3†). Substrate 1 was designed to mimic both the eastern and the western chain, as these differ exclusively in the saturation when regarding the carbon skeleton up to atom C5. We assumed that this substrate mimic choice would be a reasonable compromise to investigate CorB binding. Together with substrate mimic 2,19 these have been the substrates for the in vitro assay to test the enzymatic Claisen condensation reaction. In this way, the PPant arm is mimicked, and the molecules are activated for the subsequent reactions.Purified CorB was assayed using 1 and 2 as substrates. In the presence of freshly purified CorB, the m/z of the expected product methyl (5-(4-hydroxy-3-(2-methylbutanoyl)-2-oxo-2H-pyran-6-yl)pentyl) carbamate 3 was detected using high resolution LC-MS. (Fig. 2). To form the pyrone ring, the first of the two SNAC-substrates is covalently bound to C121 of CorB (compare with Fig. 1), whereas the other chain – in vivo ACP-tethered – is still bound to SNAC. Then, CorB catalyzes the interconnection of both chains and releases the SNAC entity (of ACP in vivo). This is followed by lactonization, yielding a closed pyrone ring and liberating the product. In control reactions, either without a substrate, or without CorB, 3 was not detected.
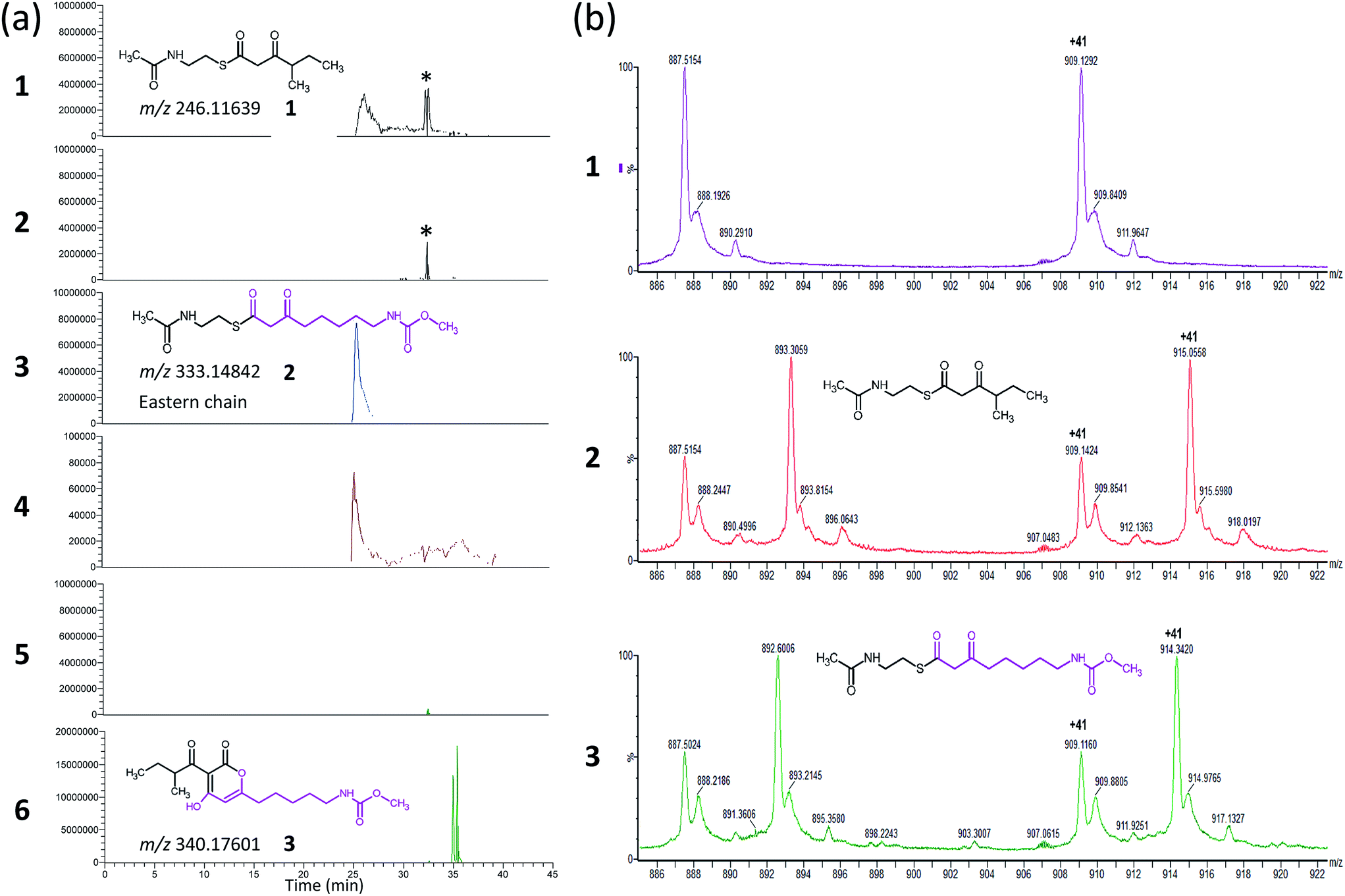 | ||
| Fig. 2 Results of the CorB in vitro activity assay (a) and ESI-MS experiment of CorB used in the crystallization experiment. (a) Extracted ion chromatograms for the assay (traces 2, 4, and 6) and the respective control reaction without CorB (traces 1, 3, and 5). The extracted m/z of substrate 1 (trace 1 and 2) and substrate 2 (trace 3 and 4), as well as for product 3 (trace 5 and 6) are given. In presence of the CorB protein the concentration of the educts decreased and the product was formed. The asterisk marks a contamination that is similar in mass but not related to substrate 1. The impurity is not present in trace 5 and 6 due to screening for a different mass. (b) Section of the ESI-MS experiments. All three samples resulted from an identical setup including the same protein and differ exclusively in the presence or absence of the substrates. The results for the wildtype CorB (panel 1), CorB incubated with 1 (panel 2), and CorB incubated with 2 (panel 3) are depicted. | ||
Phylogenetic analysis
A set of 415 highly homologous proteins was selected from the NCBI database of prokaryotic genomes. On the basis of these data, a strict consensus tree was reconstructed using maximum likelihood and parsimony algorithms (Fig. 3).20,21 A clear separation of the ketosynthases into distinct clades was observed. The clade comprising CorB includes many proteins annotated as β-ketoacyl-(acyl-carrier-protein) synthase III (FabH) proteins, and forms a sister clade to proteins related to β-ketoacyl-(acyl-carrier-protein) synthase I (FabB). Likewise, the recently biochemically analyzed ketosynthases CerJ7 and DarB22 belong to distinct clades. In the context of CorB it is notable that MyxB, the corresponding KS encoded in the myxopyronin biosynthetic gene cluster of Myxococcus fulvus,23 is not the closest homologue. This finding is surprising because of the high structural similarity of the α-pyrone antibiotics produced by the myxobacteria C. coralloides and M. fulvus. The core region of the corallopyronins and the myxopyronins is identical; solely the western chain (Fig. 1) exhibits some differences. In Cor, carbon C-24 is hydroxylated and the chain is elongated, comprising seven more carbon atoms than myxopyronin A. Even though CorB and MyxB are responsible for the chain interconnection prior to pyrone ring formation (this study and Sucipto et al., respectively), the phylogeny indicates that some minor differences exist between these enzymes. However, both enzymes harbor the Cys-His-Asn catalytic triad characteristic for FabH and type III PKSs instead of the Cys-His-His triad found in ketosynthases of type I PKSs. Twelve out of 15 ketosynthase (KS) and KS-like protein(domain)s encoded within the corallopyronin gene locus (Fig. S4†) possess this Cys-His-Asn triad.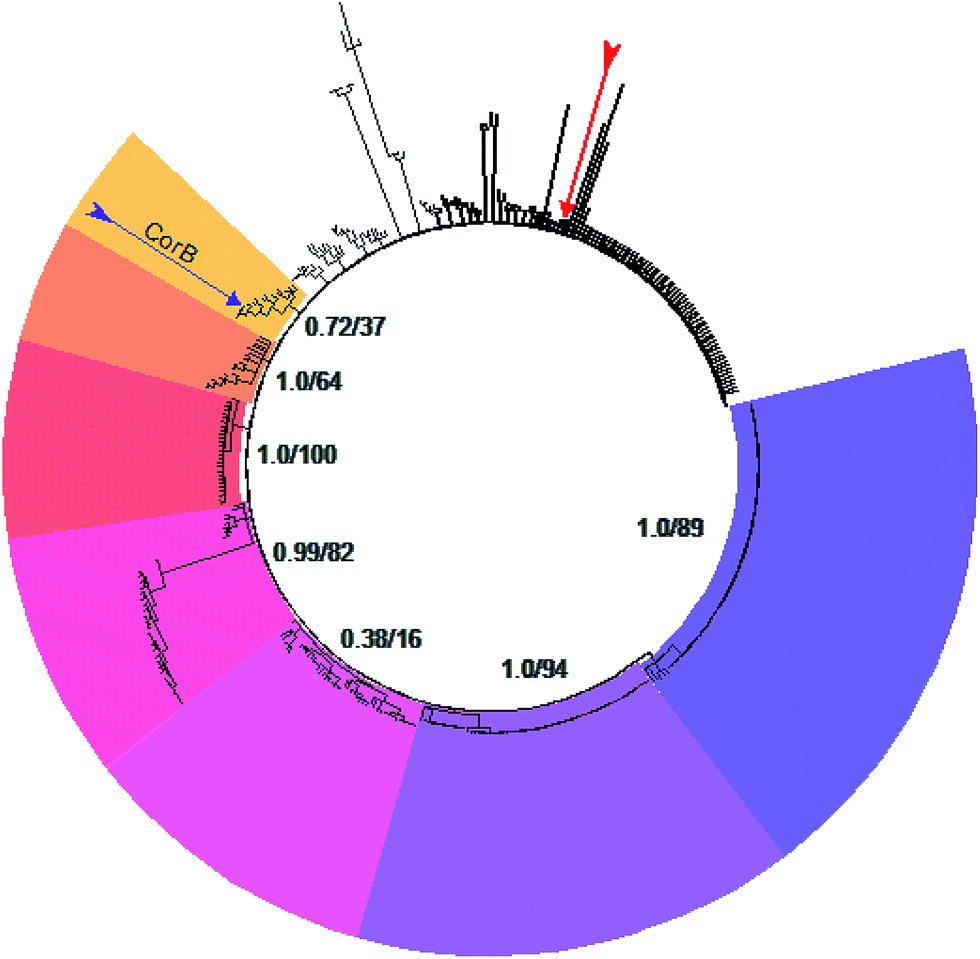 | ||
| Fig. 3 Consensus tree of selected ketosynthases. The different clades are coloured. CorB (marked by the purple arrow) is situated in the yellow clade. These proteins show highest homology to FabH. Starting in the yellow clade, the homology to FabB (dark purple clade) increases counterclockwise. The red arrow marks MyxB, the enzyme catalyzing the CorB-corresponding chain interconnection in myxopyronin biosynthesis. The support values of the respective clade (maximum likelihood and parsimony, respectively) are given. | ||
The structure of CorB
As CorB catalyzes the polyketide-interconnecting reaction, we sought to determine the structure of CorB to analyze the architecture of the active site and to gain insight into the determinants of specificity for the formation of the pyrone ring. We produced pure CorB and crystallized it for X-ray structure determination. We solved the structure of the native, dimeric protein to 1.6 Å resolution (Table S1†) by molecular replacement and confirmed that CorB assumes a typical thiolase fold (Fig. S5†).16 Additionally, we observed that the catalytic cysteine C121 was partially oxidized (CorB-ox) (Fig. S6a†). This oxidation would hamper the incorporation of our substrate mimic as it dramatically changes the charge distribution of the reactive sulfur atom, and therefore the nucleophilicity of the active cysteine. As attempts to reduce the catalytic cysteine were unsuccessful, we optimized protein production and crystallization to prevent oxidation. The resulting structure (CorB-red) did not show significant oxidation, as evidenced by the lack of additional electron density near C121 (Fig. S6B†), and we next derivatized CorB through addition of 1. The resulting structure (CorB-1) clearly showed positive electron density around the catalytic cysteine as a result of substrate binding (Fig. S6C†). Since the electron density was not clearly contoured, we confirmed the successful attachment of the substrate mimic by an ESI-mass experiment. The CorB sequence translates into a theoretical mass of 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 233.6 Da. The ESI mass spectrum of the wildtype protein (Fig. 2) results in an experimental mass of 37233.2 Da, thus confirming the integrity and identity of the CorB protein. After incubation of CorB with 1, we observed a mass increase of 244 Da by ESI-mass spectroscopy. This mass increase corresponds well with the total mass of the entire substrate 1 (theoretical mass 245 Da) (Fig. 1 and S7†), and it therefore shows that 1 attaches to CorB.
233.6 Da. The ESI mass spectrum of the wildtype protein (Fig. 2) results in an experimental mass of 37233.2 Da, thus confirming the integrity and identity of the CorB protein. After incubation of CorB with 1, we observed a mass increase of 244 Da by ESI-mass spectroscopy. This mass increase corresponds well with the total mass of the entire substrate 1 (theoretical mass 245 Da) (Fig. 1 and S7†), and it therefore shows that 1 attaches to CorB.
Structure comparison to other thiolase enzymes
As a member of the thiolase superfamily CorB features a typical thiolase fold characterized by two βαβα(β)αββ motifs. As described for CerJ,17 CorB lacks one β-strand (β8 for FabH from E. coli) in the C-term βαβα(β)αββ motif (Fig. 4 and S5†). Another feature of the dimeric CorB structure is a deep, widely accessible T-shaped channel that results mainly from a fairly open conformation of helix α4, loop β6α4, the residues connecting β3 with β4 (residues 152–166) and β1 with β2 (residues 27–46). As expected, a structural similarity search24 identified several FabH enzymes from different organisms as close structural homologs25–28 with Cα atom rms deviations of around 2.5 Å (Table S2†). The core fold of the thiolase is very well conserved among these proteins, and main differences occur in the part of the protein that is responsible for the T-shaped tunnel of the active site of CorB (see above). In addition to FabH enzymes, a structural comparison revealed several PKS enzymes29–33 (Table S3†) and 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) synthases as structurally homologous enzymes. Although the core motif is very well conserved and the quality of the CorB alignment is comparable between the two groups, the more open shape of CorB fits best to the HMG-CoA synthases. We conclude this from two observations. First, residues between β3β4 and β1β2 show an open conformation as found in HMG-CoA synthases, whereas the PKS and FabH enzymes exhibit a tighter active site as a result of a closed setup of the corresponding residues (Fig. S8†). Second, helix α5, which is tilted in CorB compared with all structurally related proteins, is also shifted towards the active site by approximately 2 Å, placing the tip of the helix (P266) at a similar position to that found in the HMG-CoA enzymes.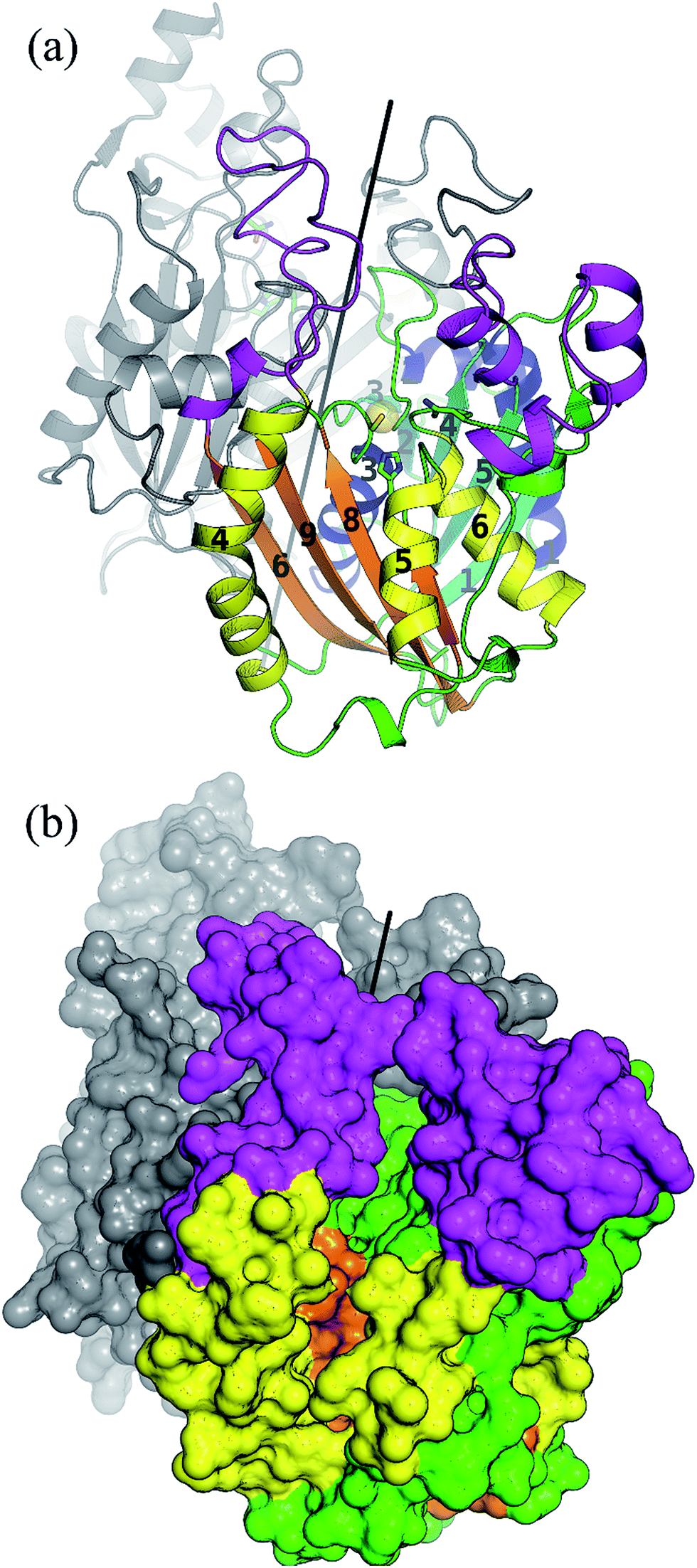 | ||
| Fig. 4 Overall structure of the dimeric CorB enzyme reveals a classical thiolase fold. Both characteristic βαβα(β)αββ motifs of this superfamily are coloured by their secondary structural elements in cyan and blue for the N-terminal motif and orange and yellow for the C-terminal motif. The regions exhibiting larger deviations to structural homologue enzymes are coloured in pink. The catalytic triad is represented as ball and sticks and the reactive sulfur atom of C121 is emphasized by a yellow sphere. The dimeric two-fold axis (parallel to the crystallographic two-fold axis) is shown in black. (b) Surface representation revealing a wide T-shaped active site cavity of CorB. | ||
Comparison of the catalytic triad
The active site of CorB contains a catalytic triad formed by C121, H264 and N294. These three residues are arranged in a clamp geometry that is reported to stabilize the carbon chain elongation process during fatty acid biosynthesis or polyketide formation.34–36 All FabH and PKS enzymes possess this catalytic triad. CerJ has to be excluded from the active site comparison as it features a sequential rather than a clamp-like catalytic triad observed in cysteine proteases.17 The structural similarity of the CorB active site especially to those of HMG-CoA synthases is remarkable and agrees well with the biochemical data available for PKS and FabH enzymes on the one hand and CorB and HMG-CoA enzymes on the other. The catalysis of FabH and PKS enzymes is accompanied by a decarboxylation step to form the nucleophilic carbon atom that is subsequently attached to the electrophilic carbonyl atom linked as a thioester to the catalytic cysteine residue. This decarboxylation step is not observed in the family of HMG-CoA.37–39 For HMG-CoA synthases, this is mainly because the third residue of the triad (Asn) is not involved in substrate stabilization. A closer look reveals the structural reason for the difference in the chemical reaction. In HMG-CoA synthases, helix α5 is shifted towards the active center by about 1.2 Å, and this helix carries a proline (P266) at its C-terminus. A search in the non-redundant UniProt database showed that this proline is strictly conserved in all HMG-CoA synthases but not found in FabH and PKS enzymes. The steric repulsion of the proline residue is the major reason for the separation of both clamp residues (His-Asn) of the catalytic triad, and thus a small but significant distance increase (Fig. 2b). A more detailed investigation comparing the distances of the triad residues (Fig. 5) in the FabH, PKS and HMG-CoA enzymes showed that the shortest mean distances are observed for the PKS and FabH enzymes. In the group of the HMG-CoA synthases, the mean distance of the asparagine to the triad members is increased by at least 1.1 Å. Such a change in the distance would substantially weaken a postulated hydrogen bond and therefore makes it plausible that the decarboxylation process is disfavoured by the loss of stabilization. Indeed, a hydrogen bond to the corresponding asparagine residue (N326) is not reported in the complex structure of the HMG-CoA synthase from Brassica juncea with acetyl-CoA.38 With a H264-(Cγ atom)-to-N292-(Cγ atom) distance of 8.8 Å, the residues of the catalytic triad of CorB-1 are even farther apart (Fig. 5). Although decarboxylation is chemically not expected due to the substrate specificity of CorB, the opened active site of CorB clearly correlates with the increased separation of the catalytic residues. | ||
| Fig. 5 Structural comparison of CorB with related enzymes. (a) Superposition of CorB with 15 non-redundant homologous enzymes. The protein chain is colored according to the mean Cα-rmsd deviation for this superposition, ranging from 0.3 Å (blue) to 4 Å (red). As expected larger deviations occur at the surface. The N-terminal βαβα(β)αββ motif is structurally more conserved compared to the C-terminal part, in good agreement with the dimeric structure of CorB that is formed by residues in the N-terminal region. The black line shows the two-fold axis that generates the biological CorB dimer. (b) The distances separating the catalytic triad residues of CorB are substantially larger compared with the decarboxylating enzymes FabH (grey) and type-III PKS enzymes (cyan), and they are more similar to the HMG-CoA synthases (violet). The Cα-atoms of the catalytic cysteines and the Cγ-atoms of the catalytic histidines or asparagines are shown as small spheres for the structural homologs. With a H264-(Cγ atom)-to-N292-(Cγ atom) distance of 8.8 Å (orange line), CorB features the largest His-Asn distance. | ||
To investigate the relevance of the members of the catalytic triad for catalysis, we performed site-directed mutagenesis of CorB, and investigated several single residue mutants (ESI†). These point mutations were introduced by two-step PCR with primers carrying the desired mutation, and the resulting constructs were verified by DNA sequencing. In particular, the three amino acids forming the catalytic triad C–H–N were mutated to C121A, C121S, H264A, H264F, and N292A. In all mutants, product formation was not observed using the established assay, in contrast to the wt enzyme. These results demonstrate that all three triad residues participate in the reaction catalyzed by CorB, and they in particular show that N292 is required for catalysis, in contrast to what has been reported for HMG-CoA synthase.37–39
Binding of the substrate mimic to CorB
The active site of CorB forms a T-shaped cavity. The anterior opening forms the entrance to the active site, which is loaded with CoA-linked acyl-entities. This is in good agreement with the available structural data of thiolase enzymes in complex with acetylated CoA.40,41 The catalytic cysteine C121 is found in the center of a deep cleft at the bottom of the cavity. Next to the catalytic cysteine, H264 and N292 are found on the lateral site. Opposed to them, the oxyanion hole is formed by the amide backbone of S324. Elongated channels can be found on both sides of the catalytic center. The proximal cavity is flanked by primarily hydrophobic residues (Fig. 6) and runs towards the two-fold dimeric interface. Residues 93 to 95 and 206 to 208 of the adjacent protomer contribute to the formation of this channel. In chalcone synthases and type II polyketide synthases, this channel harbors the so-called gatekeeper residues that are thought to control the chain length of the nascent polyketide chain.42–44 In HMG-CoA synthases, this channel is blocked by a set of rigid residues.38,39,45 The proximal cavity of CorB is also much larger compared to another reported 2-pyrone synthase.35 This is in good agreement with the reported biochemistry of this 2-pyrone synthase, which generates a rather small product, 6-methyl-4-hydroxy-2-pyrone, through the iterative condensation of malonyl entities. In addition to 2-pyrone synthases, the proximal cavity is also reported to accommodate substrates in PKS11 of Mycobacterium tuberculosis31 and the anthraniloyl-skeleton in PqsD.46 In contrast to PKS11 and PqsD, the cavity in CorB is substantially enlarged towards the dimeric interface. This differs compared to the anthranilate binding site in PsqD where the cavity is sealed in that direction by an intensive π–π-stacking of residues F205, R145 and F218. This site of the cavity is blocked by the bulky side chain of tryptophan W230 in PKS11. Interestingly, the architecture of the CorB cavity is closely related to that of FabH from Mycobacterium tuberculosis,41 and the binding pocket found for lauroyl-coenzyme A of this enzyme almost perfectly superposes with the cavity of CorB, suggesting that this pocket acts as a substrate channel for the interconnection reaction.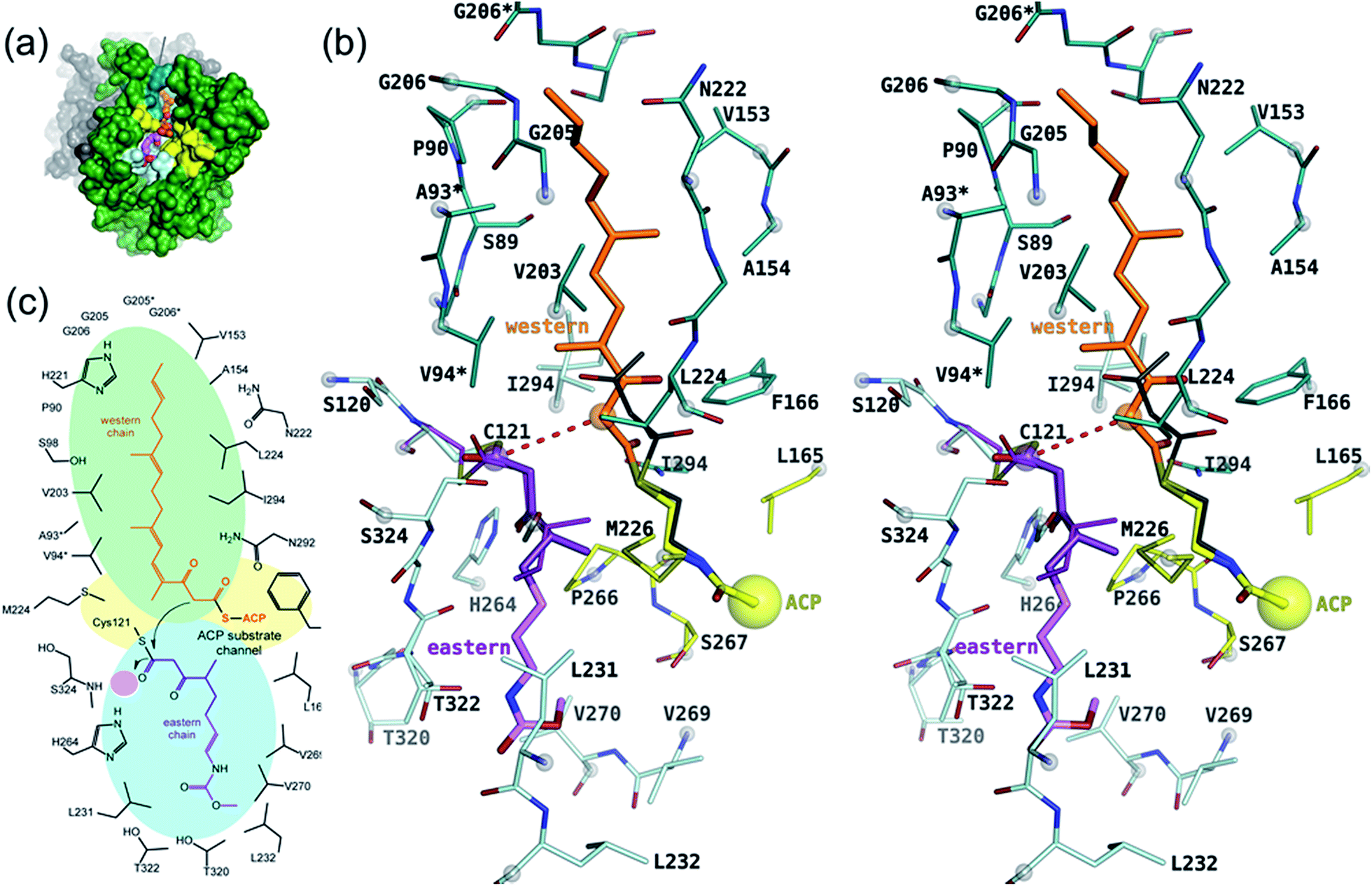 | ||
| Fig. 6 Proposed model of catalysis of CorB. (a) Overview of the binding site showing the substrates in a ball-and stick representation in the cavity of CorB. (b) Stereo representation of the binding situation. The substrate mimic 1 (dark violet) was placed into the active site on the basis of its unbiased (Fo − Fc)-difference electron density and the remaining portion of the eastern chain (light magenta) was modeled into the cavity. The western chain was modeled into the proximal cavity on the basis of a homologue 2-pyrone synthase using the pantotheine entity of CoA as an anchor point for the western chain. (c) Schematic view of (b). The oxyanion hole is highlighted by a pink circle. | ||
The electron density of derivatized CorB-1§ differs at the active site from that observed for CorB-ox and CorB-red. For CorB-ox we found that the catalytic cysteine is oxidized to cysteine-S-dioxide (Fig. S6A†). This is likely a result of crystal growth, which initially took several days and facilitated oxidation of the reactive sulfur atom of C121. Optimization of the purification and crystallization protocol then yielded crystals overnight. These crystals allowed us to obtain a structure that clearly shows a reduced cysteine at the active site (CorB-red). For CorB-1, which was also crystallized with the optimized protocol, we observed a substantial positive electron density at C121 as a result of substrate incubation prior to crystallization. We also attempted crystallization of CorB incubated with substrate 2 but unfortunately could not produce suitable crystals for structure determination. As the crystallization conditions are similar and both structures, CorB-1 and CorB-red, result from the same protein batch, we conclude that the positive density at C121 is due to substrate binding. Although all structures are virtually identical (Cα-rms deviation of 0.4 Å), the oxidation of the cysteine in CorB-ox led to drastic change in its electrochemical properties and hampered the covalent linkage to the substrate. Thus, we conclude that CorB-ox is inactive. Although the electron density for the substrate observed in CorB-1 is weak and ambiguous (Fig. S6C†), we confirmed the attachment of the substrates 1 and 2 by ESI-mass spectroscopy. The experiments were performed using the same protein batch and identical solutions, incubation times and temperatures but differ exclusively in the absence or presence of substrate 1 or 2. We observe a mass difference of 244 Da for 1 (theoretical expected mass increase: 245 Da) which corresponds well with the intact substrate (Fig. 2B and S9†). For 2, we observed a mass increase of 214 Da, which corresponds well with the theoretical expected mass increase of 214 Da of the covalently linked substrate lacking the SNAC entity (Fig. 2B and S9†). In addition to the electron density around the catalytic cysteine, no extra density features were found either in CorB-ox or CorB-red. We therefore conclude that the mass increase of CorB-1 results from the attachment of substrate 1 to C121. The exact nature of the bound substrate could not be unequivocally determined either by ESI-mass spectroscopy or by X-ray analysis. As the electron density is weak, it does not allow for proper ligand building and refinement of the substrate (Fig. S6D†). Nevertheless, the electron density is sufficiently clear to use it as a basis for modelling of substrate 1 into the CorB cavity. Several factors likely contribute to the observed weak electron density. It is possible that the active site is not fully occupied, and the substrate could also have high temperature factors. Moreover the additional state observed by ESI-MS would picture the electron density as an overlay of both states yielding a lower quality.
With respect to the difference in electron density, we placed 1 into the cavity of CorB in two slightly different conformations (Fig. S6C†). The distal channel is formed by a set of mostly hydrophobic residues (L231, L232, V269, V270 and T320). The substrate model would suggest a hydrogen bond to H264. This is in good agreement with our mutation studies, which show that a replacement of H264 with either alanine or phenylalanine would result in a loss of activity. The binding direction of 1 is comparable to the situation found in a eukaryotic HMG-CoA synthase covalently attached to an irreversible inhibitor,38 thus supporting the mode of binding of the eastern chain.
Proposed catalytic mechanism
As a ternary complex of CorB with both substrates (eastern and western chain) could not be generated, we modelled the second polyketide (western chain) into the active site of CorB-1. For this, we compared several enzymes of the thiolase family in complex with acyl-CoA molecules.35,40,41 We superposed the CoA recognition site of another 2-pyrone synthase35 (2-PS) and FabH from Mycobacterium tuberculosis41 with the corresponding residues in CorB (residues 44–47 and 161–166) and found that the acyl moiety would point into the proximal cavity (Fig. 6). On the basis of this structural comparison, we placed a model of the western chain into the proximal cavity of CorB using the CoA PPant entity of the structure relative as the anchor point for the western chain. The western chain would fit into the cavity without steric clashes.This model of substrate binding is in good agreement with the observed biochemistry. In our model, the eastern chain is covalently linked to the catalytic cysteine in the first step of catalysis and occupies the distal channel of CorB. The covalent attachment to the catalytic cysteine activates the eastern chain. In the second step, the western chain is placed into the proximal cavity, orienting the α-carbon in a position suitable for the nucleophilic attack by the activated eastern chain. The nucleophilicity of the western chain might be enhanced by an interaction with N292, which was determined to be crucial for catalysis. The resulting intermediate is stabilized by the oxyanion hole formed by the backbone amide of S234, and the polyketide connection reaction proceeds with the release of the catalytic cysteine. We suggest that the lactonization process is a spontaneous reaction that takes place in solution after dissipation as the lacton formation is accompanied with sterical requirements that are unlikely to take place in the active site of CorB.
Our insights into Cor biosynthesis will facilitate future derivatization approaches of α-pyrone antibiotics using combinatorial biosynthesis. Usually, modifications of an original natural product are advantageous in terms of pharmacokinetics. However, Cor and its derivatives represent difficult-to-synthesize compounds since chemical synthesis is not feasible on a commercial scale.47 Now, in silico and in vitro experiments become possible to determine the substrate flexibility of CorB. This forms a promising template for the rational design of optimized producer strains for the further development of the compound, e.g. as anthelmintic.
Experimental
Cultivation of strains
B035 was cultivated in casitone medium (MD1 medium, supplemented with 0.2% glucose). MD1 medium consists of casitone 3 g L−1, CaCl2 × 2H2O 0.7 g L−1, MgSO4 × 7H2O 2 g L−1. The flasks were agitated on a rotary shaker (140 rpm) at 30 °C for 7–14 days. E. coli strains were cultivated in LB medium (tryptone 10 g, yeast extract 5 g, NaCl 10 g, filled with H2O to 1 L; for solid medium 1.6% agar was added) supplemented with 100 μg mL−1 ampicillin (Amp) for selection and to maintain the constructs.Molecular-biological methods
Molecular-biological work was performed according to standard procedures. Polymerase chain reactions (PCRs) were performed with oligonucleotides obtained from eurofins genomics and are listed in Table S4.† DNA isolation from agarose gels was performed using either the Wizard® SV Gel and PCR Clean-Up System (Promega), or the QIAquick Gel Extraction Kit (Qiagen). Plasmid isolation was performed using either the PureYield™ Plasmid Miniprep System kit (Promega), or the QIAprep Spin Miniprep Kit (Qiagen).DNA manipulation
Either genomic DNA or plasmid DNA served as template for the amplification of DNA fragments. Genomic DNA isolation of C. coralloides B035 is described in Erol et al., 2010.4 In order to obtain the expression construct for CorB a fragment of 1012 bp was amplified using the primer pair CorB_TOPO_up and CorB_TOPO_dn. This fragment was introduced into the vector pET151TOPO by directional TOPO® cloning of the blunt-ended PCR products. Then, the resulting vector was transferred in E. coli One Shot®TOP10 cells by transformation. The resulting plasmid was isolated and checked for identity by sequencing. Plasmid with the desired sequence was transferred into competent cells of the expression host E. coli BL21 (DE3) Star by transformation. Subsequently, cells were plated on LB agar supplemented with ampicillin for selection.Site-directed mutagenesis
To introduce point mutations into the enzyme, a two-step PCR was applied. Two primers were designed carrying the point mutation. In a first round the point mutation was introduced into the two amplified fragments using (i) the forward primer CorB_TOPO_up and the reverse primer carrying the sequence coding for the amino acid exchange, and (ii) the forward primer carrying the sequence coding for the amino acid exchange and the reverse primer CorB_TOPO_dn (Table S4†). The two fragments obtained by the first round of PCRs served as templates for the second PCR round. In this second PCR, first 10 cycles were performed without primers in the sample, then the primers CorB_TOPO_up and CorB_TOPO_dn were added, and 20 further cycles were performed. The fragments obtained by the second round of PCR were introduced into pET151TOPO vector by directional TOPO® cloning. Then, the resulting vector was transferred in E. coli One Shot®TOP10 cells by transformation. The resulting plasmid was isolated and checked for the presence of the insert by either PCR, or restriction. The identity of positive plasmids was verified by sequencing. Plasmid with the desired sequence was transferred to competent cells of the expression host E. coli BL21 (DE3) Star by transformation. Subsequently, cells were plated on LB agar supplemented with ampicillin for selection.Protein production
For the expression of the proteins, the corresponding E. coli strains carrying the desired construct were inoculated in 9 mL LBAmp liquid medium and incubated overnight at 37 °C. With this preculture, the main culture was inoculated in LB medium (1 L) at 37 °C until an OD600 of 0.4–0.7 was reached. Then the cells were cooled down to 16 °C and protein expression was induced by adding IPTG (final concentration 0.05–0.1 mM). Growth was then continued at 16 °C overnight. Cells were harvested by centrifugation, resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0), and transferred to 50 mL Falcon tubes. The cells were then disrupted by sonification (6 cycles à 10 pulses, and cooling on ice during the cycles). After sonification cell debris was removed by centrifugation for 30–45 min at 8500 rpm at 4 °C. The protein-containing supernatant was transferred onto gravity flow columns (Qiagen, Germany), which were packed with 1 mL Ni-NTA agarose. The flow-through was collected and again loaded onto the columns 3 more times. Then 2 washing steps followed (washing buffer 1: 50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0; washing buffer 2: 50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0). Bound protein was finally eluted with 5 × 500 μL elution buffer (50 mM NaH2PO4, 300 mM NaCl, pH 8.0, stepwise increasing imidazole concentration 1 × 100 mM, 1 × 150 mM, 1 × 200 mM, 2 × 300 mM). Fractions containing the targeted protein were combined and loaded onto a spin filter column (Millipore, 10 kDa exclusion size) for desalting. This column was centrifuged at 5000 rpm at 4 °C until only one tenth of the starting volume remained. Then the protein was re-buffered in either Tris buffer (50 mM Tris, pH 8.0), or Tris buffer containing DTT and glycerol (20 mM Tris, 1 mM DTT, and 5% glycerol, pH 7.5).The protein production for crystallization experiments was essentially similar but instead E. coli Rosetta 2 cells were used for protein expression at 20 °C. The cell pellet was resuspended in His-A buffer (20 mM TrisHCl (pH 8.8), 0.5 M NaCl, 10 mM imidazole and 2 mM DTT). After further centrifugation (18000 rpm, 1 h, 4 °C) the supernatant was loaded onto a Ni2+-affinity column (5 mL HisTrapFF GE Healthcare™) in a cyclic manner. The column was connected to an Äkta purifier (GE Healthcare™) and proteins were eluted by applying a step gradient of His-B buffer (20 mM TrisHCl (pH 8.8), 0.5 M NaCl, 500 mM imidazole and 2 mM DTT). Fractions containing CorB were pooled and dialyzed against SEC buffer (20 mM TrisHCl (pH 8.8), 150 mM NaCl) for 18 h at 4 °C. After buffer exchange, TEV protease was added at a 1/10 mass ratio (m/m). The reaction was placed on a merry-go-round for 3 days at 4 °C. Protein digestion was analyzed using SDS-PAGE and the sample was applied onto a Superdex200 16/60 (GE Healthcare™). Fractions containing untagged CorB were pooled, concentrated to 3 mg mL−1 and used for crystallographic experiments or flash-frozen in liquid nitrogen for storage at −80 °C. To obtain reduced CorB all reaction buffers contained reducing agent (TCEP, 5 mM) beside His-A and His-B buffer.
Synthesis of the substrate analogues
All reagents were obtained from commercial suppliers (Sigma-Aldrich and Acros Organics) and were used without further purification. Reactions were performed under an inert atmosphere (Argon) by using the Schlenk technique. All solvents were obtained in HPLC grade and were dried prior to use by the following procedures: dichloromethane (DCM) was distilled from a calcium hydride suspension. Methanol and chloroform were distilled prior to use. For thin-layer chromatography TLC aluminum sheets silica gel 60 F254 (MERCK) were used. Open column chromatography was performed on silica gel 60; particle size 0.015–0.04 mm (Macherey-Nagel).Synthesis of S-(2-acetamidoethyl)4-methyl-3-oxohexanethioate
A solution of 2-methylbutanoic acid (200 mg, 2 mmol) in dry dichloromethane (10 mL) was cooled to −20 °C under argon. Meldrum's acid (290 mg, 2 mmol), DMAP (240 mg, 2 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (400 mg, 2 mmol) were added slowly and the temperature was kept at −20 °C for one hour. After the solution was slowly allowed to warm to room temperature, stirring was continued for an additional two hours. Water (20 mL) was added and the aqueous phase was extracted with dichloromethane (2 × 20 mL). The combined organic fractions were dried with Na2SO4 and concentrated under reduced pressure to give a yellow oil. The crude product and N-acetylcysteamine (240 mg, 2 mmol) were dissolved in toluene (20 mL) and heated under reflux for two hours. The solvent was removed under reduced pressure and the residue was purified by column chromatography over silica gel (chloroform) to give 350 mg of a clear oil. The title compound exist as a keto:enol mixture (3:2) in CDCl3. Yield: 71%. 1H NMR (300 MHz, CDCl3): δ 0.86 (3H, t, 3J = 7.4 Hz), 0.87 (3H, t, 3J = 7.4 Hz), 1.07 (3H, d, 3J = 6.8 Hz), 1.10 (3H, d 3J = 6.8 Hz), 1.33–1.75 (4H, m), 1.94 (3H, s), 1.94 (3H, s), 2.08 (1H, ddq, 3J1 = 6.9 Hz, 3J2 = 6.8 Hz, 3J3 = 6.8 Hz, enol-form), 2.53 (1H, ddq, 3J1 = 6.9 Hz, 3J2 = 6.8 Hz, 3J3 = 6.8 Hz, keto-form), 3.02–3.09 (4H, m), 3.38–3.48 (4H, m), 3.70 (2H, s, keto), 5.42 (1H, s, enol), 6.11 (2H, bs), 12.60 (1H, s, enol-OH) 13C NMR (75 MHz, CDCl3): δ 11.4, 11.7, 15.3, 17.5, 23.1, 23.1, 25.5, 27.1, 27.8, 29.1, 39.2, 39.9, 41.0, 48.4, 55.4, 98.2, 170.4, 170.6, 181.2, 192.4, 194.4, 206.0 ppm, IR (film): 997, 1085, 1190, 1286, 1544, 1610, 1655, 1718, 2876, 2968, 3288 cm−1, HRMS: (ESI+): m/z calculated for C11H20O3NS: 246.1158, found 246.1156 [M + H]+.
CorB in vitro assay
The purified protein was used for the activity assay. A typical assay (final volume 100 μL) consisted of 1 μL of each substrate (concentration of 2: 30 mM; concentration of 1: 50 mM) and 0.28 mg protein. The assay was filled up to the final volume by adding the reaction buffer. The latter consisted of 20 mM Tris, 1 mM DTT, and 5% glycerol (pH 7.5). Negative controls were performed in the same way by omitting protein. Incubation was performed at room temperature. The assay was stopped by adding 200 μL of ethyl acetate. The samples were agitated vigorously, and the following phase separation was reached by 2 min centrifugation at full speed in a table top centrifuge. The organic phase was transferred in a new vial, and subsequently dried under nitrogen flow.UPLC-MS analysis
Samples were dissolved in 20 μL 50% acetonitrile (ACN), 0.1% trifluoroacetic acid (TFA), and 49.9% H2O. 1.5 μL solution were injected onto a C18 trap column (20 mm length, 100 μm inner diameter, ReproSil-Pur 120 C18-AQ, 5 μm, Dr Maisch GmbH, Ammerbuch-Entringen, Germany) made in-house. Solvent A was 0.1% formic acid (FA), solvent B 90% ACN, 0.1% FA. The analytical column was equilibrated with 5% solvent B. The trap column was equilibrated with 1% ACN, 0.1% TFA, 98.9% H2O. Bound peptides on the trap column were eluted onto a C18 analytical column (150 mm length, 75 μm inner diameter, ReproSil-Pur 120 C18-AQ, 1.9 μm). Peptides were separated during a linear gradient of 15% to 80% solvent B within 19 min at 350 nL min−1. The nanoHPLC was coupled online to an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Ions between 190 and 600 m/z were scanned in the orbitrap detector with a resolution of 30000 (maximum fill time 400 ms, AGC target 106, lock mass 445.12003 m/z). Data analysis was performed using the Xcalibur™ Software (Thermo Scientific). The isolation width for the extracted ion chromatograms was <50 ppm.
ESI-MS analysis of derivatized CorB
To elucidate the binding of 1 to CorB, electrospray ionization mass spectrometry (ESI-MS) was performed on a Synapt G2 mass spectrometer (Waters). Experiments were conducted with CorB and CorB incubated with a 10× molar excess of 1. Prior to measurements, buffers were replaced with Milli-Q H2O by cycles of concentration and adding Milli-Q H2O. Measurement of the samples was conducted by direct infusion in ESI positive mode with a capillary voltage of 3 kV, sampling cone of 30 and extraction cone of 3.0. The instrument was operated at a resolution of 20000, a scan range from 50 to 2000 Da and a scan time of 0.5 s. Spectra of each infusion were combined.
Protein crystallization
Commercial screens were used to screen for crystallization conditions. All screens were set up at 20 °C and 4 °C in 96 well plates using the sitting drop vapor diffusion method. Based on initial hits fine screens were prepared at 20 °C using the hanging drop vapor diffusion method (1 μL × 1 μL drop size and 490 μL reservoir volume). Best crystals grew overnight in crystallization buffer (0.1 M sodium cacodylate (pH 6.5), 8% (w/v) PEG8000 and 34% (v/v) MPD). These crystals were harvested after 12–16 h and used for the structure determination of CorB-red. To obtain the structure of CorB-1, we incubated the identical protein batch overnight with 1 and performed crystallization immediately.Structure determination and refinement
Data sets were collected at the in-house X-ray source at 100 K using 0.5° rotation and 300 s exposure time per image. All crystals were grown in conditions that included precipitant concentrations in the cryoprotectant range (+30% (v/v) MPD. Data were reduced using the X-ray Detector Software (XDS) program package.48,49 A sequence analysis using HHPRED50 revealed the homology of CorB to several proteins of the thiolase superfamily to which CorB shows a sequence identity of 21% to 33%. Initial phases were obtained by molecular replacement procedure using PHASER51 and a manually adjusted model of PqsD46 (pdb-code: 3H78). Several cycles of reciprocal space rigid body refinement using REFMAC552 and real space rigid body refinement using COOT53 improved the phase information and established an initial model of CorB. This model was improved through iterative cycles of refinement including simulated annealing and TLS parameterization54 using PHENIX55 and REFMAC5, and rebuilding of the structure with COOT. Geometric parameters were validated using Molprobity.56 Figures were generated and rendered using PYMOL.57
Phylogenetic analysis
A set of homologous ketosynthases was submitted to phylogenetic reconstruction. This set comprised the amino acid sequences of CorB, the homologous MyxB, and previously studied ketosynthases,17 as well as close homologs (Bit-Score ≥ 800) obtained from sequence similarity searches using BLAT58 (v34) in prokaryotic genomes available from NCBI. On this set of homologs we used MAFFT59 (v6.851b) to generate a multiple sequence alignment (MSA) based on the chemical properties of the amino acid residues. Subsequently both a Maximum Likelihood (ML), with FastTree20 (v2.1.7), as well as a Maximum Parsimony (P), with phangorn21 (v1.99.11), phylogenetic trees were computed. The latter parsimonious approach obtained local confidence values using 100 bootstrapped versions of the MSA. Subsequently, using ape60 (v3.2), from both phylogenies a strict consensus tree was inferred. Based on this consensus tree, in a final step, we computed a tree with branch lengths applying the accelerated transformation (ACCTRAN) algorithm.Conclusions
In conclusion, we found that the interconnecting PKS CorB possesses a typical thiolase fold, and we showed by site-directed mutagenesis that all members of the catalytic triad (C121, H264, N292) are required for catalysis. The catalytic triad residues of CorB are more distant from each other compared with structurally related enzymes that facilitate a decarboxylation reaction, and this separation is linked to a shift in helix α5 that is similar to that seen in HMG-CoA synthases. Interestingly, both CorB and HMG-CoA synthases have a wider active site compared with FabH and PKS. In CorB the active site is accessible from two directions, giving rise to a wide, T-shaped cavity. We could show that a substrate mimic occupies one side of the channel and we suggest that the remarkable wide T-shaped binding site of CorB is the key feature to accommodate two polyketide chains during catalysis to interconnect the substrates by a head-to-head Claisen-like condensation reaction.The ability of CorB to catalyze chain fusions makes it a promising tool for semi-synthetic approaches, and opens up the possibility to use the enzyme as a tool in synthetic biology approaches. Future experiments will reveal the complete substrate flexibility of this pyrone-forming enzyme.61
Acknowledgements
Financial support came from the German Centre for Infection Research (DZIF), and from the German Research Foundation (Collaborative Research Center 766 and Research Unit FOR854). We gratefully acknowledge J. H. Sahner and R. Müller for supporting us with substrate 2. We thank students C. Kielkopf, A.-S. Ramms, D. Stehle, B. Marx, J. B. Heim, and M. Oelker for their contributions to protein purification and their crystallization trials, and M. Sylvester for MS measurements.Notes and references
- I. Chopra, L. Hesse and A. J. O'Neill, J. Appl. Microbiol., 2002, 92, 4S–15S CrossRef PubMed.
- C. Hertweck, Angew. Chem., Int. Ed. Engl., 2009, 48, 4688–4716 CrossRef CAS PubMed.
- J. W. Li and J. C. Vederas, Science, 2009, 325, 161–165 CrossRef PubMed.
- O. Erol, T. F. Schaberle, A. Schmitz, S. Rachid, C. Gurgui, M. El Omari, F. Lohr, S. Kehraus, J. Piel, R. Muller and G. M. Konig, ChemBioChem, 2010, 11, 1253–1265 CrossRef CAS PubMed.
- A. Schiefer, A. Schmitz, T. F. Schaberle, S. Specht, C. Lammer, K. L. Johnston, D. G. Vassylyev, G. M. Konig, A. Hoerauf and K. Pfarr, J. Infect. Dis., 2012, 206, 249–257 CrossRef CAS PubMed.
- J. Mukhopadhyay, K. Das, S. Ismail, D. Koppstein, M. Jang, B. Hudson, S. Sarafianos, S. Tuske, J. Patel, R. Jansen, H. Irschik, E. Arnold and R. H. Ebright, Cell, 2008, 135, 295–307 CrossRef CAS PubMed.
- K. Mariner, M. McPhillie, R. Trowbridge, C. Smith, A. J. O'Neill, C. W. Fishwick and I. Chopra, Antimicrob. Agents Chemother., 2011, 55, 2413–2416 CrossRef CAS PubMed.
- H. Irschik, R. Jansen, G. Hofle, K. Gerth and H. Reichenbach, J. Antibiot., 1985, 38, 145–152 CrossRef CAS PubMed.
- T. F. Schaberle, A. Schiefer, A. Schmitz, G. M. Konig, A. Hoerauf and K. Pfarr, Int. J. Med. Microbiol., 2014, 304, 72–78 CrossRef CAS PubMed.
- S. Brand, D. Holscher, A. Schierhorn, A. Svatos, J. Schroder and B. Schneider, Planta, 2006, 224, 413–428 CrossRef CAS PubMed.
- Y. Katsuyama, M. Matsuzawa, N. Funa and S. Horinouchi, J. Biol. Chem., 2007, 282, 37702–37709 CrossRef CAS PubMed.
- B. Shen, Curr. Opin. Chem. Biol., 2003, 7, 285–295 CrossRef CAS PubMed.
- S. J. Wakil, J. K. Stoops and V. C. Joshi, Annu. Rev. Biochem., 1983, 52, 537–579 CrossRef CAS PubMed.
- B. Middleton, Biochem. J., 1972, 126, 35–47 CrossRef CAS PubMed.
- A. M. Haapalainen, G. Merilainen and R. K. Wierenga, Trends Biochem. Sci., 2006, 31, 64–71 CrossRef CAS PubMed.
- C. Davies, R. J. Heath, S. W. White and C. O. Rock, Structure, 2000, 8, 185–195 CrossRef CAS PubMed.
- T. Bretschneider, G. Zocher, M. Unger, K. Scherlach, T. Stehle and C. Hertweck, Nat. Chem. Biol., 2012, 8, 154–161 CrossRef CAS PubMed.
- Q. L. He, X. Y. Jia, M. C. Tang, Z. H. Tian, G. L. Tang and W. Liu, ChemBioChem, 2009, 10, 813–819 CrossRef CAS PubMed.
- H. Sucipto, J. H. Sahner, E. Prusov, S. C. Wenzel, R. W. Hartmann, J. Koehnke and R. Muller, Chem. Sci., 2015, 6, 5076–5085 RSC.
- M. N. Price, P. S. Dehal and A. P. Arkin, Mol. Biol. Evol., 2009, 26, 1641–1650 CrossRef CAS PubMed.
- K. P. Schliep, Bioinformatics, 2011, 27, 592–593 CrossRef CAS PubMed.
- S. W. Fuchs, K. A. Bozhuyuk, D. Kresovic, F. Grundmann, V. Dill, A. O. Brachmann, N. R. Waterfield and H. B. Bode, Angew. Chem., Int. Ed. Engl., 2013, 52, 4108–4112 CrossRef CAS PubMed.
- H. Sucipto, S. C. Wenzel and R. Muller, ChemBioChem, 2013, 14, 1581–1589 CrossRef CAS PubMed.
- L. Holm and C. Sander, Trends Biochem. Sci., 1995, 20, 478–480 CrossRef CAS PubMed.
- K. S. Gajiwala, S. Margosiak, J. Lu, J. Cortez, Y. Su, Z. Nie and K. Appelt, FEBS Lett., 2009, 583, 2939–2946 CrossRef CAS PubMed.
- S. D. Pegan, K. Rukseree, G. C. Capodagli, E. A. Baker, O. Krasnykh, S. G. Franzblau and A. D. Mesecar, Biochemistry, 2013, 52, 912–925 CrossRef CAS PubMed.
- X. Qiu, A. E. Choudhry, C. A. Janson, M. Grooms, R. A. Daines, J. T. Lonsdale and S. S. Khandekar, Protein Sci., 2005, 14, 2087–2094 CrossRef CAS PubMed.
- X. Qiu, C. A. Janson, W. W. Smith, M. Head, J. Lonsdale and A. K. Konstantinidis, J. Mol. Biol., 2001, 307, 341–356 CrossRef CAS PubMed.
- M. B. Austin, M. E. Bowman, J. L. Ferrer, J. Schroder and J. P. Noel, Chem. Biol., 2004, 11, 1179–1194 CrossRef CAS PubMed.
- M. B. Austin, T. Saito, M. E. Bowman, S. Haydock, A. Kato, B. S. Moore, R. R. Kay and J. P. Noel, Nat. Chem. Biol., 2006, 2, 494–502 CrossRef CAS PubMed.
- K. Gokulan, S. E. O'Leary, W. K. Russell, D. H. Russell, M. Lalgondar, T. P. Begley, T. R. Ioerger and J. C. Sacchettini, J. Biol. Chem., 2013, 288, 16484–16494 CrossRef CAS PubMed.
- H. Morita, S. Kondo, S. Oguro, H. Noguchi, S. Sugio, I. Abe and T. Kohno, Chem. Biol., 2007, 14, 359–369 CrossRef CAS PubMed.
- S. B. Rubin-Pitel, H. Zhang, T. Vu, J. S. Brunzelle, H. Zhao and S. K. Nair, Chem. Biol., 2008, 15, 1079–1090 CrossRef CAS PubMed.
- M. He, M. Varoglu and D. H. Sherman, J. Bacteriol., 2000, 182, 2619–2623 CrossRef CAS PubMed.
- J. M. Jez, M. B. Austin, J. Ferrer, M. E. Bowman, J. Schroder and J. P. Noel, Chem. Biol., 2000, 7, 919–930 CrossRef CAS PubMed.
- J. G. Olsen, A. Kadziola, P. von Wettstein-Knowles, M. Siggaard-Andersen, Y. Lindquist and S. Larsen, FEBS Lett., 1999, 460, 46–52 CrossRef CAS PubMed.
- N. Campobasso, M. Patel, I. E. Wilding, H. Kallender, M. Rosenberg and M. N. Gwynn, J. Biol. Chem., 2004, 279, 44883–44888 CrossRef CAS PubMed.
- F. Pojer, J. L. Ferrer, S. B. Richard, D. A. Nagegowda, M. L. Chye, T. J. Bach and J. P. Noel, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11491–11496 CrossRef CAS PubMed.
- N. Shafqat, A. Turnbull, J. Zschocke, U. Oppermann and W. W. Yue, J. Mol. Biol., 2010, 398, 497–506 CrossRef CAS PubMed.
- M. M. Alhamadsheh, F. Musayev, A. A. Komissarov, S. Sachdeva, H. T. Wright, N. Scarsdale, G. Florova and K. A. Reynolds, Chem. Biol., 2007, 14, 513–524 CrossRef CAS PubMed.
- F. Musayev, S. Sachdeva, J. N. Scarsdale, K. A. Reynolds and H. T. Wright, J. Mol. Biol., 2005, 346, 1313–1321 CrossRef CAS PubMed.
- M. B. Austin and J. P. Noel, Nat. Prod. Rep., 2003, 20, 79–110 RSC.
- J. M. Jez, M. E. Bowman and J. P. Noel, Biochemistry, 2001, 40, 14829–14838 CrossRef CAS PubMed.
- A. T. Keatinge-Clay, D. A. Maltby, K. F. Medzihradszky, C. Khosla and R. M. Stroud, Nat. Struct. Mol. Biol., 2004, 11, 888–893 Search PubMed.
- C. N. Steussy, A. A. Vartia, J. W. Burgner 2nd, A. Sutherlin, V. W. Rodwell and C. V. Stauffacher, Biochemistry, 2005, 44, 14256–14267 CrossRef CAS PubMed.
- A. K. Bera, V. Atanasova, H. Robinson, E. Eisenstein, J. P. Coleman, E. C. Pesci and J. F. Parsons, Biochemistry, 2009, 48, 8644–8655 CrossRef CAS PubMed.
- A. Rentsch and M. Kalesse, Angew. Chem., Int. Ed. Engl., 2012, 51, 11381–11384 CrossRef CAS PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 133–144 CrossRef CAS PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- M. Remmert, A. Biegert, A. Hauser and J. Soding, Nat. Methods, 2012, 9, 173–175 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- M. D. Winn, M. N. Isupov and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 122–133 CrossRef CAS.
- P. D. Adams, P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L. W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS PubMed.
- V. B. Chen, W. B. Arendall 3rd, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12–21 CrossRef CAS PubMed.
- Schrodinger, LLC, unpublished work.
- W. J. Kent, Genome Res., 2002, 12, 656–664 CrossRef CAS PubMed.
- K. Katoh, K. Misawa, K. Kuma and T. Miyata, Nucleic Acids Res., 2002, 30, 3059–3066 CrossRef CAS PubMed.
- E. Paradis, J. Claude and K. Strimmer, Bioinformatics, 2004, 20, 289–290 CrossRef CAS PubMed.
- C. Hertweck, Trends Biochem. Sci., 2015, 40, 189–199 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Result of the feeding experiments, synthesis of 1 including 1H NMR and 13C NMR, sequence analysis, X-ray data statistics, topology plot, structure comparison, density representation, primer sequences and strains and plasmids. See DOI: 10.1039/c5sc02488a |
| ‡ These authors contributed equally to the work. |
| § Structure factor amplitudes and atomic coordinates were deposited in the Protein Data Bank with entry code 4YUF for CorB-ox and 4YUC for CorB-1 and 5C1J for CorB-red, respectively. |
| This journal is © The Royal Society of Chemistry 2015 |